
Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis


Abstract
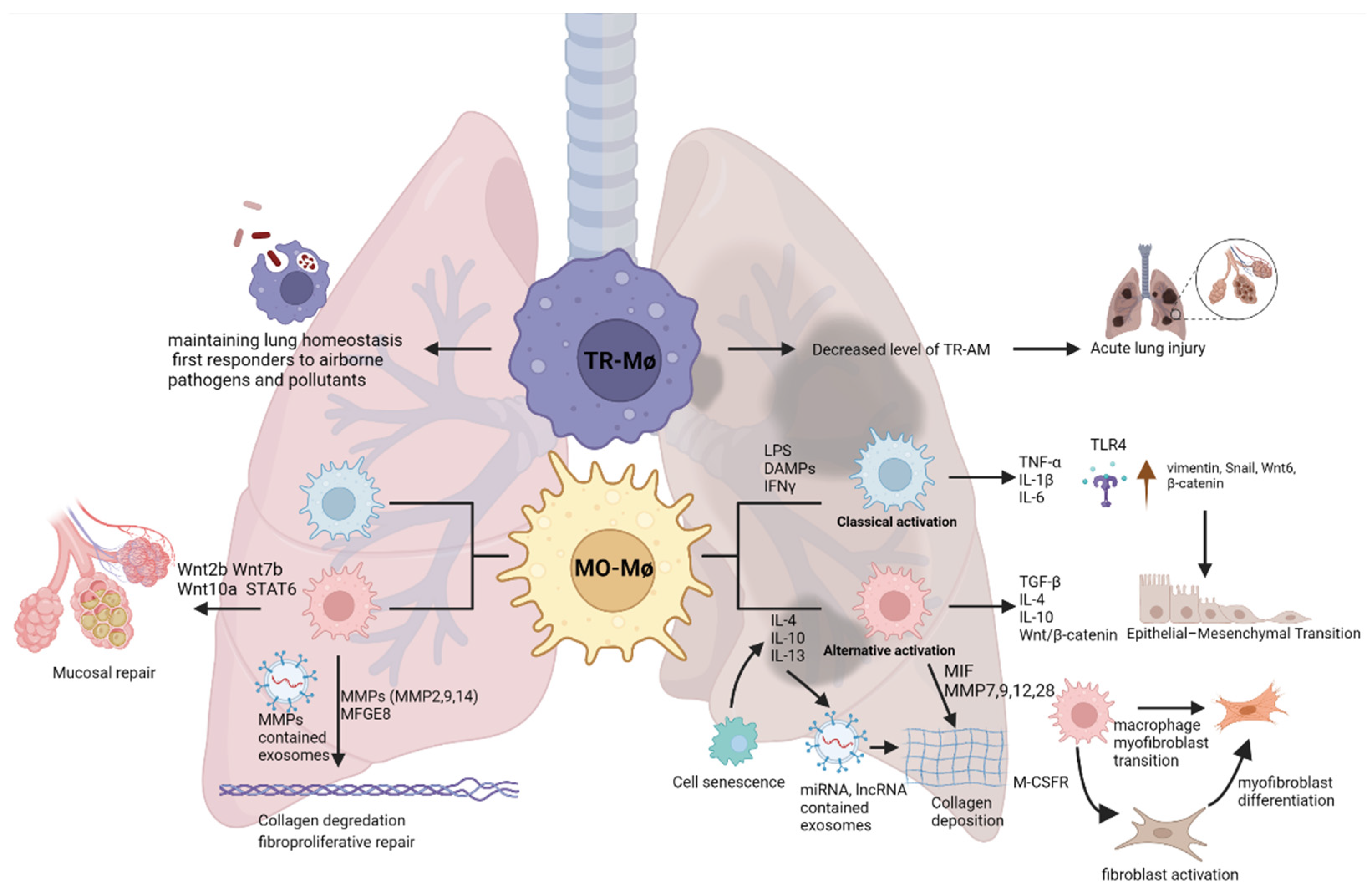
1. Introduction
2. Development of Pharmacological Treatment for IPF
3. Macrophages in Fibrosis
3.1. Cytokines, TGF-β and Wnt/β-Catenin Signaling
3.2. Role of Macrophages in Fibrogenesis, Myofibroblasts Differentiation and Epithelial–Mesenchymal Transition
3.3. Role of Macrophages on ECM
3.4. Macrophage-Derived Extracellular Vesicles
3.5. Cell Senescence and Genetic Factors
4. Macrophages as a Target
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMs | alveolar macrophages |
| ARDS | acute respiratory distress syndrome |
| ATII | alveolar epithelial cell type 2 |
| BAL | bronchoalveolar lavage |
| CCR8 | chemokine receptor 8 |
| CDH11 | cadherin-11 |
| CSF1 | colony-stimulating factor 1 |
| CTGF | connective tissue growth factor |
| DAMPs | damage-associated molecular patterns |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| EVs | exosomes and extracellular vesicles |
| FGF-1 | fibroblast growth factor receptor-1 |
| IFIGENIA | controlled Idiopathic Pulmonary Fibrosis International Group Exploring N-Acetylcysteine I Annual |
| ILD | interstitial lung disease |
| IMs | interstitial macrophages |
| IPF | idiopathic pulmonary fibrosis |
| Jmjd | Jumonji domain containing |
| M1 | classically activated macrophages |
| M2 | alternatively activated macrophages |
| M-CSFR | colony-stimulating factor receptor |
| MFGE8 | milk fat globule EGF 8 |
| MIF | macrophage migration inhibitory factor |
| miRNA | microRNA |
| MMP | matrix metalloprotease |
| MMT | myofibroblast transition |
| Mo-AMs | monocyte-derived alveolar macrophages |
| mRNA | messenger RNA |
| NAC | N-acetylcysteine |
| PANTHER | Prednisone, Azathioprine, and N-Acetylcysteine: A Study That Evaluates Response |
| PAMPs | pathogen-associated molecular patterns |
| PDGF-R | platelet-derived growth factor receptor |
| PFT | pulmonary function test |
| RIF | radiation-induced lung fibrosis |
| rRNA | ribosomal RNA |
| SAP | serum amyloid P |
| TGF-β | transforming growth factor beta |
| TLR4 | Toll-like receptor 4 |
| TR-Mφ | tissue resident macrophages |
| tRNA | transfer RNA |
| UIP | usual interstitial pneumonia |
| VEGFR-2 | vascular endothelial growth factor receptor-2 |
References
- Kumar, A.; Kapnadak, S.G.; Girgis, R.E.; Raghu, G. Lung transplantation in idiopathic pulmonary fibrosis. Expert Rev. Respir. Med. 2018, 12, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.-F.; Flaherty, K.R.; Lasky, J.A.; et al. An Official ATS/ERS/JRS/ALAT Statement: Idiopathic Pulmonary Fibrosis: Evidence-based Guidelines for Diagnosis and Management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.P.; McKeever, T.M.; Fogarty, A.W.; Navaratnam, V.; Hubbard, R.B. Increasing Global Mortality from Idiopathic Pulmonary Fibrosis in the Twenty-First Century. Ann. Am. Thorac. Soc. 2014, 11, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Chen, S.-Y.; Yeh, W.-S.; Maroni, B.; Li, Q.; Lee, Y.-C.; Collard, H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001–2011. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Vancheri, C.; Failla, M.; Crimi, N.; Raghu, G. Idiopathic pulmonary fibrosis: A disease with similarities and links to cancer biology. Eur. Respir. J. 2010, 35, 496–504. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Jablonski, R.P.; Kim, S.; Cheresh, P.; Williams, D.B.; Morales-Nebreda, L.; Cheng, Y.; Yeldandi, A.; Bhorade, S.; Pardo, A.; Selman, M.; et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017, 31, 2520–2532. [Google Scholar] [CrossRef]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Cell. Mol. Physiol. 2010, 298, L715–L731. [Google Scholar] [CrossRef]
- Shochet, G.E.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L1025–L1034. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, J.C.; Kropski, J.A.; Blackwell, T.S. Idiopathic pulmonary fibrosis: Epithelial-mesenchymal interactions and emerging therapeutic targets. Matrix Biol. 2018, 71–72, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Du Bois, R.M.; Wells, A.U. Cryptogenic fibrosing alveolitis/idiopathic pulmonary fibrosis. Eur. Respir. J. Suppl. 2001, 32, 43s–55s. [Google Scholar] [PubMed]
- Rudd, R.M.; Haslam, P.L.; Turner-Warwick, M. Cryptogenic fibrosing alveolitis. Relationships of pulmonary physiology and bronchoalveolar lavage to response to treatment and prognosis. Am. Rev. Respir. Dis. 1981, 124, 1–8. [Google Scholar] [CrossRef]
- Raghu, G.; DePaso, W.J.; Cain, K.; Hammar, S.P.; Wetzel, C.E.; Dreis, D.F.; Hutchinson, J.; Pardee, N.E.; Winterbauer, R.H. Azathioprine Combined with Prednisone in the Treatment of Idiopathic Pulmonary Fibrosis: A Prospective Double-blind, Randomized, Placebo-controlled Clinical Trial. Am. Rev. Respir. Dis. 1991, 144, 291–296. [Google Scholar] [CrossRef]
- Behr, J.; Maier, K.; Degenkolb, B.; Krombach, F.; Vogelmeier, C. Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am. J. Respir. Crit. Care Med. 1997, 156, 1897–1901. [Google Scholar] [CrossRef]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-Dose Acetylcysteine in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research, N.; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef]
- King, T.E.; Albera, C.; Bradford, W.Z.; Costabel, U.; Hormel, P.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; Szwarcberg, J.; Thomeer, M.; et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): A multicentre, randomised, placebo-controlled trial. Lancet 2009, 374, 222–228. [Google Scholar] [CrossRef]
