

Interactions between the NLRP3-Dependent IL-1β and the Type I Interferon Pathways in Human Plasmacytoid Dendritic Cells

 ,
, 
 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
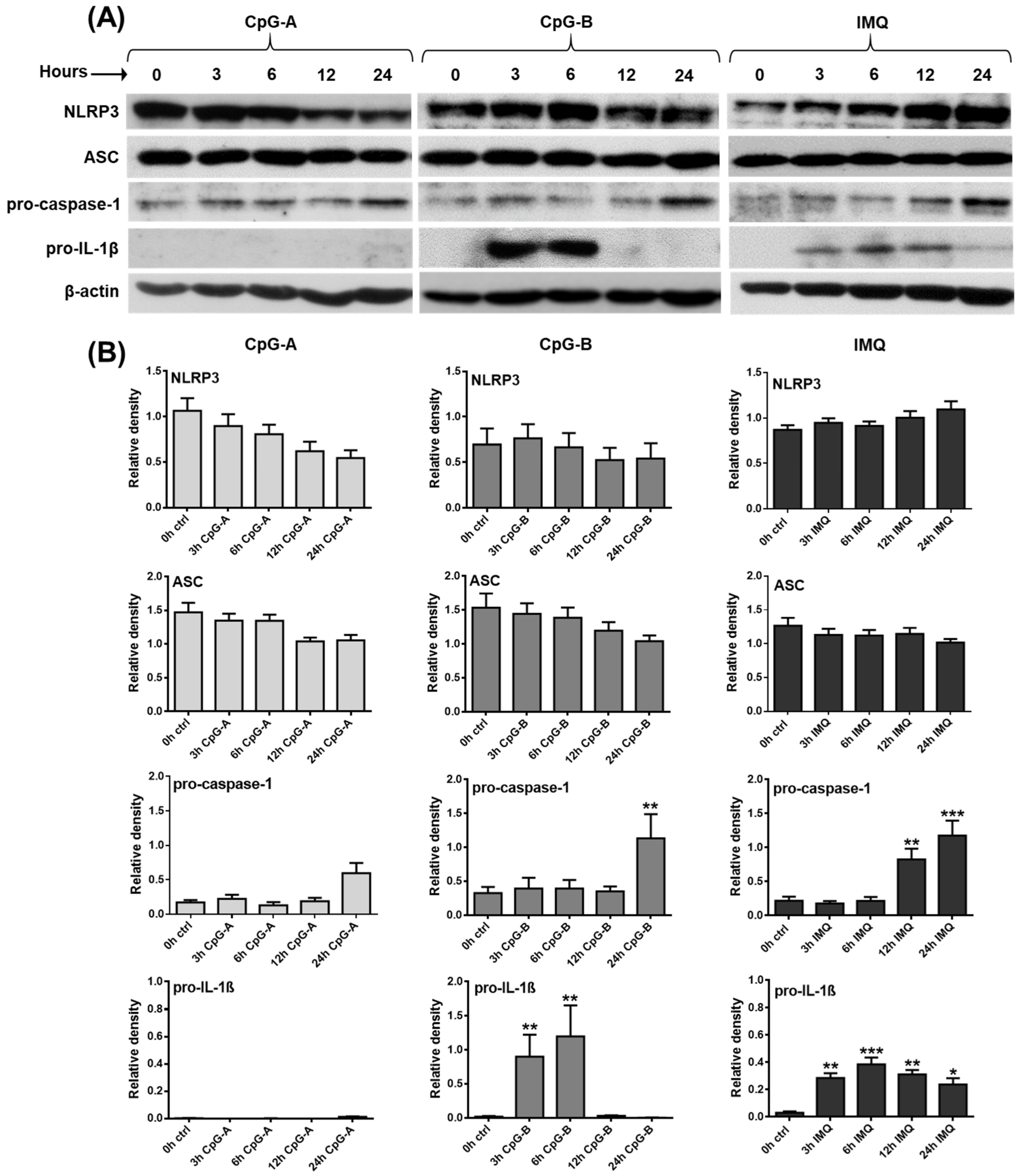
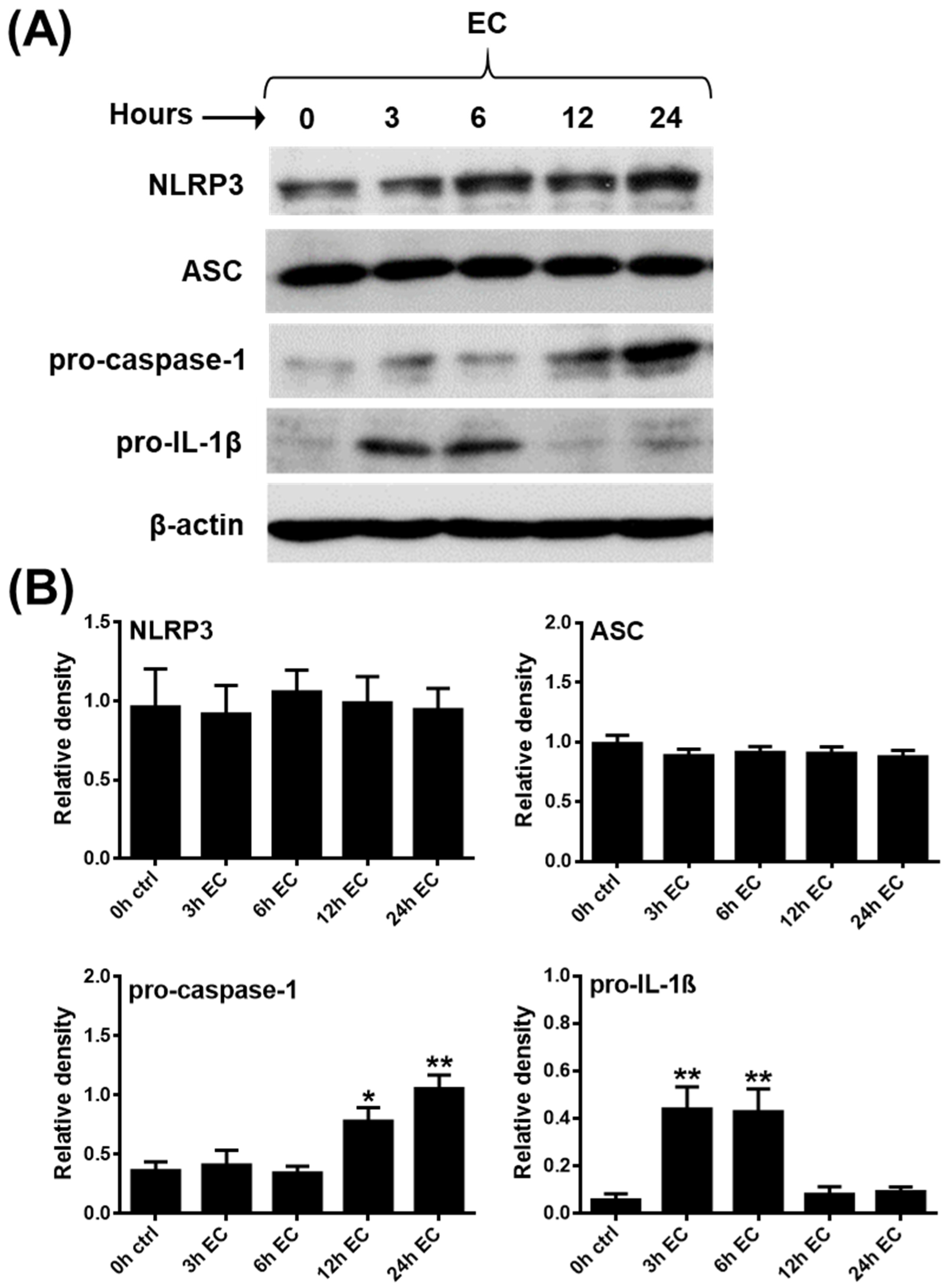
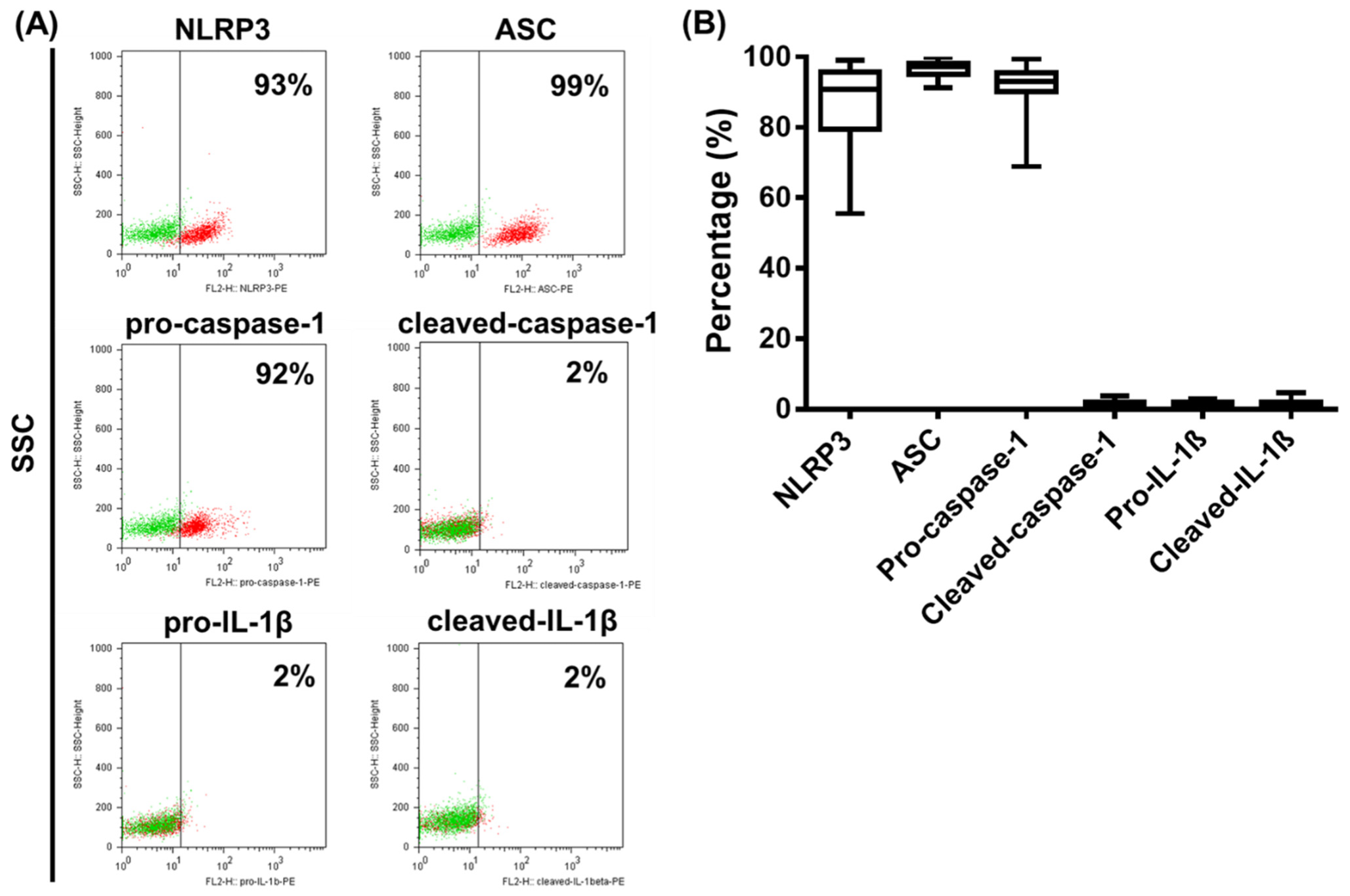
2.1. Investigating the Expression Profile of NLRP3 Pathway Components in a Human pDC Cell Line
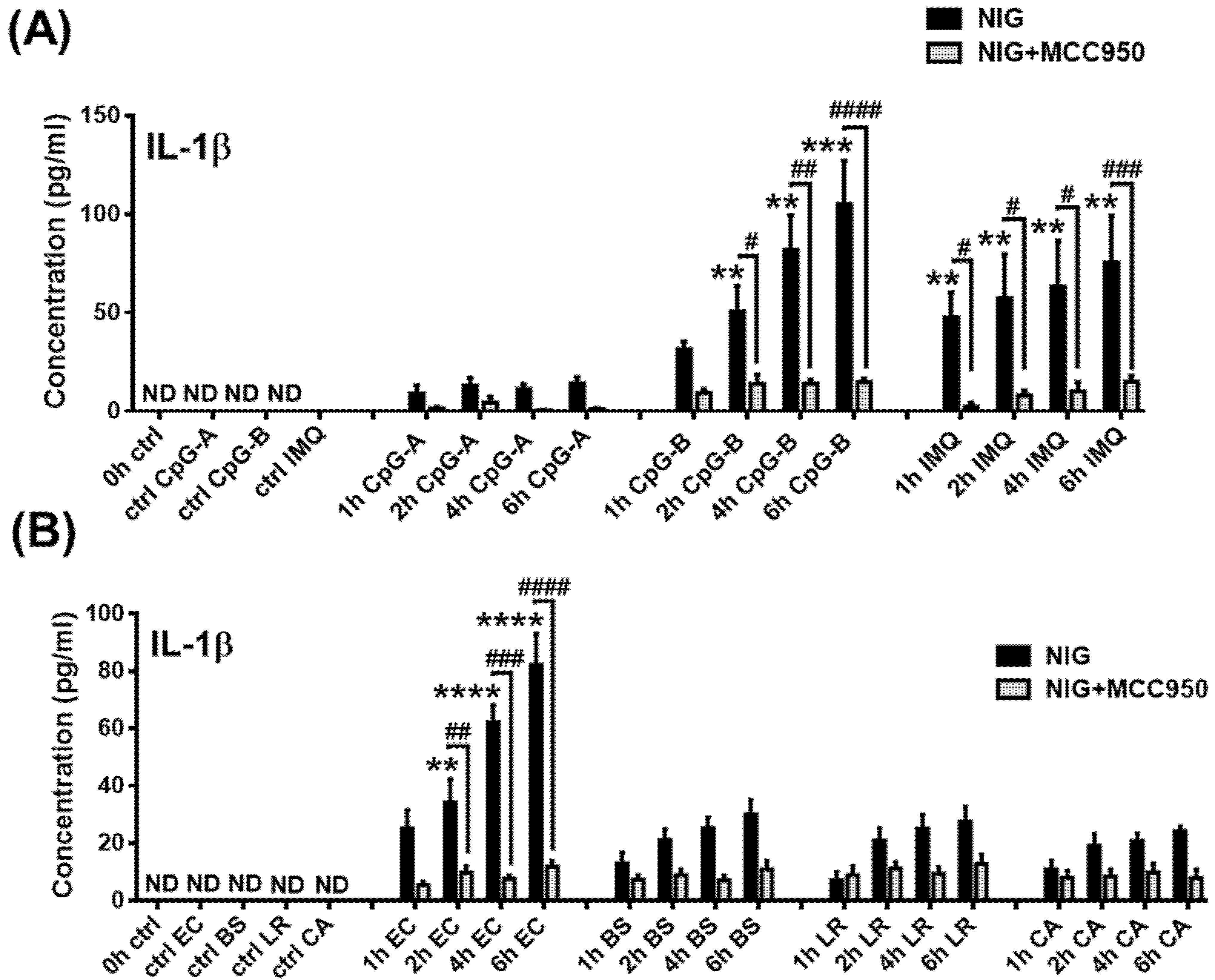
2.2. Assessment of Mature IL-1β Secretion in a Human pDC Cell Line
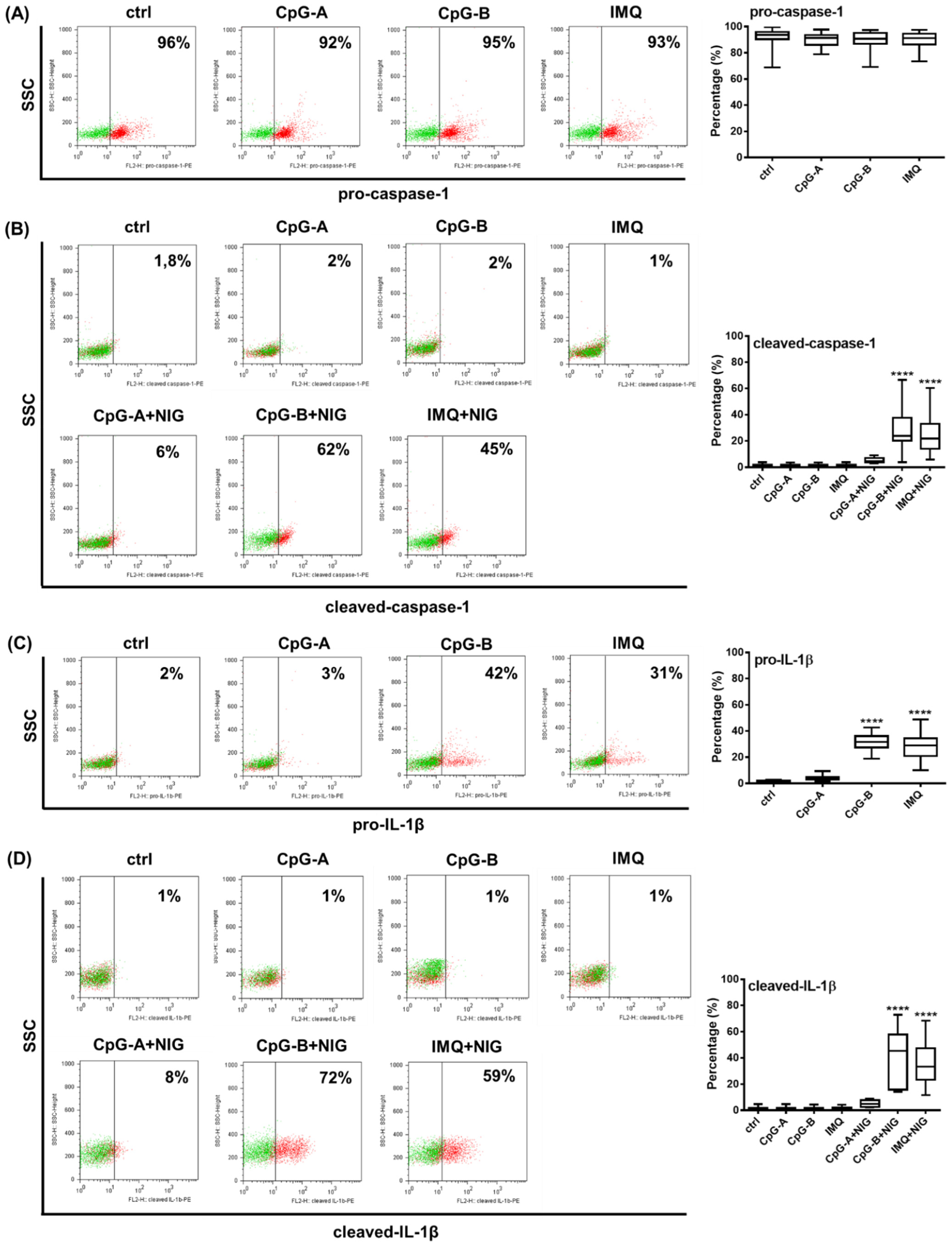
2.3. Analysis of NLRP3 Pathway Activity in Primary Human pDCs
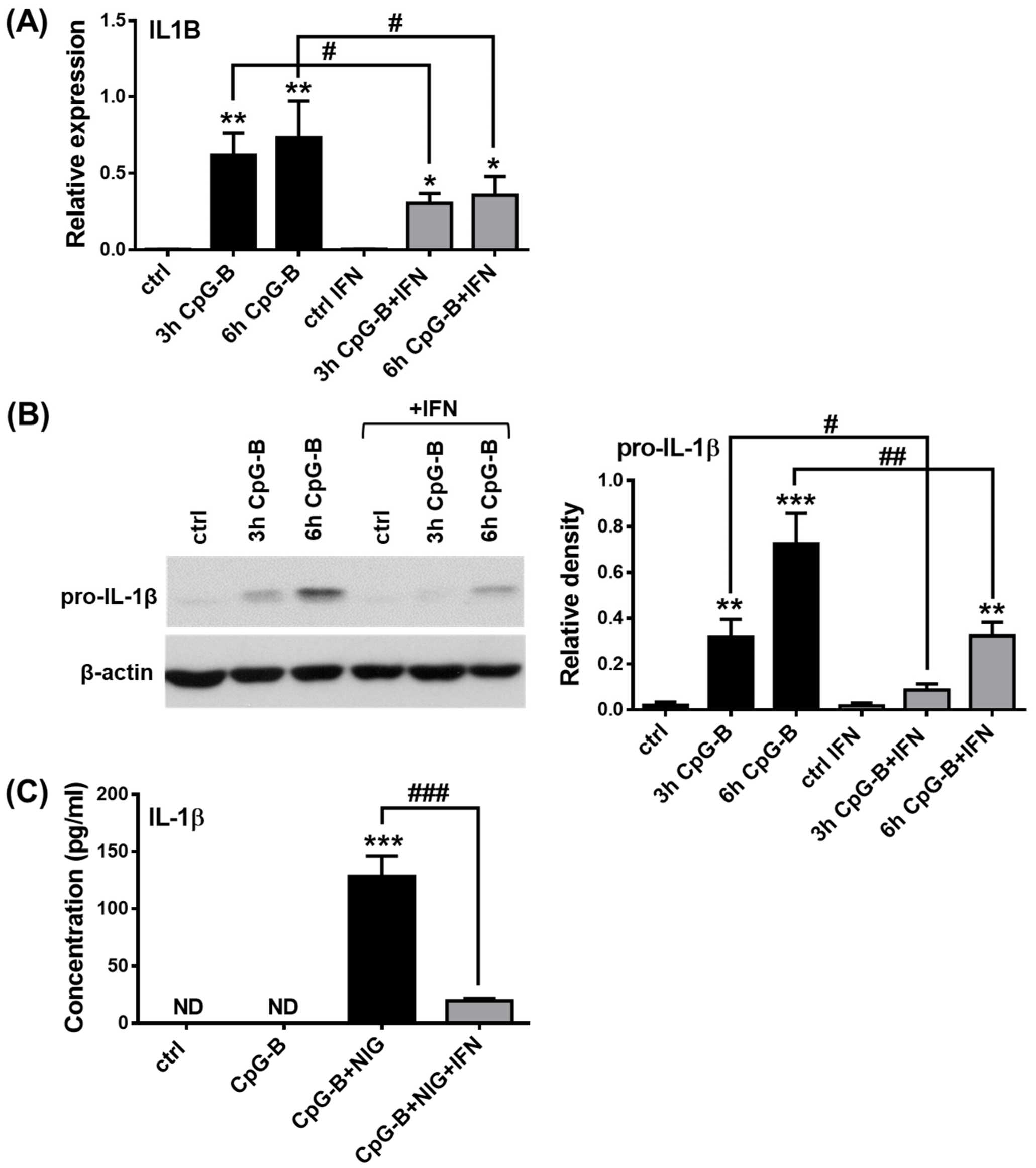
2.4. Analysis of NLRP3 Pathway Activity in the Presence of Type I IFNs in pDCs
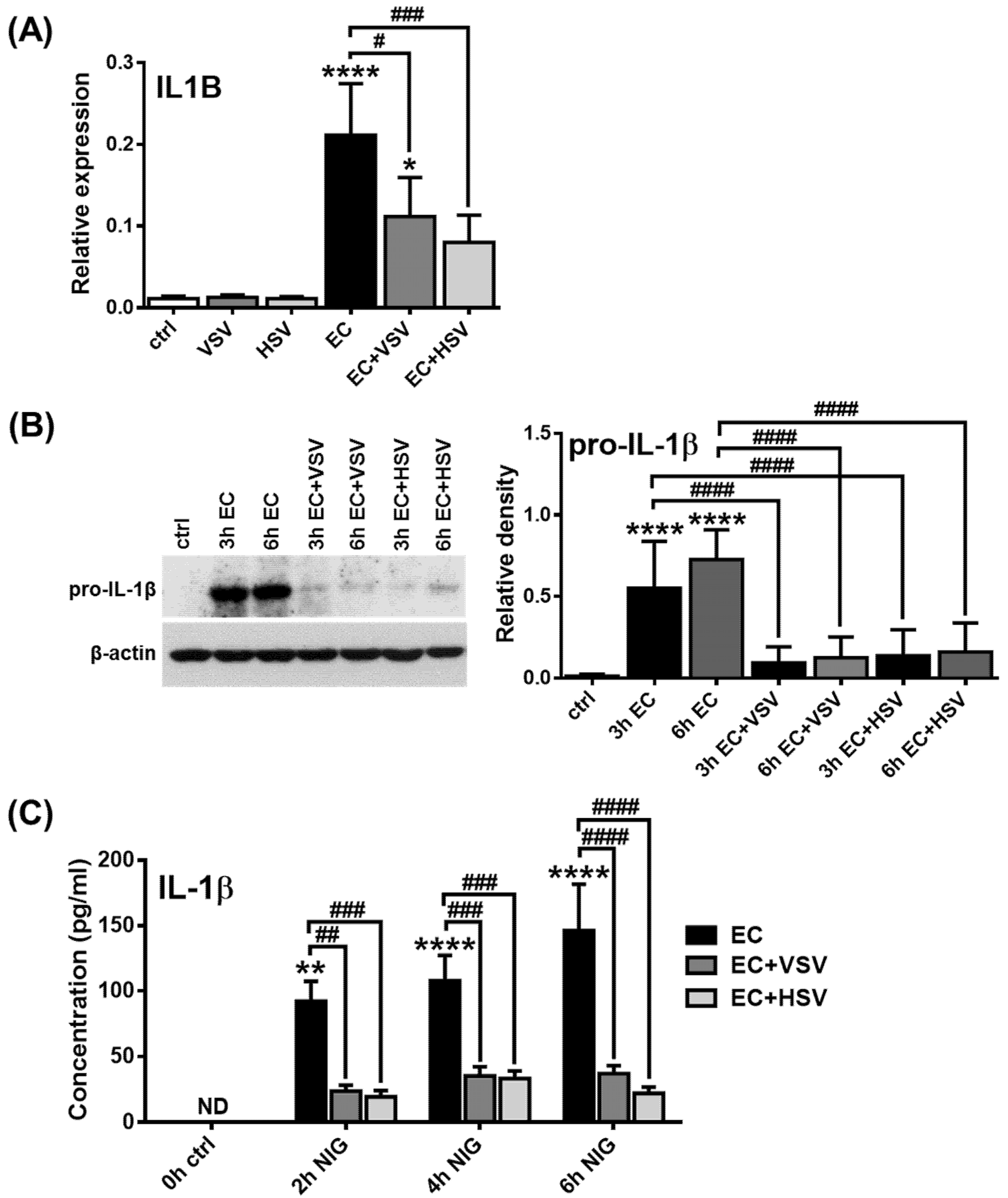
2.5. Assessing the NLRP3 Activity in pDCs in the Presence of Viruses with High Type I IFN Inducing Capacity
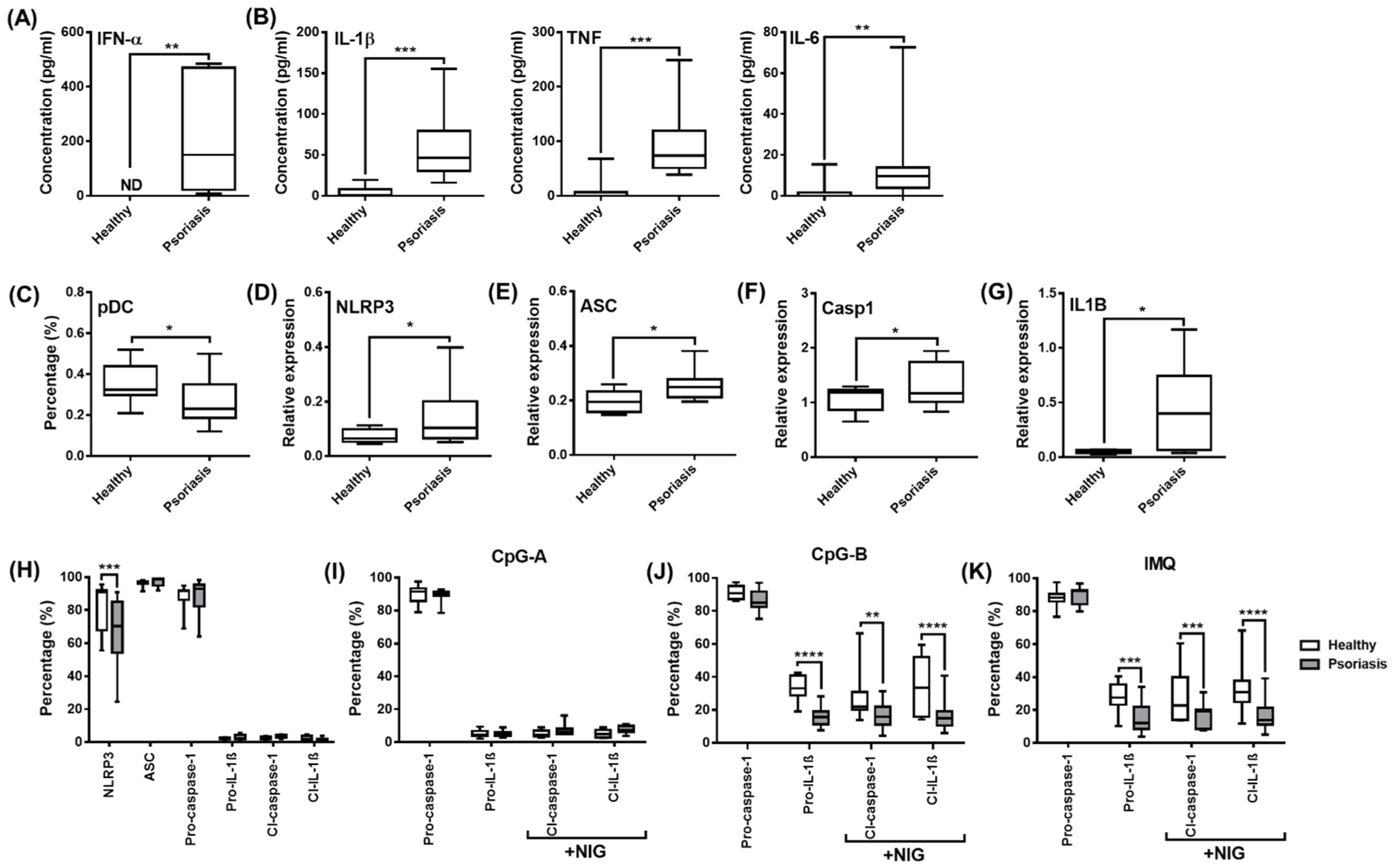
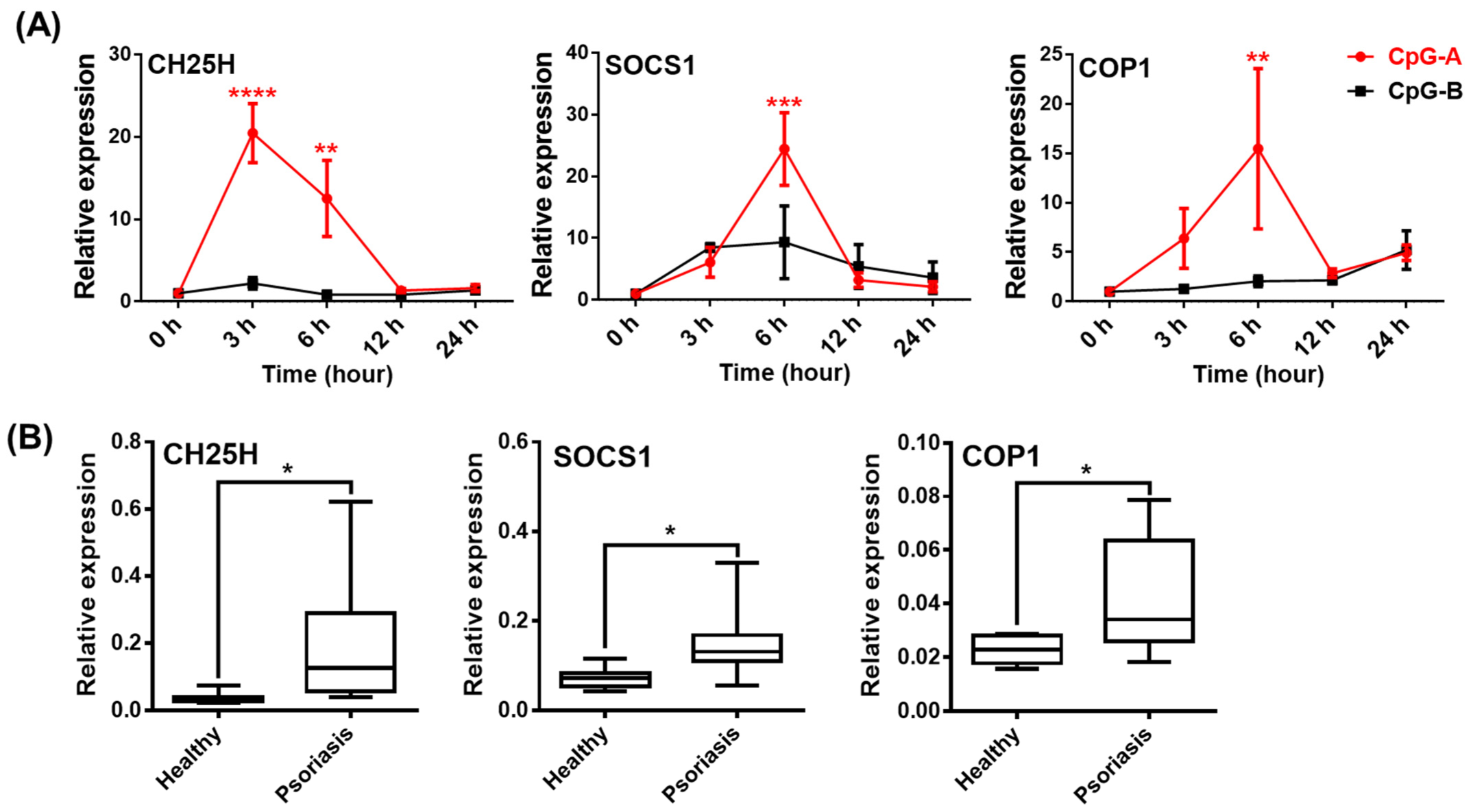
2.6. Analysis of NLRP3-Dependent IL-1β Pathway Activity in pDCs from Psoriasis Patients Associated with High IFN Signature
3. Discussion
4. Materials and Methods
4.1. GEN2.2 Cell Line
4.2. Collection of Human Blood Samples and Isolation of PBMCs
4.3. Stimulation and Treatments of the Cells
4.4. Generation and Activation of Primary Macrophages
4.5. Flow Cytometric Analysis
4.6. Confocal Microscopy
4.7. Quantitative Real-Time PCR
4.8. Western Blotting
4.9. ELISA
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef]
- Lande, R.; Gilliet, M. Plasmacytoid dendritic cells: Key players in the initiation and regulation of immune responses. Ann. N. Y. Acad. Sci. 2010, 1183, 89–103. [Google Scholar] [CrossRef]
- Ronnblom, L.; Eloranta, M.L.; Alm, G.V. Role of natural interferon-alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity 2003, 36, 463–472. [Google Scholar] [CrossRef]
- Bencze, D.; Fekete, T.; Pazmandi, K. Type I Interferon Production of Plasmacytoid Dendritic Cells under Control. Int. J. Mol. Sci. 2021, 22, 4190. [Google Scholar] [CrossRef]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef]
- Bao, M.; Liu, Y.J. Regulation of TLR7/9 signaling in plasmacytoid dendritic cells. Protein Cell 2013, 4, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Szabo, A.; Magyarics, Z.; Pazmandi, K.; Gopcsa, L.; Rajnavolgyi, E.; Bacsi, A. TLR ligands upregulate RIG-I expression in human plasmacytoid dendritic cells in a type I IFN-independent manner. Immunol. Cell Biol. 2014, 92, 671–678. [Google Scholar] [CrossRef]
- Fekete, T.; Bencze, D.; Szabo, A.; Csoma, E.; Biro, T.; Bacsi, A.; Pazmandi, K. Regulatory NLRs Control the RLR-Mediated Type I Interferon and Inflammatory Responses in Human Dendritic Cells. Front. Immunol. 2018, 9, 2314. [Google Scholar] [CrossRef]
- Deb, P.; Dai, J.; Singh, S.; Kalyoussef, E.; Fitzgerald-Bocarsly, P. Triggering of the cGAS-STING Pathway in Human Plasmacytoid Dendritic Cells Inhibits TLR9-Mediated IFN Production. J. Immunol. 2020, 205, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Dorfleutner, A.; Bryan, N.B.; Yun, C.; Radian, A.D.; de Almeida, L.; Rojanasakul, Y.; Stehlik, C. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012, 36, 464–476. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 2022, 606, 585–593. [Google Scholar] [CrossRef]
- Lei-Leston, A.C.; Murphy, A.G.; Maloy, K.J. Epithelial Cell Inflammasomes in Intestinal Immunity and Inflammation. Front. Immunol. 2017, 8, 1168. [Google Scholar] [CrossRef]
- Hatscher, L.; Amon, L.; Heger, L.; Dudziak, D. Inflammasomes in dendritic cells: Friend or foe? Immunol. Lett. 2021, 234, 16–32. [Google Scholar] [CrossRef]
