
Towards an Ideal In Cell Hybridization-Based Strategy to Discover Protein Interactomes of Selected RNA Molecules


 , and
, and
Abstract
:1. Introduction
2. Experimental Design
2.1. Capture Oligonucleotide Design
2.2. Cell Number and Cell Type Choices
3. Purification Procedure
3.1. Crosslinking
3.2. Cell Lysis and Lysate Preparation for Hybridization
3.3. Hybridization
3.4. Bead Coupling and Washing
3.5. Elution
4. Post-Purification
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Rissland, O.S. The organization and regulation of mRNA–protein complexes. Wiley Interdiscip. Rev. RNA 2017, 8, e1369. [Google Scholar] [CrossRef]
- Marchese, D.; de Groot, N.S.; Lorenzo Gotor, N.; Livi, C.M.; Tartaglia, G.G. Advances in the characterization of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2016, 7, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Zealy, R.W.; Wrenn, S.P.; Davila, S.; Min, K.W.; Yoon, J.H. MicroRNA-binding proteins: Specificity and function. Wiley Intediscip. Rev. RNA 2017, 8. [Google Scholar] [CrossRef]
- Ferrè, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein-lncRNA interaction. Brief. Bioinform. 2016, 17, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, D.; Xie, J. Aberrant splicing in neurological diseases. Wiley Interdiscip. Rev. RNA 2013, 4, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, K.; Calin, G.A.; Pichler, M. RNA-Binding Proteins as Important Regulators of Long Non-Coding RNAs in Cancer. Int. J. Mol. Sci. 2020, 21, 2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasinski-Bergner, S.; Steven, A.; Seliger, B. The Role of the RNA-Binding Protein Family MEX-3 in Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 5209. [Google Scholar] [CrossRef] [PubMed]
- Libner, C.D.; Salapa, H.E.; Levin, M.C. The Potential Contribution of Dysfunctional RNA-Binding Proteins to the Patho-genesis of Neurodegeneration in Multiple Sclerosis and Relevant Models. Int. J. Mol. Sci. 2020, 21, 4571. [Google Scholar] [CrossRef]
- Lim, Y.W.; James, D.; Huang, J.; Lee, M. The Emerging Role of the RNA-Binding Protein SFPQ in Neuronal Function and Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 7151. [Google Scholar] [CrossRef]
- Suñé-Pou, M.; Prieto-Sánchez, S.; Boyero-Corral, S.; Moreno-Castro, C.; El Yousfi, Y.; Suñé-Negre, J.M.; Hernández-Munain, C.; Suñé, C. Targeting Splicing in the Treatment of Human Disease. Genes 2017, 8, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, J.; Miscianinov, V.; Sluimer, J.C.; Newby, D.E.; Baker, A.H. Targeting Non-coding RNA in Vascular Biology and Disease. Front. Physiol. 2018, 9, 1655. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.T.; Han, C.; Sun, Y.M.; Chen, T.Q.; Chen, Y.Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55. [Google Scholar] [CrossRef]
- Bell, J.L.; Hagemann, S.; Holien, J.K.; Liu, T.; Nagy, Z.; Schulte, J.H.; Misiak, D.; Hüttelmaier, S. Identification of RNA-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma. Int. J. Mol. Sci. 2020, 21, 5098. [Google Scholar] [CrossRef]
- Glaß, M.; Michl, P.; Hüttelmaier, A.S. RNA Binding Proteins as Drivers and Therapeutic Target Candidates in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 4190. [Google Scholar] [CrossRef] [PubMed]