- Raghu, G.; Brown, K.K.; Bradford, W.Z.; Starko, K.; Noble, P.W.; Schwartz, D.A.; King, T.E. A Placebo-Controlled Trial of Interferon Gamma-1b in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2004, 350, 125–133. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Raghu, G.; Rochwerg, B.; Zhang, Y.; Cuello-Garcia, C.; Azuma, A.; Behr, J.; Brozek, J.L.; Collard, H.R.; Cunningham, W.; Homma, S.; et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2015, 192, e3–e19. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.J.; Ruhrmund, D.W.; Pan, L.; Seiwert, S.D.; Kossen, K. Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 2011, 20, 85–97. [Google Scholar] [CrossRef]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Distler, O.; Highland, K.B.; Gahlemann, M.; Azuma, A.; Fischer, A.; Mayes, M.D.; Raghu, G.; Sauter, W.; Girard, M.; Alves, M.; et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. N. Engl. J. Med. 2019, 380, 2518–2528. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Eapen, M.S.; Hansbro, P.M.; McAlinden, K.; Kim, R.Y.; Ward, C.; Hackett, T.L.; Walters, E.H.; Sohal, S.S. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci. Rep. 2017, 7, 13392. [Google Scholar] [CrossRef]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Torres-González, E.; Rojas, M.; Corredor, C.; Ritzenthaler, J.; Xu, J.; Roman, J.; Brigham, K.; Stecenko, A. Activation of Alveolar Macrophages via the Alternative Pathway in Herpesvirus-Induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2006, 35, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.M.; Hunt, D.A.; Wakefield, L.M.; McCartney-Francis, N.; Wahl, L.M.; Roberts, A.B.; Sporn, M.B. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc. Natl. Acad. Sci. USA 1987, 84, 5788–5792. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; LaVine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Araki, N.; Hatae, T.; Furukawa, A.; Swanson, J.A. Phosphoinositide-3-kinase-independent contractile activities associated with Fcγ-receptor-mediated phagocytosis and macropinocytosis in macrophages. J. Cell. Sci. 2003, 116 Pt 2, 247–257. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Gregory, C.D.; Devitt, A. The macrophage and the apoptotic cell: An innate immune interaction viewed simplistically? Immunology 2004, 113, 1–14. [Google Scholar] [CrossRef]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef]
- Van Furth, R. Macrophage activity and clinical immunology. Origin and kinetics of mononuclear phagocytes. Ann. N. Y. Acad. Sci. 1976, 278, 161–175. [Google Scholar] [CrossRef]
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Bowden, D.H.; Adamson, I.Y. The pulmonary interstitial cell as immediate precursor of the alveolar macrophage. Am. J. Pathol. 1972, 68, 521–537. [Google Scholar] [PubMed]
- Bedoret, D.; Wallemacq, H.; Marichal, T.; Desmet, C.; Quesada Calvo, F.; Henry, E.; Closset, R.; Dewals, B.G.; Thielen, C.; Gustin, P.; et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Investig. 2009, 119, 3723–3738. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Sugimoto, C.; Arainga, M.; Alvarez-Hernandez, X.; Didier, E.; Kuroda, M.J. In Vivo Characterization of Alveolar and Interstitial Lung Macrophages in Rhesus Macaques: Implications for Understanding Lung Disease in Humans. J. Immunol. 2014, 192, 2821–2829. [Google Scholar] [CrossRef]
- Franke-Ullmann, G.; Pförtner, C.; Walter, P.; Steinmüller, C.; Lohmann-Matthes, M.L.; Kobzik, L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J. Immunol. 1996, 157, 3097–3104. [Google Scholar]
- Martin, T.R.; Frevert, C.W. Innate immunity in the lungs. Proc. Am. Thorac. Soc. 2005, 2, 403–411. [Google Scholar] [CrossRef]
- Schneberger, D.; Aharonson-Raz, K.; Singh, B. Monocyte and macrophage heterogeneity and Toll-like receptors in the lung. Cell Tissue Res. 2011, 343, 97–106. [Google Scholar] [CrossRef]
- Puttur, F.; Gregory, L.G.; Lloyd, C.M. Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 2019, 97, 246–257. [Google Scholar] [CrossRef]
- Tan, S.; Krasnow, M.A. Developmental origin of lung macrophage diversity. Development 2016, 143, 1318–1327. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.-I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, H.; Sun, H.; Peng, X.; Tang, C.; Gan, Y.; Chen, X.; Mathur, A.; Hu, B.; Slade, M.D.; et al. Chitinase 3–Like 1 Suppresses Injury and Promotes Fibroproliferative Responses in Mammalian Lung Fibrosis. Sci. Transl. Med. 2014, 6, 240ra76. [Google Scholar] [CrossRef]
- He, C.; Larson-Casey, J.L.; Gu, L.; Ryan, A.J.; Murthy, S.; Carter, A.B. Cu,Zn–Superoxide Dismutase–Mediated Redox Regulation of Jumonji Domain Containing 3 Modulates Macrophage Polarization and Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 58–71. [Google Scholar] [CrossRef]
- Murthy, S.; Larson-Casey, J.L.; Ryan, A.J.; He, C.; Kobzik, L.; Carter, A.B. Alternative activation of macrophages and pulmonary fibrosis are modulated by scavenger receptor, macrophage receptor with collagenous structure. FASEB J. 2015, 29, 3527–3536. [Google Scholar] [CrossRef]
- Meziani, L.; Mondini, M.; Petit, B.; Boissonnas, A.; De Montpreville, V.T.; Mercier, O.; Vozenin, M.-C.; Deutsch, E. CSF1R inhibition prevents radiation pulmonary fibrosis by depletion of interstitial macrophages. Eur. Respir. J. 2018, 51, 1702120. [Google Scholar] [CrossRef]
- Joshi, N.; Watanabe, S.; Verma, R.; Jablonski, R.P.; Chen, C.-I.; Cheresh, P.; Markov, N.; Reyfman, P.A.; McQuattie-Pimentel, A.C.; Sichizya, L.; et al. A spatially restricted fibrotic niche in pulmonary fibrosis is sustained by M-CSF/M-CSFR signalling in monocyte-derived alveolar macrophages. Eur. Respir. J. 2020, 55, 1900646. [Google Scholar] [CrossRef]
- Ji, W.-J.; Ma, Y.-Q.; Zhou, X.; Zhang, Y.-D.; Lu, R.-Y.; Sun, H.-Y.; Guo, Z.-Z.; Zhang, Z.; Li, Y.-M.; Wei, L.-Q. Temporal and spatial characterization of mononuclear phagocytes in circulating, lung alveolar and interstitial compartments in a mouse model of bleomycin-induced pulmonary injury. J. Immunol. Methods 2014, 403, 7–16. [Google Scholar] [CrossRef]
- Naik, P.N.; Horowitz, J.C.; Moore, T.A.; Wilke, C.A.; Toews, G.B.; Moore, B.B. Pulmonary fibrosis induced by γ-herpesvirus in aged mice is associated with increased fibroblast responsiveness to transforming growth factor-β. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Mathie, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary macrophages: Key players in the innate defence of the airways. Thorax 2015, 70, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Barleon, B.; Sozzani, S.; Zhou, D.; Weich, H.A.; Mantovani, A.; Marme, D. Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87, 3336–3343. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Yu, Q.; Liu, D.; Chen, Z.; Zhang, Y.; Lu, J.; Ma, X.; Huang, F.; Han, J.; Wei, L.; et al. Epithelial–mesenchymal Transition of Peritoneal Mesothelial Cells Is Enhanced by M2c Macrophage Polarization. Immunol. Investig. 2022, 51, 301–315. [Google Scholar] [CrossRef]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef]