- Jha, S.; Brickey, W.J.; Ting, J.P. Inflammasomes in Myeloid Cells: Warriors Within. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Gritsenko, A.; Yu, S.; Martin-Sanchez, F.; Diaz-Del-Olmo, I.; Nichols, E.M.; Davis, D.M.; Brough, D.; Lopez-Castejon, G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020, 11, 565924. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, R.; Terlizzi, M.; Di Crescenzo, V.G.; Popolo, A.; Pecoraro, M.; Perillo, G.; Galderisi, A.; Pinto, A. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1alpha in an AIM2 inflammasome-dependent manner. Am. J. Pathol. 2015, 185, 3115–3124. [Google Scholar] [CrossRef]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Nunez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Xing, Y.; Yao, X.; Li, H.; Xue, G.; Guo, Q.; Yang, G.; An, L.; Zhang, Y.; Meng, G. Cutting Edge: TRAF6 Mediates TLR/IL-1R Signaling-Induced Nontranscriptional Priming of the NLRP3 Inflammasome. J. Immunol. 2017, 199, 1561–1566. [Google Scholar] [CrossRef]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Groslambert, M.; Py, B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018, 11, 359–374. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; O’Connell, R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J. Immunol. 2007, 179, 6933–6942. [Google Scholar] [CrossRef] [PubMed]
- Masumoto, J.; Zhou, W.; Morikawa, S.; Hosokawa, S.; Taguchi, H.; Yamamoto, T.; Kurata, M.; Kaneko, N. Molecular biology of autoinflammatory diseases. Inflamm. Regen. 2021, 41, 33. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; McDermott, M.F.; Kanneganti, T.D. Inflammasomes and autoimmunity. Trends Mol. Med. 2011, 17, 57–64. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Bi, L. Role of the NLRP3 inflammasome in autoimmune diseases. Biomed. Pharmacother. 2020, 130, 110542. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W.; Li, W.; Zhao, Y. NLRP3 Inflammasome: Checkpoint Connecting Innate and Adaptive Immunity in Autoimmune Diseases. Front. Immunol. 2021, 12, 732933. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.S. Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J. Physiol. Pharmacol. 2018, 22, 1–15. [Google Scholar] [CrossRef]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Type I Interferons in Autoimmune Disease. Annu. Rev. Pathol. 2019, 14, 369–393. [Google Scholar] [CrossRef]
- Chasset, F.; Dayer, J.M.; Chizzolini, C. Type I Interferons in Systemic Autoimmune Diseases: Distinguishing Between Afferent and Efferent Functions for Precision Medicine and Individualized Treatment. Front. Pharmacol. 2021, 12, 633821. [Google Scholar] [CrossRef]
- Di Domizio, J.; Cao, W. Fueling autoimmunity: Type I interferon in autoimmune diseases. Expert Rev. Clin. Immunol. 2013, 9, 201–210. [Google Scholar] [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Forster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef]
- Labzin, L.I.; Lauterbach, M.A.; Latz, E. Interferons and inflammasomes: Cooperation and counterregulation in disease. J. Allergy Clin. Immunol. 2016, 138, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Kopitar-Jerala, N. The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 2017, 8, 873. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Robinson, K.M.; McHugh, K.J.; Scheller, E.V.; Mandalapu, S.; Chen, C.; Di, Y.P.; Clay, M.E.; Enelow, R.I.; Dubin, P.J.; et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L158–L167. [Google Scholar] [CrossRef]
- Robinson, K.M.; Choi, S.M.; McHugh, K.J.; Mandalapu, S.; Enelow, R.I.; Kolls, J.K.; Alcorn, J.F. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J. Immunol. 2013, 191, 5153–5159. [Google Scholar] [CrossRef] [PubMed]
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011, 186, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.M.; McHugh, K.J.; Mandalapu, S.; Clay, M.E.; Lee, B.; Scheller, E.V.; Enelow, R.I.; Chan, Y.R.; Kolls, J.K.; Alcorn, J.F. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J. Infect. Dis. 2014, 209, 865–875. [Google Scholar] [CrossRef]
- Li, W.; Moltedo, B.; Moran, T.M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef]
- Tian, Z.; Shen, X.; Feng, H.; Gao, B. IL-1 beta attenuates IFN-alpha beta-induced antiviral activity and STAT1 activation in the liver: Involvement of proteasome-dependent pathway. J. Immunol. 2000, 165, 3959–3965. [Google Scholar] [CrossRef]