- Colantoni, A.; Rupert, J.; Vandelli, A.; Tartaglia, G.G.; Zacco, E. Zooming in on RNA–protein interactions: A multi-level workflow to identify interaction partners. Biochem. Soc. Trans. 2020, 48, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Gerber, A.P. RNA-Centric Approaches to Profile the RNA–Protein Interaction Landscape on Selected RNAs. Non-Coding RNA 2021, 7, 11. [Google Scholar] [CrossRef]
- Smith, J.M.; Sandow, J.J.; Webb, A.I. The search for RNA-binding proteins: A technical and interdisciplinary challenge. Biochem. Soc. Trans. 2021, 49, 393–403. [Google Scholar] [CrossRef]
- Masuda, A.; Kawachi, T.; Ohno, K. Rapidly Growing Protein-Centric Technologies to Extensively Identify RNA–Protein Interactions: Application to the Analysis of Co-Transcriptional RNA Processing. Int. J. Mol. Sci. 2021, 22, 5312. [Google Scholar] [CrossRef]
- Moore, K.S.; ‘t Hoen, P. Computational approaches for the analysis of RNA–protein interactions: A primer for biologists. J. Biol. Chem. 2019, 294, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theil, K.; Imami, K.; Rajewsky, N. Identification of proteins and miRNAs that specifically bind an mRNA in vivo. Nat. Commun. 2019, 10, 4205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.R.; Luo, H.; Vari, H.K.; Cox, B.J.; Simmonds, A.J.; Krause, H.M.; Lipshitz, H.D.; Smibert, C.A. A multiprotein complex that mediates translational enhancement in Drosophila. J. Biol. Chem. 2007, 282, 34031–34038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, J.R.; Collins, K. Human Y5 RNA specializes a Ro ribonucleoprotein for 5S ribosomal RNA quality control. Genes Dev. 2007, 21, 3067–3072. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, M.; Porter, D.F.; Khavari, P.A. Methods to study RNA–protein interactions. Nat. Methods 2019, 16, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Slobodin, B.; Gerst, J.E. A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA 2010, 16, 2277–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samra, N.; Atir-Lande, A.; Pnueli, L.; Arava, Y. The elongation factor eEF3 (Yef3) interacts with mRNA in a translation independent manner. BMC Mol. Biol. 2015, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative profiling of in vivo-assembled RNA–protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteom. 2011, 10, M110.007385. [Google Scholar] [CrossRef] [Green Version]
- Iadevaia, V.; Wouters, M.D.; Kanitz, A.; Matia-González, A.M.; Laing, E.E.; Gerber, A.P. Tandem RNA isolation reveals functional rearrangement of RNA-binding proteins on CDKN1B/p27Kip1 3’UTRs in cisplatin treated cells. RNA Biol. 2020, 17, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Popp, M.W.; Maquat, L.E. Biochemical analysis of long non-coding RNA-containing ribonucleoprotein complexes. Methods 2012, 58, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Gumireddy, K.; Li, A.; Yan, J.; Setoyama, T.; Johannes, G.J.; Orom, U.A.; Tchou, J.; Liu, Q.; Zhang, L.; Speicher, D.W.; et al. Identification of a long non-coding RNA-associated RNP complex regulating metastasis at the translational step. EMBO J. 2013, 32, 2672–2684. [Google Scholar] [CrossRef] [PubMed]