- Ballinger, M.N.; Newstead, M.W.; Zeng, X.; Bhan, U.; Mo, X.M.; Kunkel, S.L.; Moore, B.B.; Flavell, R.; Christman, J.W.; Standiford, T.J. IRAK-M Promotes Alternative Macrophage Activation and Fibroproliferation in Bleomycin-Induced Lung Injury. J. Immunol. 2015, 194, 1894–1904. [Google Scholar] [CrossRef]
- Srivastava, M.; Saqib, U.; Naim, A.; Roy, A.; Liu, D.; Bhatnagar, D.; Ravinder, R.; Baig, M.S. The TLR4-NOS1-AP1 signaling axis regulates macrophage polarization. Inflamm. Res. 2017, 66, 323–334. [Google Scholar] [CrossRef]
- Bolourani, S.; Sari, E.; Brenner, M.; Wang, P. Extracellular CIRP Induces an Inflammatory Phenotype in Pulmonary Fibroblasts via TLR4. Front. Immunol. 2021, 12, 721970. [Google Scholar] [CrossRef]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018, 102, 168–172. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Müller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis—Mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef]
- Thorley, A.J.; Ford, P.A.; Giembycz, M.A.; Goldstraw, P.; Young, A.; Tetley, T.D. Differential Regulation of Cytokine Release and Leukocyte Migration by Lipopolysaccharide-Stimulated Primary Human Lung Alveolar Type II Epithelial Cells and Macrophages. J. Immunol. 2007, 178, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, N.; Clauberg, S.; Essmann, F.; Liebmann, J.; Kolb-Bachofen, V. Human Skin Endothelial Cells Can Express All 10 TLR Genes and Respond to Respective Ligands. Clin. Vaccine Immunol. 2008, 15, 138–146. [Google Scholar] [CrossRef]
- Pinhal-Enfield, G.; Ramanathan, M.; Hasko, G.; Vogel, S.N.; Salzman, A.L.; Boons, G.-J.; Leibovich, S.J. An Angiogenic Switch in Macrophages Involving Synergy between Toll-Like Receptors 2, 4, 7, and 9 and Adenosine A2A Receptors. Am. J. Pathol. 2003, 163, 711–721. [Google Scholar] [CrossRef]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen Stimulates Macrophage Chemokine Secretion Through Toll-Like Receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G.; Freeze, H.H. Endogenous Damage-Associated Molecular Pattern Molecules at the Crossroads of Inflammation and Cancer. Neoplasia 2009, 11, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Swanson, L.; Katkar, G.D.; Tam, J.; Pranadinata, R.F.; Chareddy, Y.; Coates, J.; Anandachar, M.S.; Castillo, V.; Olson, J.; Nizet, V.; et al. TLR4 signaling and macrophage inflammatory responses are dampened by GIV/Girdin. Proc. Natl. Acad. Sci. USA 2020, 117, 26895–26906. [Google Scholar] [CrossRef]
- Barbarin, V.; Xing, Z.; Delos, M.; Lison, M.; Huaux, F. Pulmonary overexpression of IL-10 augments lung fibrosis and Th2 responses induced by silica particles. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, L841–L848. [Google Scholar] [CrossRef]
- Büttner, C.; Skupin, A.; Reimann, T.; Rieber, E.P.; Unteregger, G.; Geyer, P.; Frank, K.-H. Local Production of Interleukin-4 During Radiation-induced Pneumonitis and Pulmonary Fibrosis in Rats: Macrophages as a Prominent Source of Interleukin-4. Am. J. Respir. Cell Mol. Biol. 1997, 17, 315–325. [Google Scholar] [CrossRef]
- He, C.; Ryan, A.J.; Murthy, S.; Carter, A.B. Accelerated Development of Pulmonary Fibrosis via Cu,Zn-superoxide Dismutase-induced Alternative Activation of Macrophages. J. Biol. Chem. 2013, 288, 20745–20757. [Google Scholar] [CrossRef]
- García-Fojeda, B.; Minutti, C.M.; Montero-Fernández, C.; Stamme, C.; Casals, C. Signaling Pathways That Mediate Alveolar Macrophage Activation by Surfactant Protein A and IL-4. Front. Immunol. 2022, 13, 860262. [Google Scholar] [CrossRef]
- Fichtner-Feigl, S.; Strober, W.; Kawakami, K.; Puri, R.K.; Kitani, A. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 2006, 12, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Kaviratne, M.; Hesse, M.; Leusink, M.; Cheever, A.W.; Davies, S.J.; McKerrow, J.H.; Wakefield, L.M.; Letterio, J.J.; Wynn, T.A. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-β independent. J. Immunol. 2004, 173, 4020–4029. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Homer, R.J.; Zhu, Z.; Lanone, S.; Wang, X.; Koteliansky, V.; Shipley, J.M.; Gotwals, P.; Noble, P.; Chen, Q.; et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor β1. J. Exp. Med. 2001, 194, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Sari, E.; Oztay, F.; Tasci, A.E. Vitamin D modulates E-cadherin turnover by regulating TGF-β and Wnt signalings during EMT-mediated myofibroblast differentiation in A459 cells. J. Steroid Biochem. Mol. Biol. 2020, 202, 105723. [Google Scholar] [CrossRef]
- Letterio, J.J.; Roberts, A.B. Regulation of immune responses by TGF-β. Annu. Rev. Immunol. 1998, 16, 137–161. [Google Scholar] [CrossRef]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-β induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127 Pt 2, 2021–2036. [Google Scholar] [CrossRef]
- Roberts, A.B.; Anzano, M.A.; Wakefield, L.M.; Roche, N.S.; Stern, D.F.; Sporn, M.B. Type beta transforming growth factor: A bifunctional regulator of cellular growth. Proc. Natl. Acad. Sci. USA 1985, 82, 119–123. [Google Scholar] [CrossRef]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Heine, U.I.; Liotta, L.A.; Falanga, V.; Kehrl, J.H. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef]
- Yildirim, M.; Kayalar, O.; Atahan, E.; Oztay, F. Atorvastatin attenuates pulmonary fibrosis in mice and human lung fibroblasts, by the regulation of myofibroblast differentiation and apoptosis. J. Biochem. Mol. Toxicol. 2022, 36, 23074. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, M.; Oztay, F.; Kayalar, O.; Tasci, A.E. Effect of long noncoding RNAs on epithelial-mesenchymal transition in A549 cells and fibrotic human lungs. J. Cell. Biochem. 2021, 122, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Kopp, J.B. TGF-β and fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef]