- Kohase, M.; Zhang, Y.H.; Lin, J.X.; Yamazaki, S.; Sehgal, P.B.; Vilcek, J. Interleukin-1 can inhibit interferon-beta synthesis and its antiviral action: Comparison with tumor necrosis factor. J. Interferon Res. 1988, 8, 559–570. [Google Scholar] [CrossRef]
- Veckman, V.; Julkunen, I. Streptococcus pyogenes activates human plasmacytoid and myeloid dendritic cells. J. Leukoc. Biol. 2008, 83, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Parcina, M.; Wendt, C.; Goetz, F.; Zawatzky, R.; Zahringer, U.; Heeg, K.; Bekeredjian-Ding, I. Staphylococcus aureus-induced plasmacytoid dendritic cell activation is based on an IgG-mediated memory response. J. Immunol. 2008, 181, 3823–3833. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.L.; Morris, J.L.; Rush, C.M.; Ketheesan, N. Plasmacytoid dendritic cell bactericidal activity against Burkholderia pseudomallei. Microbes Infect. 2015, 17, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Raieli, S.; Trichot, C.; Korniotis, S.; Pattarini, L.; Soumelis, V. TLR1/2 orchestrate human plasmacytoid predendritic cell response to gram+ bacteria. PLoS Biol. 2019, 17, e3000209. [Google Scholar] [CrossRef]
- Romani, L.; Bistoni, F.; Gaziano, R.; Bozza, S.; Montagnoli, C.; Perruccio, K.; Pitzurra, L.; Bellocchio, S.; Velardi, A.; Rasi, G.; et al. Thymosin alpha 1 activates dendritic cells for antifungal Th1 resistance through toll-like receptor signaling. Blood 2004, 103, 4232–4239. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Ortiz, Z.G.; Specht, C.A.; Wang, J.P.; Lee, C.K.; Bartholomeu, D.C.; Gazzinelli, R.T.; Levitz, S.M. Toll-like receptor 9-dependent immune activation by unmethylated CpG motifs in Aspergillus fumigatus DNA. Infect. Immun. 2008, 76, 2123–2129. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Wang, J.P.; Lee, C.K.; Levitz, S.M. Cooperative stimulation of dendritic cells by Cryptococcus neoformans mannoproteins and CpG oligodeoxynucleotides. PLoS ONE 2008, 3, e2046. [Google Scholar] [CrossRef]
- Ramirez-Ortiz, Z.G.; Lee, C.K.; Wang, J.P.; Boon, L.; Specht, C.A.; Levitz, S.M. A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe 2011, 9, 415–424. [Google Scholar] [CrossRef]
- Loures, F.V.; Rohm, M.; Lee, C.K.; Santos, E.; Wang, J.P.; Specht, C.A.; Calich, V.L.; Urban, C.F.; Levitz, S.M. Recognition of Aspergillus fumigatus hyphae by human plasmacytoid dendritic cells is mediated by dectin-2 and results in formation of extracellular traps. PLoS Pathog. 2015, 11, e1004643. [Google Scholar] [CrossRef]
- Hole, C.R.; Leopold Wager, C.M.; Mendiola, A.S.; Wozniak, K.L.; Campuzano, A.; Lin, X.; Wormley, F.L., Jr. Antifungal Activity of Plasmacytoid Dendritic Cells against Cryptococcus neoformans In Vitro Requires Expression of Dectin-3 (CLEC4D) and Reactive Oxygen Species. Infect. Immun. 2016, 84, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Van de Veerdonk, F.L.; Joosten, L.A.; Netea, M.G. The interplay between inflammasome activation and antifungal host defense. Immunol. Rev. 2015, 265, 172–180. [Google Scholar] [CrossRef]
- Skeldon, A.; Saleh, M. The inflammasomes: Molecular effectors of host resistance against bacterial, viral, parasitic, and fungal infections. Front. Microbiol. 2011, 2, 15. [Google Scholar] [CrossRef]
- Tavares, A.H.; Burgel, P.H.; Bocca, A.L. Turning Up the Heat: Inflammasome Activation by Fungal Pathogens. PLoS Pathog. 2015, 11, e1004948. [Google Scholar] [CrossRef] [PubMed]
- Vladimer, G.I.; Marty-Roix, R.; Ghosh, S.; Weng, D.; Lien, E. Inflammasomes and host defenses against bacterial infections. Curr. Opin. Microbiol. 2013, 16, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Brewer, S.M.; Brubaker, S.W.; Monack, D.M. Host inflammasome defense mechanisms and bacterial pathogen evasion strategies. Curr. Opin. Immunol. 2019, 60, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Hanagata, N. CpG oligodeoxynucleotide nanomedicines for the prophylaxis or treatment of cancers, infectious diseases, and allergies. Int. J. Nanomed. 2017, 12, 515–531. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Jo, E.K. NLRP3 inflammasome and host protection against bacterial infection. J. Korean Med. Sci. 2013, 28, 1415–1423. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Agod, Z.; Fekete, T.; Budai, M.M.; Varga, A.; Szabo, A.; Moon, H.; Boldogh, I.; Biro, T.; Lanyi, A.; Bacsi, A.; et al. Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells. Redox Biol. 2017, 13, 633–645. [Google Scholar] [CrossRef]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch. Pharmacol. 2000, 362, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Schneider, E.; Rissiek, A.; Winzer, R.; Puig, B.; Rissiek, B.; Haag, F.; Mittrucker, H.W.; Magnus, T.; Tolosa, E. Generation and Function of Non-cell-bound CD73 in Inflammation. Front. Immunol. 2019, 10, 1729. [Google Scholar] [CrossRef]
- Diaz-Garcia, E.; Garcia-Tovar, S.; Alfaro, E.; Zamarron, E.; Mangas, A.; Galera, R.; Ruiz-Hernandez, J.J.; Sole-Violan, J.; Rodriguez-Gallego, C.; Van-Den-Rym, A.; et al. Role of CD39 in COVID-19 Severity: Dysregulation of Purinergic Signaling and Thromboinflammation. Front. Immunol. 2022, 13, 847894. [Google Scholar] [CrossRef]
- Morello, S.; Capone, M.; Sorrentino, C.; Giannarelli, D.; Madonna, G.; Mallardo, D.; Grimaldi, A.M.; Pinto, A.; Ascierto, P.A. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J. Transl. Med. 2017, 15, 244. [Google Scholar] [CrossRef]
- Coade, S.B.; Pearson, J.D. Metabolism of adenine nucleotides in human blood. Circ. Res. 1989, 65, 531–537. [Google Scholar] [CrossRef]
- Greaney, A.J.; Leppla, S.H.; Moayeri, M. Bacterial Exotoxins and the Inflammasome. Front. Immunol. 2015, 6, 570. [Google Scholar] [CrossRef] [PubMed]
- Eder, C. Mechanisms of interleukin-1beta release. Immunobiology 2009, 214, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Pizzirani, C.; Ferrari, D.; Chiozzi, P.; Adinolfi, E.; Sandona, D.; Savaglio, E.; Di Virgilio, F. Stimulation of P2 receptors causes release of IL-1beta-loaded microvesicles from human dendritic cells. Blood 2007, 109, 3856–3864. [Google Scholar] [CrossRef] [PubMed]
- Hanagata, N. Structure-dependent immunostimulatory effect of CpG oligodeoxynucleotides and their delivery system. Int. J. Nanomed. 2012, 7, 2181–2195. [Google Scholar] [CrossRef]
- Stehlik, C.; Dorfleutner, A. COPs and POPs: Modulators of inflammasome activity. J. Immunol. 2007, 179, 7993–7998. [Google Scholar] [CrossRef]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef]
- Reboldi, A.; Dang, E.V.; McDonald, J.G.; Liang, G.; Russell, D.W.; Cyster, J.G. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 2014, 345, 679–684. [Google Scholar] [CrossRef]
- Inoue, M.; Williams, K.L.; Oliver, T.; Vandenabeele, P.; Rajan, J.V.; Miao, E.A.; Shinohara, M.L. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci. Signal. 2012, 5, ra38. [Google Scholar] [CrossRef] [PubMed]
- Druilhe, A.; Srinivasula, S.M.; Razmara, M.; Ahmad, M.; Alnemri, E.S. Regulation of IL-1beta generation by Pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ. 2001, 8, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Fekri, S.Z.; Sigurdardottir, G.; Bivik Eding, C.; Sandin, C.; Enerback, C. Enhanced Inflammasome Activity in Patients with Psoriasis Promotes Systemic Inflammation. J. Investig. Dermatol. 2021, 141, 586–595.E5. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Peng, C.; Chen, W.; Sun, Z.; Zheng, J.; Zhang, S.; Ding, Y.; Shi, Y. Circulating Metabolomic Signature in Generalized Pustular Psoriasis Blunts Monocyte Hyperinflammation by Triggering Amino Acid Response. Front. Immunol. 2021, 12, 739514. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.C.; Paixao, C.S.; Viana, D.L.; Rocha, B.O.; Saldanha, M.; da Mota, L.M.H.; Machado, P.R.L.; Pagliari, C.; de Oliveira, M.F.; Arruda, S.; et al. Mononuclear Phagocyte Activation Is Associated With the Immunopathology of Psoriasis. Front. Immunol. 