- Graindorge, A.; Pinheiro, I.; Nawrocka, A.; Mallory, A.C.; Tsvetkov, P.; Gil, N.; Carolis, C.; Buchholz, F.; Ulitsky, I.; Heard, E.; et al. In-cell identification and measurement of RNA–protein interactions. Nat. Commun. 2019, 10, 5317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Sun, W.; Shi, T.; Lu, P.; Zhuang, M.; Liu, J.L. Capturing RNA–protein interaction via CRUIS. Nucleic Acids Res. 2020, 48, e52. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.D. Insight into lncRNA biology using hybridization capture analyses. Biochim. Biophys. Acta 2016, 1859, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Mazar, J.; Zhao, W.; Khalil, A.M.; Lee, B.; Shelley, J.; Govindarajan, S.S.; Yamamoto, F.; Ratnam, M.; Aftab, M.N.; Collins, S.; et al. The functional characterization of long noncoding RNA SPRY4-IT1 in human melanoma cells. Oncotarget 2014, 5, 8959–8969. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [Green Version]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [Green Version]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Zielinski, J.; Kilk, K.; Peritz, T.; Kannanayakal, T.; Miyashiro, K.Y.; Eiríksdóttir, E.; Jochems, J.; Langel, U.; Eberwine, J. In vivo identification of ribonucleoprotein–RNA interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 1557–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoener, R.A.; Becker, J.T.; Scalf, M.; Sherer, N.M.; Smith, L.M. Elucidating the in vivo interactome of HIV-1 RNA by hybridization capture and mass spectrometry. Sci. Rep. 2017, 7, 16965. [Google Scholar] [CrossRef] [PubMed]
- Knoener, R.; Evans, E., 3rd; Becker, J.T.; Scalf, M.; Benner, B.; Sherer, N.M.; Smith, L.M. Identification of host proteins dif-ferentially associated with HIV-1 RNA splice variants. eLife 2021, 10, e62470. [Google Scholar] [CrossRef] [PubMed]
- Henke, K.B.; Miller, R.M.; Knoener, R.A.; Scalf, M.; Spiniello, M.; Smith, L.M. Identifying Protein Interactomes of Target RNAs Using HyPR-MS. Methods Mol. Biol. 2022, 2404, 219–244. [Google Scholar] [CrossRef] [PubMed]
- Spiniello, M.; Knoener, R.A.; Steinbrink, M.I.; Yang, B.; Cesnik, A.J.; Buxton, K.E.; Scalf, M.; Jarrard, D.F.; Smith, L.M. HyPR-MS for Multiplexed Discovery of MALAT1, NEAT1, and NORAD lncRNA Protein Interactomes. J. Proteome Res. 2018, 17, 3022–3038. [Google Scholar] [CrossRef] [PubMed]
- Spiniello, M.; Steinbrink, M.I.; Cesnik, A.J.; Miller, R.M.; Scalf, M.; Shortreed, M.R.; Smith, L.M. Comprehensive in vivo identification of the c-Myc mRNA protein interactome using HyPR-MS. RNA 2019, 25, 1337–1352. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Kennedy-Darling, J.; Holden, M.T.; Shortreed, M.R.; Smith, L.M. Multiplexed programmable release of captured DNA. CheBioChem 2014, 15, 2353–2356. [Google Scholar] [CrossRef] [Green Version]
- Urdaneta, E.C.; Beckmann, B.M. Fast and unbiased purification of RNA–protein complexes after UV cross-linking. Methods 2020, 178, 72–82. [Google Scholar] [CrossRef]