- Coker, R.K.; Laurent, G.J.; Shahzeidi, S.; Lympany, P.A.; Du Bois, R.M.; Jeffery, P.K.; McAnulty, R.J. Transforming growth factors-β1, -β2, and -β3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am. J. Pathol. 1997, 150, 981–991. [Google Scholar] [PubMed]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef]
- Yu, X.; Buttgereit, A.; Lelios, I.; Utz, S.G.; Cansever, D.; Becher, B.; Greter, M. The Cytokine TGF-β Promotes the Development and Homeostasis of Alveolar Macrophages. Immunity 2017, 47, 903–912.e4. [Google Scholar] [CrossRef]
- Königshoff, M.; Balsara, N.; Pfaff, E.-M.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt Signaling Is Increased in Idiopathic Pulmonary Fibrosis. PLoS ONE 2008, 3, e2142. [Google Scholar] [CrossRef]
- Morrisey, E.E. Wnt Signaling and Pulmonary Fibrosis. Am. J. Pathol. 2003, 162, 1393–1397. [Google Scholar] [CrossRef]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.S.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt Coreceptor Lrp5 Is a Driver of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef]
- Murray, L.A.; Chen, Q.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Peng, X.; Gulati, M.; Homer, R.J.; Russell, T.; van Rooijen, N.; et al. TGF-β driven lung fibrosis is macrophage dependent and blocked by Serum amyloid P. Int. J. Biochem. Cell Biol. 2011, 43, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, L.; Barron, L.D.; Hart, K.M.; Vannella, K.M.; Thompson, R.W.; Oland, S.; Cheever, A.W.; Sciurba, J.; Ramalingam, T.R.; Fisher, A.J.; et al. Macrophages are critical to the maintenance of IL-13-dependent lung inflammation and fibrosis. Mucosal Immunol. 2016, 9, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, M.; Sahin, K.B.; West, Z.E.; Murray, R.Z. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int. J. Mol. Sci. 2017, 18, 1545. [Google Scholar] [CrossRef] [PubMed]
- Nacu, N.; Luzina, I.G.; Highsmith, K.; Lockatell, V.; Pochetuhen, K.; Cooper, Z.A.; Gillmeister, M.P.; Todd, N.W.; Atamas, S.P. Macrophages produce TGF-β-induced (β-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts. J. Immunol. 2008, 180, 5036–5044. [Google Scholar] [CrossRef]
- Hay, E.D. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 2005, 233, 706–720. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.-H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-β-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef]
- Miyazono, K. Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B 2009, 85, 314–323. [Google Scholar] [CrossRef]
- Tan, T.K.; Zheng, G.; Hsu, T.-T.; Lee, S.R.; Zhang, J.; Zhao, Y.; Tian, X.; Wang, Y.; Wang, Y.M.; Cao, Q.; et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab. Investig. 2013, 93, 434–449. [Google Scholar] [CrossRef]
- Bednarczyk, R.B.; Tuli, N.Y.; Hanly, E.K.; Ben Rahoma, G.; Maniyar, R.; Mittelman, A.; Geliebter, J.; Tiwari, R.K. Macrophage inflammatory factors promote epithelial-mesenchymal transition in breast cancer. Oncotarget 2018, 9, 24272–24282. [Google Scholar] [CrossRef]
- Macias-Ceja, D.C.; Coll, S.; Bauset, C.; Seco-Cervera, M.; Gisbert-Ferrandiz, L.; Navarro, F.; Cosin-Roger, J.; Calatayud, S.; Barrachina, M.D.; Ortiz-Masia, D. IFNγ-Treated Macrophages Induce EMT through the WNT Pathway: Relevance in Crohn’s Disease. Biomedicines 2022, 10, 1093. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.T.; Dai, Z.; Song, K.; Zhang, Z.J.; Zhou, Z.J.; Zhou, S.L.; Zhao, Y.M.; Xiao, Y.S.; Sun, Q.M.; Ding, Z.B.; et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int. J. Oncol. 2015, 46, 587–596. [Google Scholar] [CrossRef] [PubMed]
- An, M.; Li, D.; Yuan, M.; Li, Q.; Zhang, L.; Wang, G. Different macrophages equally induce EMT in endometria of adenomyosis and normal. Reproduction 2017, 154, 79–92. [Google Scholar] [CrossRef]
- Shi, J.; Li, Q.; Sheng, M.; Zheng, M.; Yu, M.; Zhang, L. The Role of TLR4 in M1 Macrophage-Induced Epithelial-Mesenchymal Transition of Peritoneal Mesothelial Cells. Cell. Physiol. Biochem. 2016, 40, 1538–1548. [Google Scholar] [CrossRef]
- Hu, Y.; He, M.-Y.; Zhu, L.-F.; Yang, C.-C.; Zhou, M.-L.; Wang, Q.; Zhang, W.; Zheng, Y.-Y.; Wang, D.-M.; Xu, Z.-Q.; et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, F.; Zhang, J.; Wang, L.; Zheng, Y.; Wu, X.; Wang, J.; Huang, Q.; Lai, M. Tumor-associated macrophages remodeling EMT and predicting survival in colorectal carcinoma. OncoImmunology 2018, 7, e1380765. [Google Scholar] [CrossRef]
- Hesse, M.; Modolell, M.; La Flamme, A.C.; Schito, M.; Fuentes, J.M.; Cheever, A.W.; Pearce, E.J.; Wynn, T.A. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: Granulomatous pathology is shaped by the pattern of L-arginine metabolism. J. Immunol. 2001, 167, 6533–6544. [Google Scholar] [CrossRef]
- Song, E.; Ouyang, N.; Hörbelt, M.; Antus, B.; Wang, M.; Exton, M.S. Influence of Alternatively and Classically Activated Macrophages on Fibrogenic Activities of Human Fibroblasts. Cell. Immunol. 2000, 204, 19–28. [Google Scholar] [CrossRef]
- Vierhout, M.; Ayoub, A.; Naiel, S.; Yazdanshenas, P.; Revill, S.D.; Reihani, A.; Dvorkin-Gheva, A.; Shi, W.; Ask, K. Monocyte and macrophage derived myofibroblasts: Is it fate? A review of the current evidence. Wound Repair Regen. 2021, 29, 548–562. [Google Scholar] [CrossRef]
- Shook, B.A.; Wasko, R.R.; Rivera-Gonzalez, G.C.; Salazar-Gatzimas, E.; López-Giráldez, F.; Dash, B.C.; Muñoz-Rojas, A.R.; Aultman, K.D.; Zwick, R.K.; Lei, V.; et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science 2018, 362, eaar2971. [Google Scholar] [CrossRef]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S.; Kiebalo, M.; Kaarteenaho, R.; Glogauer, M.; Kapoor, M.; et al. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-β. Sci. Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Ball, M.S.; Martyanov, V.; Popovich, D.; Schaafsma, E.; Han, S.; ElTanbouly, M.; Orzechowski, N.M.; Carns, M.; Arroyo, E.; et al. Profibrotic Activation of Human Macrophages in Systemic Sclerosis. Arthritis Rheumatol. 2020, 72, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jin, H.; Ullenbruch, M.; Hu, B.; Hashimoto, N.; Moore, B.; McKenzie, A.; Lukacs, N.W.; Phan, S.H. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: Role of IL-4/IL-13 and mediation via STAT-6. J. Immunol. 2004, 173, 3425–3431. [Google Scholar] [CrossRef] [PubMed]