2020, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Tzvetkov, E.; Pereira, A.; Wu, Y.; Kasar, S.; Przetak, M.M.; Vlach, J.; Niewold, T.B.; Jensen, M.A.; Okitsu, S.L. TLR7 and TLR8 Differentially Activate the IRF and NF-kappaB Pathways in Specific Cell Types to Promote Inflammation. Immunohorizons 2020, 4, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Congy-Jolivet, N.; Cenac, C.; Dellacasagrande, J.; Puissant-Lubrano, B.; Apoil, P.A.; Guedj, K.; Abbas, F.; Laffont, S.; Sourdet, S.; Guyonnet, S.; et al. Monocytes are the main source of STING-mediated IFN-alpha production. EBioMedicine 2022, 80, 104047. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, C.W.; Oh, D.S.; Jung, H.E.; Lee, H.K. Monocytes Contribute to IFN-beta Production via the MyD88-Dependent Pathway and Cytotoxic T-Cell Responses against Mucosal Respiratory Syncytial Virus Infection. Immune Netw. 2021, 21, e27. [Google Scholar] [CrossRef] [PubMed]
- de Marcken, M.; Dhaliwal, K.; Danielsen, A.C.; Gautron, A.S.; Dominguez-Villar, M. TLR7 and TLR8 activate distinct pathways in monocytes during RNA virus infection. Sci. Signal. 2019, 12, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Giannessi, F.; Percario, Z.A.; Affabris, E. The involvement of plasmacytoid cells in HIV infection and pathogenesis. Cytokine Growth Factor Rev. 2018, 40, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Lewis, K.L.; Firner, S.; Thiel, V.; Hugues, S.; Reith, W.; Ludewig, B.; Reizis, B. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc. Natl. Acad. Sci. USA 2012, 109, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, J.; Zhu, S.; Liu, Y.J.; Chen, J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol. 2017, 8, 1268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lawrence, T.; Liang, Y. The Role of Plasmacytoid Dendritic Cells in Cancers. Front. Immunol. 2021, 12, 749190. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Gaugler, B.; Mohty, M.; Malard, F. Plasmacytoid dendritic cell biology and its role in immune-mediated diseases. Clin. Transl. Immunol. 2020, 9, e1139. [Google Scholar] [CrossRef]
- Panda, S.K.; Kolbeck, R.; Sanjuan, M.A. Plasmacytoid dendritic cells in autoimmunity. Curr. Opin. Immunol. 2017, 44, 20–25. [Google Scholar] [CrossRef]
- Crozat, K.; Vivier, E.; Dalod, M. Crosstalk between components of the innate immune system: Promoting anti-microbial defenses and avoiding immunopathologies. Immunol. Rev. 2009, 227, 129–149. [Google Scholar] [CrossRef]
- Worah, K.; Mathan, T.S.M.; Vu Manh, T.P.; Keerthikumar, S.; Schreibelt, G.; Tel, J.; Duiveman-de Boer, T.; Skold, A.E.; van Spriel, A.B.; de Vries, I.J.M.; et al. Proteomics of Human Dendritic Cell Subsets Reveals Subset-Specific Surface Markers and Differential Inflammasome Function. Cell Rep. 2016, 16, 2953–2966. [Google Scholar] [CrossRef]
- Hatscher, L.; Lehmann, C.H.K.; Purbojo, A.; Onderka, C.; Liang, C.; Hartmann, A.; Cesnjevar, R.; Bruns, H.; Gross, O.; Nimmerjahn, F.; et al. Select hyperactivating NLRP3 ligands enhance the TH1- and TH17-inducing potential of human type 2 conventional dendritic cells. Sci. Signal. 2021, 14, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Chaperot, L.; Blum, A.; Manches, O.; Lui, G.; Angel, J.; Molens, J.P.; Plumas, J. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 2006, 176, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Barr, D.P.; Belz, G.T.; Reading, P.C.; Wojtasiak, M.; Whitney, P.G.; Heath, W.R.; Carbone, F.R.; Brooks, A.G. A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of NK cells following HSV-1 infection. Eur. J. Immunol. 2007, 37, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Lousberg, E.L.; Diener, K.R.; Fraser, C.K.; Phipps, S.; Foster, P.S.; Chen, W.; Uematsu, S.; Akira, S.; Robertson, S.A.; Brown, M.P.; et al. Antigen-specific T-cell responses to a recombinant fowlpox virus are dependent on MyD88 and interleukin-18 and independent of Toll-like receptor 7 (TLR7)- and TLR9-mediated innate immune recognition. J. Virol. 2011, 85, 3385–3396. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Colafrancesco, S.; Emmi, G.; Imazio, M.; Lopalco, G.; Maggio, M.C.; Sota, J.; Dinarello, C.A. Interleukin 1alpha: A comprehensive review on the role of IL-1alpha in the pathogenesis and treatment of autoimmune and inflammatory diseases. Autoimmun. Rev. 2021, 20, 102763. [Google Scholar] [CrossRef]