- Vasse, G.F.; Nizamoglu, M.; Heijink, I.H.; Schleputz, M.; van Rijn, P.; Thomas, M.J.; Burgess, J.K.; Melgert, B.N. Macrophage-stroma interactions in fibrosis: Biochemical, biophysical, and cellular perspectives. J. Pathol. 2021, 254, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Little, K.; Llorián-Salvador, M.; Tang, M.; Du, X.; Marry, S.; Chen, M.; Xu, H. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J. Neuroinflamm. 2020, 17, 355. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Wang, S.; Huang, X.-R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.-F.; Nikolic-Paterson, D.; Lan, H.-Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Jiang, H.; Pan, J.; Huang, X.-R.; Wang, Y.-C.; Huang, H.-F.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y.; Chen, J.-H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef]
- Cui, H.; Xie, N.; Banerjee, S.; Ge, J.; Jiang, D.; Dey, T.; Mathews, Q.; Liu, R.-M.; Liu, G. Lung Myofibroblast Promotes Macrophage Pro-Fibrotic Activity through Lactate Induced Histone Lactylation. Am. J. Respir. Cell Mol. Biol. 2021, 64, 115–125. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Aimes, R.T.; Quigley, J.P. Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J. Biol. Chem. 1995, 270, 5872–5876. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, E.; Imai, K.; Fujii, Y.; Sato, H.; Seiki, M.; Okada, Y. Membrane Type 1 Matrix Metalloproteinase Digests Interstitial Collagens and Other Extracellular Matrix Macromolecules. J. Biol. Chem. 1997, 272, 2446–2451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, J.; Sun, H.; Zhang, Y.; Zou, D. New insights into fibrosis from the ECM degradation perspective: The macrophage-MMP-ECM interaction. Cell Biosci. 2022, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, S.; Gaxiola, M.; Arreola, J.L.; Ramírez, R.; Jara, P.; D’Armiento, J.; Richards, T.; Selman, M.; Pardo, A. Overexpression of MMP9 in macrophages attenuates pulmonary fibrosis induced by bleomycin. Int. J. Biochem. Cell Biol. 2007, 39, 2324–2338. [Google Scholar] [CrossRef]
- Eickelberg, O.; Kohler, E.; Reichenberger, F.; Bertschin, S.; Woodtli, T.; Erne, P.; Perruchoud, A.P.; Roth, M. Extracellular matrix deposition by primary human lung fibroblasts in response to TGF-β1 and TGF-β3. Am. J. Physiol. 1999, 276, L814–L824. [Google Scholar] [CrossRef]
- Fujishima, S.; Shiomi, T.; Yamashita, S.; Yogo, Y.; Nakano, Y.; Inoue, T.; Nakamura, M.; Tasaka, S.; Hasegawa, N.; Aikawa, N.; et al. Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch. Pathol. Lab. Med. 2010, 134, 1136–1142. [Google Scholar] [CrossRef]
- Gharib, S.A.; Johnston, L.K.; Huizar, I.; Birkland, T.P.; Hanson, J.; Wang, Y.; Parks, W.C.; Manicone, A.M. MMP28 promotes macrophage polarization toward M2 cells and augments pulmonary fibrosis. J. Leukoc. Biol. 2014, 95, 9–18. [Google Scholar] [CrossRef]
- Madala, S.K.; Pesce, J.T.; Ramalingam, T.R.; Wilson, M.S.; Minnicozzi, S.; Cheever, A.W.; Thompson, R.W.; Mentink-Kane, M.M.; Wynn, T.A. Matrix Metalloproteinase 12-Deficiency Augments Extracellular Matrix Degrading Metalloproteinases and Attenuates IL-13–Dependent Fibrosis. J. Immunol. 2010, 184, 3955–3963. [Google Scholar] [CrossRef]
- Atabai, K.; Jame, S.; Azhar, N.; Kuo, A.; Lam, M.; McKleroy, W.; DeHart, G.; Rahman, S.; Xia, D.D.; Melton, A.C.; et al. Mfge8 diminishes the severity of tissue fibrosis in mice by binding and targeting collagen for uptake by macrophages. J. Clin. Investig. 2009, 119, 3713–3722. [Google Scholar] [CrossRef]
- Van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A Common Pathway for a Specialized Function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef] [PubMed]
- Artlett, C.M.; Sassi-Gaha, S.; Hope, J.L.; Feghali-Bostwick, C.A.; Katsikis, P.D. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res. Ther. 2017, 19, 144. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Santos-Álvarez, J.C.; Velázquez-Enríquez, J.M.; García-Carrillo, R.; Rodríguez-Beas, C.; Ramírez-Hernández, A.A.; Reyes-Jiménez, E.; González-García, K.; López-Martínez, A.; Mayoral, L.P.-C.; Aguilar-Ruiz, S.R.; et al. miRNAs Contained in Extracellular Vesicles Cargo Contribute to the Progression of Idiopathic Pulmonary Fibrosis: An In Vitro Aproach. Cells 2022, 11, 1112. [Google Scholar] [CrossRef]
- Aston, C.; Jagirdar, J.; Lee, T.C.; Hur, T.; Hintz, R.L.; Rom, W. Enhanced insulin-like growth factor molecules in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1597–1603. [Google Scholar] [CrossRef]
- Elliot, S.; Periera-Simon, S.; Xia, X.; Catanuto, P.; Rubio, G.; Shahzeidi, S.; El Salem, F.; Shapiro, J.; Briegel, K.; Korach, K.S.; et al. MicroRNA let-7 Downregulates Ligand-Independent Estrogen Receptor–mediated Male-Predominant Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1246–1257. [Google Scholar] [CrossRef]
- Banerjee, S.; Xie, N.; Cui, H.; Tan, Z.; Yang, S.; Icyuz, M.; Abraham, E.; Liu, G. MicroRNA let-7c Regulates Macrophage Polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef]
- Su, S.; Zhao, Q.; He, C.; Huang, D.; Liu, J.; Chen, F.; Chen, J.; Liao, J.-Y.; Cui, X.; Zeng, Y.; et al. miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program. Nat. Commun. 2015, 6, 8523. [Google Scholar] [CrossRef]
- Kishore, A.; Petrek, M. Roles of Macrophage Polarization and Macrophage-Derived miRNAs in Pulmonary Fibrosis. Front. Immunol. 2021, 12, 678457. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Zhang, H.; Han, B.; Ye, Y.; Zhang, M.; Wang, Y.; Xue, J.; Wang, C. Transforming Growth Factor-β1 Promotes M1 Alveolar Macrophage Polarization in Acute Lung Injury by Up-Regulating DNMT1 to Mediate the microRNA-124/PELI1/IRF5 Axis. Front. Cell. Infect. Microbiol. 2021, 11, 693981. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, N.; Cheng, Q.; Zhang, H.; Liu, F.; Shang, Y. MiR-21-5p in Macrophage-Derived Exosomes Targets Smad7 to Promote Epithelial Mesenchymal Transition of Airway Epithelial Cells. J. Asthma Allergy 2021, 14, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Bandari, S.K.; Purushothaman, A.; Ramani, V.C.; Brinkley, G.J.; Chandrashekar, D.S.; Varambally, S.; Mobley, J.A.; Zhang, Y.; Brown, E.E.; Vlodavsky, I.; et al. Chemotherapy induces secretion of exosomes loaded with heparanase that degrades extracellular matrix and impacts tumor and host cell behavior. Matrix Biol. 2018, 65, 104–118. [Google Scholar] [CrossRef]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell. Biochem. 2008, 105, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Placido, L.; Romero, Y.; Maldonado, M.; Toscano-Marquez, F.; Ramírez, R.; Calyeca, J.; Mora, A.; Selman, M.; Pardo, A. Loss of MT1-MMP in Alveolar Epithelial Cells Exacerbates Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 22, 2923. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Bandari, S.K.; Vlodavsky, I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019, 75–76, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, M.; Khokha, R. Proteolytic factors in exosomes. Proteomics 2013, 13, 1624–1636. [Google Scholar] [CrossRef]
- Duffy, M.J.; Mullooly, M.; O’Donovan, N.; Sukor, S.; Crown, J.; Pierce, A.; McGowan, P.M. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin. Proteom. 2011, 8, 9. [Google Scholar] [CrossRef]
- Lee, H.D.; Koo, B.; Kim, Y.H.; Jeon, O.; Kim, D. Exosome release of ADAM15 and the functional implications of human macrophage-derived ADAM15 exosomes. FASEB J. 2012, 26, 3084–3095. [Google Scholar] [CrossRef]
- Yan, Y.; Shirakabe, K.; Werb, Z. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein–coupled receptors. J. Cell Biol. 2002, 158, 221–226. [Google Scholar] [CrossRef]
- Solomon, J.B. Induction of antibody formation to goat erythrocytes in the developing chick embryo and effects of maternal antibody. Immunology 1966, 11, 89–96. [Google Scholar] [PubMed]
- Lagares, D.; Ghassemi-Kakroodi, P.; Tremblay, C.; Santos, A.; Probst, C.K.; Franklin, A.; Santos, D.M.; Grasberger, P.; Ahluwalia, N.; Montesi, S.B.; et al. ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis. Nat. Med. 2017, 23, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and β-catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef] [PubMed]
- Najy, A.J.; Day, K.C.; Day, M.L. The Ectodomain Shedding of E-cadherin by ADAM15 Supports ErbB Receptor Activation. J. Biol. Chem. 2008, 283, 18393–18401. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, D.; Higham, A.; Wolosianka, S.; Gai, X.; Zhou, L.; Petersen, H.; Pinto-Plata, V.; Divo, M.; Silverman, E.K.; et al. ADAM15 expression is increased in lung CD8+ T cells, macrophages, and bronchial epithelial cells in patients with COPD and is inversely related to airflow obstruction. Respir. Res. 2020, 21, 188. [Google Scholar] [CrossRef]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]
- Minagawa, S.; Araya, J.; Numata, T.; Nojiri, S.; Hara, H.; Yumino, Y.; Kawaishi, M.; Odaka, M.; Morikawa, T.; Nishimura, S.L.; et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L391–L401. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef]
- Faner, R.; Rojas, M.; MacNee, W.; Agustí, A. Abnormal Lung Aging in Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 306–313. [Google Scholar] [CrossRef]
- Canan, C.H.; Gokhale, N.S.; Carruthers, B.; Lafuse, W.P.; Schlesinger, L.S.; Torrelles, J.B.; Turner, J. Characterization of lung inflammation and its impact on macrophage function in aging. J. Leukoc. Biol. 2014, 96, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Kwon, S.; Reedy, J.L.; White, A.O.; Song, J.S.; Hwang, I.; Chung, J.Y.; Ylaya, K.; Hewitt, S.M.; Citrin, D.E. IGF-1 Receptor Signaling Regulates Type II Pneumocyte Senescence and Resulting Macrophage Polarization in Lung Fibrosis. Int. J. Radiat. Oncol. 2021, 110, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Jiang, C.; Liu, G.; Miyata, T.; Antony, V.; Thannickal, V.J.; Liu, R.M. PAI-1 Regulation of TGF-β1-induced Alveolar Type II Cell Senescence, SASP Secretion, and SASP-mediated Activation of Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2020, 62, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.; Khan, A.A.; Yin, J.; Ferguson, T.A.; Apte, R.S. Senescence regulates macrophage activation and angiogenic fate at sites of tissue injury in mice. J. Clin. Investig. 2007, 117, 3421–3426. [Google Scholar] [CrossRef]
- Bargagli, E.; Olivieri, C.; Nikiforakis, N.; Cintorino, M.; Magi, B.; Perari, M.; Vagaggini, C.; Spina, D.; Prasse, A.; Rottoli, P. Analysis of macrophage migration inhibitory factor (MIF) in patients with idiopathic pulmonary fibrosis. Respir. Physiol. Neurobiol. 2009, 167, 261–267. [Google Scholar] [CrossRef]
- Mathew, B.; Jacobson, J.R.; Siegler, J.H.; Moitra, J.; Blasco, M.; Xie, L.; Unzueta, C.; Zhou, T.; Evenoski, C.; Al-Sakka, M.; et al. Role of Migratory Inhibition Factor in Age-Related Susceptibility to Radiation Lung Injury via NF-E2–Related Factor–2 and Antioxidant Regulation. Am. J. Respir. Cell Mol. Biol. 2013, 49, 269–278. [Google Scholar] [CrossRef]
- Gibbons, M.A.; MacKinnon, A.C.; Ramachandran, P.; Dhaliwal, K.; Duffin, R.; Phythian-Adams, A.T.; van Rooijen, N.; Haslett, C.; Howie, S.E.; Simpson, A.J.; et al. Ly6Chi Monocytes Direct Alternatively Activated Profibrotic Macrophage Regulation of Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 569–581. [Google Scholar] [CrossRef]
- He, C.; Larson-Casey, J.L.; Davis, D.; Hanumanthu, V.S.; Longhini, A.L.F.; Thannickal, V.J.; Gu, L.; Carter, A.B. NOX4 modulates macrophage phenotype and mitochondrial biogenesis in asbestosis. JCI Insight 2019, 4, e126551. [Google Scholar] [CrossRef]
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Ruven, H.J.; Kruit, A.; Heron, M.; Bosch, J.M.V.D.; Grutters, J.C. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 106–113. [Google Scholar] [CrossRef]