- Gross, C.J.; Mishra, R.; Schneider, K.S.; Medard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.A.; Fromm, S.; et al. K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef]
- Budai, M.M.; Tozser, J.; Benko, S. Different dynamics of NLRP3 inflammasome-mediated IL1beta production in GM-CSF- and M-CSF-differentiated human macrophages. J. Leukoc. Biol. 2017, 101, 1335–1347. [Google Scholar] [CrossRef]
- Ipseiz, N.; Pickering, R.J.; Rosas, M.; Tyrrell, V.J.; Davies, L.C.; Orr, S.J.; Czubala, M.A.; Fathalla, D.; Robertson, A.A.; Bryant, C.E.; et al. Tissue-resident macrophages actively suppress IL-1beta release via a reactive prostanoid/IL-10 pathway. EMBO J. 2020, 39, e103454. [Google Scholar] [CrossRef]
- Piccini, A.; Carta, S.; Tassi, S.; Lasiglie, D.; Fossati, G.; Rubartelli, A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc. Natl. Acad. Sci. USA 2008, 105, 8067–8072. [Google Scholar] [CrossRef]
- Kovacs, E.G.; Alatshan, A.; Budai, M.M.; Czimmerer, Z.; Biro, E.; Benko, S. Caffeine Has Different Immunomodulatory Effect on the Cytokine Expression and NLRP3 Inflammasome Function in Various Human Macrophage Subpopulations. Nutrients 2021, 13, 2409. [Google Scholar] [CrossRef]
- Schon, M.; Bong, A.B.; Drewniok, C.; Herz, J.; Geilen, C.C.; Reifenberger, J.; Benninghoff, B.; Slade, H.B.; Gollnick, H.; Schon, M.P. Tumor-selective induction of apoptosis and the small-molecule immune response modifier imiquimod. J. Natl. Cancer Inst. 2003, 95, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Riol-Blanco, L.; Ordovas-Montanes, J.; Perro, M.; Naval, E.; Thiriot, A.; Alvarez, D.; Paust, S.; Wood, J.N.; von Andrian, U.H. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 2014, 510, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Liu, X.; Tan, X.Z.; Jiang, C.X.; Chen, S.W.; Liang, G.N.; He, X.M.; Wu, J.; Chen, T.; Xu, Y. ROS-induced NLRP3 inflammasome priming and activation mediate PCB 118- induced pyroptosis in endothelial cells. Ecotoxicol. Environ. Saf. 2020, 189, 109937. [Google Scholar] [CrossRef]
- Netea, M.G.; Nold-Petry, C.A.; Nold, M.F.; Joosten, L.A.; Opitz, B.; van der Meer, J.H.; van de Veerdonk, F.L.; Ferwerda, G.; Heinhuis, B.; Devesa, I.; et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009, 113, 2324–2335. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, Z.S.; Xue, W.; Bai, Z.F.; Wang, Q.Y.; Dai, J.; Liu, X.; Huang, Y.J.; Cai, H.; Zhan, X.Y.; et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol. Cell 2017, 68, 185–197.E6. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Hornung, V. Alternative inflammasome activation enables IL-1beta release from living cells. Curr. Opin. Immunol. 2017, 44, 7–13. [Google Scholar] [CrossRef]
- Zhang, W.; An, E.K.; Hwang, J.; Jin, J.O. Mice Plasmacytoid Dendritic Cells Were Activated by Lipopolysaccharides Through Toll-Like Receptor 4/Myeloid Differentiation Factor 2. Front. Immunol. 2021, 12, 727161. [Google Scholar] [CrossRef] [PubMed]
- Ohradanova-Repic, A.; Machacek, C.; Fischer, M.B.; Stockinger, H. Differentiation of human monocytes and derived subsets of macrophages and dendritic cells by the HLDA10 monoclonal antibody panel. Clin. Transl. Immunol. 2016, 5, e55. [Google Scholar] [CrossRef]
- Ferrari, D.; La Sala, A.; Chiozzi, P.; Morelli, A.; Falzoni, S.; Girolomoni, G.; Idzko, M.; Dichmann, S.; Norgauer, J.; Di Virgilio, F. The P2 purinergic receptors of human dendritic cells: Identification and coupling to cytokine release. FASEB J. 2000, 14, 2466–2476. [Google Scholar] [CrossRef]
- See, P.; Dutertre, C.A.; Chen, J.; Gunther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Handler, K.; Duan, K.; et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science 2017, 7, 69. [Google Scholar] [CrossRef]
- Matsui, T.; Connolly, J.E.; Michnevitz, M.; Chaussabel, D.; Yu, C.I.; Glaser, C.; Tindle, S.; Pypaert, M.; Freitas, H.; Piqueras, B.; et al. CD2 distinguishes two subsets of human plasmacytoid dendritic cells with distinct phenotype and functions. J. Immunol. 2009, 182, 6815–6823. [Google Scholar] [CrossRef] [PubMed]
- Burgueno-Bucio, E.; Mier-Aguilar, C.A.; Soldevila, G. The multiple faces of CD5. J. Leukoc. Biol. 2019, 105, 891–904. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef]
- Hagen, S.H.; Henseling, F.; Hennesen, J.; Savel, H.; Delahaye, S.; Richert, L.; Ziegler, S.M.; Altfeld, M. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human pDC Level. Cell Rep. 2020, 33, 108485. [Google Scholar] [CrossRef] [PubMed]
- Onodi, F.; Bonnet-Madin, L.; Meertens, L.; Karpf, L.; Poirot, J.; Zhang, S.Y.; Picard, C.; Puel, A.; Jouanguy, E.; Zhang, Q.; et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J. Exp. Med. 2021, 218, e20201387. [Google Scholar] [CrossRef]
- Alculumbre, S.G.; Saint-Andre, V.; Di Domizio, J.; Vargas, P.; Sirven, P.; Bost, P.; Maurin, M.; Maiuri, P.; Wery, M.; Roman, M.S.; et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat. Immunol. 2018, 19, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Llufrio, E.M.; Wang, L.; Naser, F.J.; Patti, G.J. Sorting cells alters their redox state and cellular metabolome. Redox Biol. 2018, 16, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Magyarics, Z.; Csillag, A.; Pazmandi, K.; Rajnavolgyi, E.; Bacsi, A. Identification of plasmacytoid pre-dendritic cells by one-color flow cytometry for phenotype screening. Cytometry A 2008, 73, 254–258. [Google Scholar] [CrossRef]
- Yu, C.F.; Peng, W.M.; Oldenburg, J.; Hoch, J.; Bieber, T.; Limmer, A.; Hartmann, G.; Barchet, W.; Eis-Hubinger, A.M.; Novak, N. Human plasmacytoid dendritic cells support Th17 cell effector function in response to TLR7 ligation. J. Immunol. 2010, 184, 1159–1167. [Google Scholar] [CrossRef]
- Rodriguez Rodrigues, C.; Cabrini, M.; Remes Lenicov, F.; Sabatte, J.; Ceballos, A.; Jancic, C.; Raiden, S.; Ostrowski, M.; Silberstein, C.; Geffner, J. Epithelial cells activate plasmacytoid dendritic cells improving their anti-HIV activity. PLoS ONE 2011, 6, e28709. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald-Bocarsly, P.; Feng, D. The role of type I interferon production by dendritic cells in host defense. Biochimie 2007, 89, 843–855. [Google Scholar] [CrossRef]
- Corrales, L.; Woo, S.R.; Williams, J.B.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. Antagonism of the STING Pathway via Activation of the AIM2 Inflammasome by Intracellular DNA. J. Immunol. 2016, 196, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Du, Y.; Cai, C.; Cai, B.; Zhu, M.; Xing, C.; Tan, P.; Lin, M.; Wu, J.; Li, J.; et al. Inflammasome activation negatively regulates MyD88-IRF7 type I IFN signaling and anti-malaria immunity. Nat. Commun. 2018, 9, 4964. [Google Scholar] [CrossRef]
- Yang, C.A.; Chiang, B.L. Inflammasomes and human autoimmunity: A comprehensive review. J. Autoimmun. 2015, 61, 1–8. [Google Scholar] [CrossRef]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef]
- Wang, A.; Bai, Y. Dendritic cells: The driver of psoriasis. J. Dermatol. 2020, 47, 104–113. [Google Scholar] [CrossRef]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [CrossRef]
- Boonstra, A.; Rajsbaum, R.; Holman, M.; Marques, R.; Asselin-Paturel, C.; Pereira, J.P.; Bates, E.E.; Akira, S.; Vieira, P.; Liu, Y.J.; et al. Macrophages and myeloid dendritic cells, but not plasmacytoid dendritic cells, produce IL-10 in response to MyD88- and TRIF-dependent TLR signals, and TLR-independent signals. J. Immunol. 2006, 177, 7551–7558. [Google Scholar] [CrossRef]
- Morita, R.; Uchiyama, T.; Hori, T. Nitric oxide inhibits IFN-alpha production of human plasmacytoid dendritic cells partly via a guanosine 3’,5’-cyclic monophosphate-dependent pathway. J. Immunol. 2005, 175, 806–812. [Google Scholar] [CrossRef]
- Johansen, C.; Moeller, K.; Kragballe, K.; Iversen, L. The activity of caspase-1 is increased in lesional psoriatic epidermis. J. Invest. Dermatol. 2007, 127, 2857–2864. [Google Scholar] [CrossRef]