- Murray, L.A.; Rosada, R.; Moreira, A.P.; Joshi, A.; Kramer, M.S.; Hesson, D.P.; Argentieri, R.L.; Mathai, S.; Gulati, I.; Herzog, E.L.; et al. Serum Amyloid P Therapeutically Attenuates Murine Bleomycin-Induced Pulmonary Fibrosis via Its Effects on Macrophages. PLoS ONE 2010, 5, e9683. [Google Scholar] [CrossRef]
- Pilling, D.; Roife, D.; Wang, M.; Ronkainen, S.D.; Crawford, J.R.; Travis, E.L.; Gomer, R. Reduction of Bleomycin-Induced Pulmonary Fibrosis by Serum Amyloid P. J. Immunol. 2007, 179, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Pignatti, P.; Brunetti, G.; Moretto, D.; Yacoub, M.-R.; Fiori, M.; Balbi, B.; Balestrino, A.; Cervio, G.; Nava, S.; Moscato, G. Role of the Chemokine Receptors CXCR3 and CCR4 in Human Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 173, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, G.; O’Connor, E.C.; Kunkel, S.L.; Hogaboam, C.M. A Novel Mechanism for CCR4 in the Regulation of Macrophage Activation in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Pathol. 2008, 172, 1209–1221. [Google Scholar] [CrossRef]
- Okuma, T.; Terasaki, Y.; Kaikita, K.; Kobayashi, H.; Kuziel, W.A.; Kawasuji, M.; Takeya, M. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J. Pathol. 2004, 204, 594–604. [Google Scholar] [CrossRef]
- He, C.; Carter, A.B. The Metabolic Prospective and Redox Regulation of Macrophage Polarization. J. Clin. Cell. Immunol. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Myllarniemi, M.; Kaarteenaho, R. Pharmacological treatment of idiopathic pulmonary fibrosis—Preclinical and clinical studies of pirfenidone, nintedanib, and N-acetylcysteine. Eur. Clin. Respir. J. 2015, 2, 26385. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Toda, M.; Mizuguchi, S.; Minamiyama, Y.; Yamamoto-Oka, H.; Aota, T.; Kubo, S.; Nishiyama, N.; Shibata, T.; Takemura, S. Pirfenidone suppresses polarization to M2 phenotype macrophages and the fibrogenic activity of rat lung fibroblasts. J. Clin. Biochem. Nutr. 2018, 63, 58–65. [Google Scholar] [CrossRef]
- Bellamri, N.; Morzadec, C.; Joannes, A.; Lecureur, V.; Wollin, L.; Jouneau, S.; Vernhet, L. Alteration of human macrophage phenotypes by the anti-fibrotic drug nintedanib. Int. Immunopharmacol. 2019, 72, 112–123. [Google Scholar] [CrossRef]
- Woods, P.S.; Kimmig, L.M.; Sun, K.A.; Meliton, A.Y.; Shamaa, O.R.; Tian, Y.; Cetin-Atalay, R.; Sharp, W.W.; Hamanaka, R.B.; Mutlu, G.M. HIF-1α induces glycolytic reprograming in tissue-resident alveolar macrophages to promote cell survival during acute lung injury. Elife 2022, 11, e77457. [Google Scholar] [CrossRef]
- Melo, A.C.; Cattani-Cavalieri, I.; Barroso, M.V.; Quesnot, N.; Gitirana, L.B.; Lanzetti, M.; Valença, S.S. Atorvastatin dose-dependently promotes mouse lung repair after emphysema induced by elastase. Biomed. Pharmacother. 2018, 102, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Hayashi, S.; Mukae, H.; Vincent, R.; Hogg, J.C.; van Eeden, S.F. Effect of Atorvastatin on PM10-induced Cytokine Production by Human Alveolar Macrophages and Bronchial Epithelial Cells. Int. J. Toxicol. 2009, 28, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Morad, H.O.J.; Luqman, S.; Pinto, L.G.; Cunningham, K.P.; Vilar, B.; Clayton, G.; Shankar-Hari, M.; McNaughton, P.A. Artemisinin inhibits neutrophil and macrophage chemotaxis, cytokine production and NET release. Sci. Rep. 2022, 12, 1–18. [Google Scholar] [CrossRef]
- Konkimalla, B.; Blunder, M.; Korn, B.; Soomro, S.A.; Jansen, H.; Chang, W.; Posner, G.H.; Bauer, R.; Efferth, T. Effect of artemisinins and other endoperoxides on nitric oxide-related signaling pathway in RAW 264.7 mouse macrophage cells. Nitric Oxide 2008, 19, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.H.; Choi, J.H.; Byun, M.S.; Jue, D.M. Chloroquine inhibits production of TNF-α, IL-1β and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology 2006, 45, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Tan, L.; Rodrigues, D.A.; Prestes, E.B.; Gomes, C.P.; Gama, A.M.; Oliveira, P.L.; Paiva, C.N.; Manoury, B.; Bozza, M.T. Chloroquine inhibits pro-inflammatory effects of heme on macrophages and invivo. Free Radic. Biol. Med. 2021, 173, 104–116. [Google Scholar] [CrossRef]
- Higham, A.; Scott, T.; Li, J.; Gaskell, R.; Dikwa, A.B.; Shah, R.; Montero-Fernandez, M.A.; Lea, S.; Singh, D. Effects of corticosteroids on COPD lung macrophage phenotype and function. Clin. Sci. 2020, 134, 751–763. [Google Scholar] [CrossRef]
- Roghanian, A.; Hu, G.; Fraser, C.; Singh, M.; Foxall, R.B.; Meyer, M.J.; Lees, E.; Huet, H.; Glennie, M.J.; Beers, S.A.; et al. Cyclophosphamide Enhances Cancer Antibody Immunotherapy in the Resistant Bone Marrow Niche by Modulating Macrophage FcγR Expression. Cancer Immunol. Res. 2019, 7, 1876–1890. [Google Scholar] [CrossRef]
- Ozanne, J.; Prescott, A.R.; Clark, K. The clinically approved drugs dasatinib and bosutinib induce anti-inflammatory macrophages by inhibiting the salt-inducible kinases. Biochem. J. 2015, 465, 271–279. [Google Scholar] [CrossRef]
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, Y.; He, J.; Yang, Z.; Zhang, R.; Li, L.; Luo, Z.; Ye, Y.; Sun, Q. Hydroxychloroquine Inhibits Macrophage Activation and Attenuates Renal Fibrosis After Ischemia-Reperfusion Injury. Front. Immunol. 2021, 12, 645100. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.-Q.; Birkett, R.; Doyle, R.; Shi, B.; Roberts, E.L.; Mao, Q.; Pope, R.M. The Role of Macrophages in the Response to TNF Inhibition in Experimental Arthritis. J. Immunol. 2018, 200, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Park-Min, K.H.; Serbina, N.V.; Yang, W.; Ma, X.; Krystal, G.; Neel, B.G.; Nutt, S.L.; Hu, X.; Ivashkiv, L.B. FcγRIII-dependent inhibition of interferon-γ responses mediates suppressive effects of intravenous immune globulin. Immunity 2007, 26, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.F.; Zhang, W.; Zuo, W.; Tan, H.Q.; Bai, W. Inhibition of the PD-1/PD-L1 signaling pathway enhances innate immune response of alveolar macrophages to mycobacterium tuberculosis in mice. Pulm. Pharmacol. Ther. 2020, 60, 101842. [Google Scholar] [CrossRef] [PubMed]
- Leoni, F.; Zaliani, A.; Bertolini, G.; Porro, G.; Pagani, P.; Pozzi, P.; Donà, G.; Fossati, G.; Sozzani, S.; Azam, T.; et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Mizuta, M.; Okamoto, N.; Yasumi, T.; Iwata, N.; Umebayashi, H.; Okura, Y.; Kinjo, N.; Kubota, T.; Nakagishi, Y.; et al. Tocilizumab modifies clinical and laboratory features of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Pediatr. Rheumatol. 2020, 18, 2. [Google Scholar] [CrossRef]
- Li, X.; Wei, Y.; Li, S.; Liang, J.; Liu, Z.; Cui, Y.; Gao, J.; Yang, Z.; Li, L.; Zhou, H.; et al. Zanubrutinib ameliorates lipopolysaccharide-induced acute lung injury via regulating macrophage polarization. Int. Immunopharmacol. 2022, 111, 109138. [Google Scholar] [CrossRef]
- El-Mohandes, E.M.; Moustafa, A.M.; Khalaf, H.A.; Hassan, Y.F. The role of mast cells and macrophages in amiodarone induced pulmonary fibrosis and the possible attenuating role of atorvastatin. Biotech. Histochem. 2017, 92, 467–480. [Google Scholar] [CrossRef]
- Zitnik, R.J.; Cooper, J.A., Jr.; Rankin, J.A.; Sussman, J. Effects of in vitro amiodarone exposure on alveolar macrophage inflammatory mediator production. Am. J. Med. Sci. 1992, 304, 352–356. [Google Scholar] [CrossRef]
- Santosuosso, M.; Divangahi, M.; Zganiacz, A.; Xing, Z. Reduced tissue macrophage population in the lung by anticancer agent cyclophosphamide: Restoration by local granulocyte macrophage–colony-stimulating factor gene transfer. Blood 2002, 99, 1246–1252. [Google Scholar] [CrossRef]
- Guan, W.; Hu, J.; Yang, L.; Tan, P.; Tang, Z.; West, B.L.; Bollag, G.; Xu, H.; Wu, L. Inhibition of TAMs improves the response to docetaxel in castration-resistant prostate cancer. Endocr. Relat. Cancer 2019, 26, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Imokawa, S.; Colby, T.; Leslie, K.; Helmers, R. Methotrexate pneumonitis: Review of the literature and histopathological findings in nine patients. Eur. Respir. J. 2000, 15, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Thangam, M.; Nathan, S.; Petrovica, M.; Kar, B.; Patel, M.; Loyalka, P.; Buja, L.M.; Gregoric, I.D. Procainamide-induced pulmonary fibrosis after orthotopic heart transplantation: A case report and literature review. Cardiovasc. Pathol. 2015, 24, 250–253. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Treatment | Mechanisms of Action | Effect on Macrophages |
|---|---|---|
| Atorvastatin | Lipid-decreasing statin | Reduces mediator production of AM (IL-1β, IL-6, and TNF-A-α) and macrophage recruitment [191,192] |
| Artemisinin | Antiviral, antimalarial, and anti-inflammatory | Inhibits macrophage chemotaxis and cytokine production [193] |
| Artesunate | Antimalarial | Attenuates proinflammatory effects of monocytes/macrophages [194] |
| Chloroquine | Immunosuppressive and Anti-parasite | Reduces TNF-A-α, IL-1-β and IL-6 [195,196] |
| Corticosteroid | Anti-inflammatory | Reduces macrophage CD64, CD80 and CD86 expression, controls the phenotype of alveolar macrophages [197]. |
| Cyclophosphamide | Chemotherapy and Immunosuppressive | Activates and enhances macrophage phagocytosis [198] |
| Dasatinib | Chemotherapy | Elevates production of IL-10 while suppressing the production of IL-6, IL-12p40 and TNF-α in response to TLR stimulation [199] |
| HDAC6 Nexturastat A | HDAC inhibitor | Reduces pro-tumorigenic M2 macrophages [200]. |
| Hydroxychloroquine | Immunosuppressive and Anti-parasite | Promotes apoptosis of macrophages and inhibits activation of macrophages, especially M2 macrophages [201] |
| Infliximab | Immunosuppressive | Induces apoptosis of Ly6C+ macrophages, decreases migration of monocytes into the ankles, and reduces CCL2 [202] |
| Intravenous Immunoglobulin (IVIG) | Therapy treatment for patients with antibody deficiencies | Inhibits the activation of monocytes and macrophages, inhibition of macrophage responses to IFN-γ [203] |
| PD-1/PD-L1 signaling blocker | Checkpoint inhibitor anticancer drug | Decreases TNF-A-α, IL-6, IFN-γ and ROS from alveolar macrophages [204] |
| Suberoylanilide hydroxamic acid (SAHA), Vorinostat | Chemotherapy | Reduces TNF-α, IL-1-β, IL-12, and IFN-γ [205] |
| Tocilizumab | Immunosuppressive | Anti-IL-6 receptor, modifies macrophage activation [206] |
| Zanubrutinib | Kinase inhibitor | Inhibits M1 macrophage polarization and promotes M2 macrophage polarization [207] |
| The Name of Drug | The Type of Drug | Effect on Macrophages |
|---|---|---|
| Amiodarone | Antiarrhythmic | Induces alveolar macrophages to secrete more TNF-α and superoxide anions [208,209]. |
| Bleomycin | Chemotherapy | Recruits pro-fibrotic M2 cells and induces myofibroblast differentiation [31,66] |
| Cyclophosphamide | Chemotherapy and Immunosuppressive | Decreases spontaneous proliferation and reduces the ability to proliferate upon stimulation with GM-CSF [210] |
| Docetaxel | Chemotherapy | Induces M2 cells recruitment [211] |
| Methotrexate | Chemotherapy and Immunosuppressive drug | Macrophage recruitment [212] |
| Procainamide | Antiarrhythmic | Induces macrophage recruitment [213] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sari, E.; He, C.; Margaroli, C. Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis. Int. J. Mol. Sci. 2022, 23, 11443. https://doi.org/10.3390/ijms231911443
Sari E, He C, Margaroli C. Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis. International Journal of Molecular Sciences. 2022; 23(19):11443. https://doi.org/10.3390/ijms231911443
Chicago/Turabian StyleSari, Ezgi, Chao He, and Camilla Margaroli. 2022. "Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis" International Journal of Molecular Sciences 23, no. 19: 11443. https://doi.org/10.3390/ijms231911443
APA StyleSari, E., He, C., & Margaroli, C. (2022). Plasticity towards Rigidity: A Macrophage Conundrum in Pulmonary Fibrosis. International Journal of Molecular Sciences, 23(19), 11443. https://doi.org/10.3390/ijms231911443