- Tervaniemi, M.H.; Katayama, S.; Skoog, T.; Siitonen, H.A.; Vuola, J.; Nuutila, K.; Sormunen, R.; Johnsson, A.; Linnarsson, S.; Suomela, S.; et al. NOD-like receptor signaling and inflammasome-related pathways are highlighted in psoriatic epidermis. Sci. Rep. 2016, 6, 22745. [Google Scholar] [CrossRef]
- Ciazynska, M.; Olejniczak-Staruch, I.; Sobolewska-Sztychny, D.; Narbutt, J.; Skibinska, M.; Lesiak, A. The Role of NLRP1, NLRP3, and AIM2 Inflammasomes in Psoriasis: Review. Int. J. Mol. Sci. 2021, 22, 5898. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Dubyak, G.R. Differing caspase-1 activation states in monocyte versus macrophage models of IL-1beta processing and release. J. Leukoc. Biol. 2004, 76, 676–684. [Google Scholar] [CrossRef]
- Ito, T.; Kanzler, H.; Duramad, O.; Cao, W.; Liu, Y.J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 2006, 107, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Azar, P.; Mejia, J.E.; Cenac, C.; Shaiykova, A.; Youness, A.; Laffont, S.; Essat, A.; Izopet, J.; Passaes, C.; Muller-Trutwin, M.; et al. TLR7 dosage polymorphism shapes interferogenesis and HIV-1 acute viremia in women. JCI Insight 2020, 5, e136047. [Google Scholar] [CrossRef]
- Lacerte, P.; Brunet, A.; Egarnes, B.; Duchene, B.; Brown, J.P.; Gosselin, J. Overexpression of TLR2 and TLR9 on monocyte subsets of active rheumatoid arthritis patients contributes to enhance responsiveness to TLR agonists. Arthritis Res. Ther. 2016, 18, 10. [Google Scholar] [CrossRef]
- Juarez, E.; Nunez, C.; Sada, E.; Ellner, J.J.; Schwander, S.K.; Torres, M. Differential expression of Toll-like receptors on human alveolar macrophages and autologous peripheral monocytes. Respir. Res. 2010, 11, 2. [Google Scholar] [CrossRef]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef]
- Shamaa, O.R.; Mitra, S.; Gavrilin, M.A.; Wewers, M.D. Monocyte Caspase-1 Is Released in a Stable, Active High Molecular Weight Complex Distinct from the Unstable Cell Lysate-Activated Caspase-1. PLoS ONE 2015, 10, e0142203. [Google Scholar] [CrossRef]
- Mitra, S.; Wewers, M.D.; Sarkar, A. Mononuclear Phagocyte-Derived Microparticulate Caspase-1 Induces Pulmonary Vascular Endothelial Cell Injury. PLoS ONE 2015, 10, e0145607. [Google Scholar] [CrossRef]
- Bacsi, A.; Ebbesen, P.; Szabo, J.; Beck, Z.; Andirko, I.; Csoma, E.; Toth, F.D. Pseudotypes of vesicular stomatitis virus-bearing envelope antigens of certain HIV-1 strains permissively infect human syncytiotrophoblasts cultured in vitro: Implications for in vivo infection of syncytiotrophoblasts by cell-free HIV-1. J. Med. Virol. 2001, 64, 387–397. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bencze, D.; Fekete, T.; Pfliegler, W.; Szöőr, Á.; Csoma, E.; Szántó, A.; Tarr, T.; Bácsi, A.; Kemény, L.; Veréb, Z.; et al. Interactions between the NLRP3-Dependent IL-1β and the Type I Interferon Pathways in Human Plasmacytoid Dendritic Cells. Int. J. Mol. Sci. 2022, 23, 12154. https://doi.org/10.3390/ijms232012154
Bencze D, Fekete T, Pfliegler W, Szöőr Á, Csoma E, Szántó A, Tarr T, Bácsi A, Kemény L, Veréb Z, et al. Interactions between the NLRP3-Dependent IL-1β and the Type I Interferon Pathways in Human Plasmacytoid Dendritic Cells. International Journal of Molecular Sciences. 2022; 23(20):12154. https://doi.org/10.3390/ijms232012154
Chicago/Turabian StyleBencze, Dóra, Tünde Fekete, Walter Pfliegler, Árpád Szöőr, Eszter Csoma, Antónia Szántó, Tünde Tarr, Attila Bácsi, Lajos Kemény, Zoltán Veréb, and et al. 2022. "Interactions between the NLRP3-Dependent IL-1β and the Type I Interferon Pathways in Human Plasmacytoid Dendritic Cells" International Journal of Molecular Sciences 23, no. 20: 12154. https://doi.org/10.3390/ijms232012154
APA StyleBencze, D., Fekete, T., Pfliegler, W., Szöőr, Á., Csoma, E., Szántó, A., Tarr, T., Bácsi, A., Kemény, L., Veréb, Z., & Pázmándi, K. (2022). Interactions between the NLRP3-Dependent IL-1β and the Type I Interferon Pathways in Human Plasmacytoid Dendritic Cells. International Journal of Molecular Sciences, 23(20), 12154. https://doi.org/10.3390/ijms232012154