- Au, P.C.; Helliwell, C.; Wang, M.B. Characterizing RNA–protein interaction using cross-linking and metabolite supplemented nuclear RNA-immunoprecipitation. Mol. Biol. Rep. 2014, 41, 2971–2977. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Russell, P.; Guttman, M. Methods for comprehensive experimental identification of RNA–protein interactions. Genome Biol. 2014, 15, 203. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, E.C.; van Nostrand, E.L.; Yeo, G.W. Advances and challenges in the detection of transcriptome-wide RNA–protein interactions. Wiley Interdiscip. Rev. RNA 2018, 9, e1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Pratt, G.; Yeo, G.W.; Moore, M.J. The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu. Rev. Biochem. 2015, 84, 325–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariza-Mateos, A.; Prieto-Vega, S.; Díaz-Toledano, R.; Birk, A.; Szeto, H.; Mena, I.; Berzal-Herranz, A.; Gómez, J. RNA self-cleavage activated by ultraviolet light-induced oxidation. Nucleic Acids Res. 2012, 40, 1748–1766. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.A.; Frey, B.L.; Smith, L.M.; Auble, D.T. Formaldehyde crosslinking: A tool for the study of chromatin complexes. J. Biol. Chem. 2015, 290, 26404–26411. [Google Scholar] [CrossRef] [Green Version]
- Machyna, M.; Simon, M.D. Catching RNAs on chromatin using hybridization capture methods. Brief. Funct. Genom. 2018, 17, 96–103. [Google Scholar] [CrossRef]
- Zhang, P.; Lehmann, B.D.; Shyr, Y.; Guo, Y. The Utilization of Formalin Fixed-Paraffin-Embedded Specimens in High Throughput Genomic Studies. Int. J. Genom. 2017, 2017, 1926304. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.; Mason, J.O.; Lathe, R. Hybridization parameters revisited: Solutions containing SDS. BioTechniques 2002, 33, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Søe, M.J.; Møller, T.; Dufva, M.; Holmstrøm, K. A sensitive alternative for microRNA in situ hybridizations using probes of 2′-O-methyl RNA + LNA. J. Histochem. Cytochem. 2011, 59, 661–672. [Google Scholar] [CrossRef]
- Sinigaglia, C.; Thiel, D.; Hejnol, A.; Houliston, E.; Leclère, L. A safer, urea-based in situ hybridization method improves de-tection of gene expression in diverse animal species. Dev. Biol. 2018, 434, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lim, H.J.; Son, A. Characterization of denaturation and renaturation of DNA for DNA hybridization. Env. Health Toxicol. 2014, 29, e2014007. [Google Scholar] [CrossRef]
- Oeffinger, M. Two steps forward--one step back: Advances in affinity purification mass spectrometry of macromolecular complexes. Proteomics 2012, 12, 1591–1608. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Ruepp, A.; Brauner, B.; Dunger-Kaltenbach, I.; Frishman, G.; Montrone, C.; Stransky, M.; Waegele, B.; Schmidt, T.; Doudieu, O.N.; Stümpflen, V.; et al. CORUM: The comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008, 36, D646–D650. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhou, W.; Velculescu, V.E.; Kern, S.E.; Hruban, R.H.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Gene expression profiles in normal and cancer cells. Science 1997, 276, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
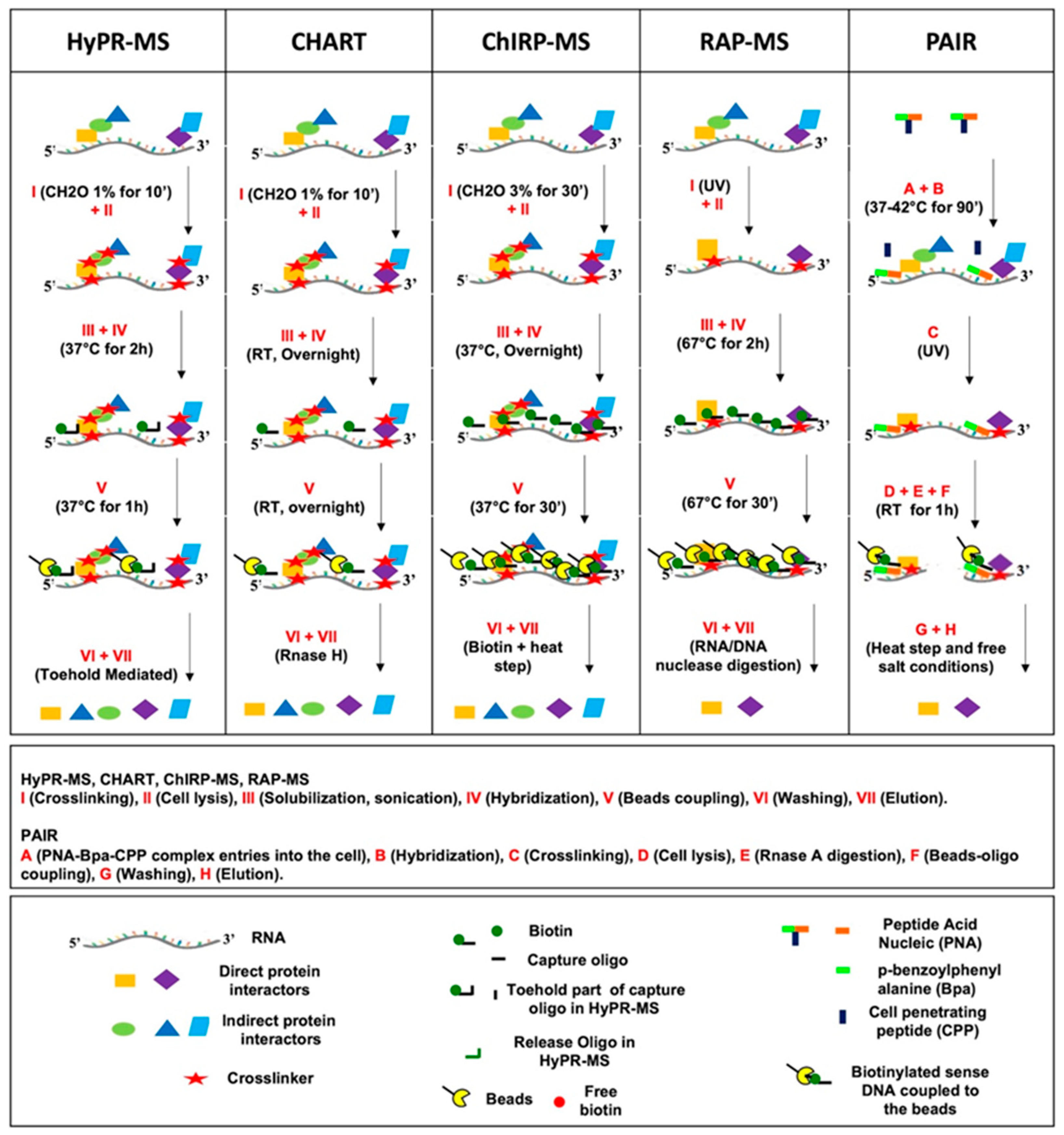

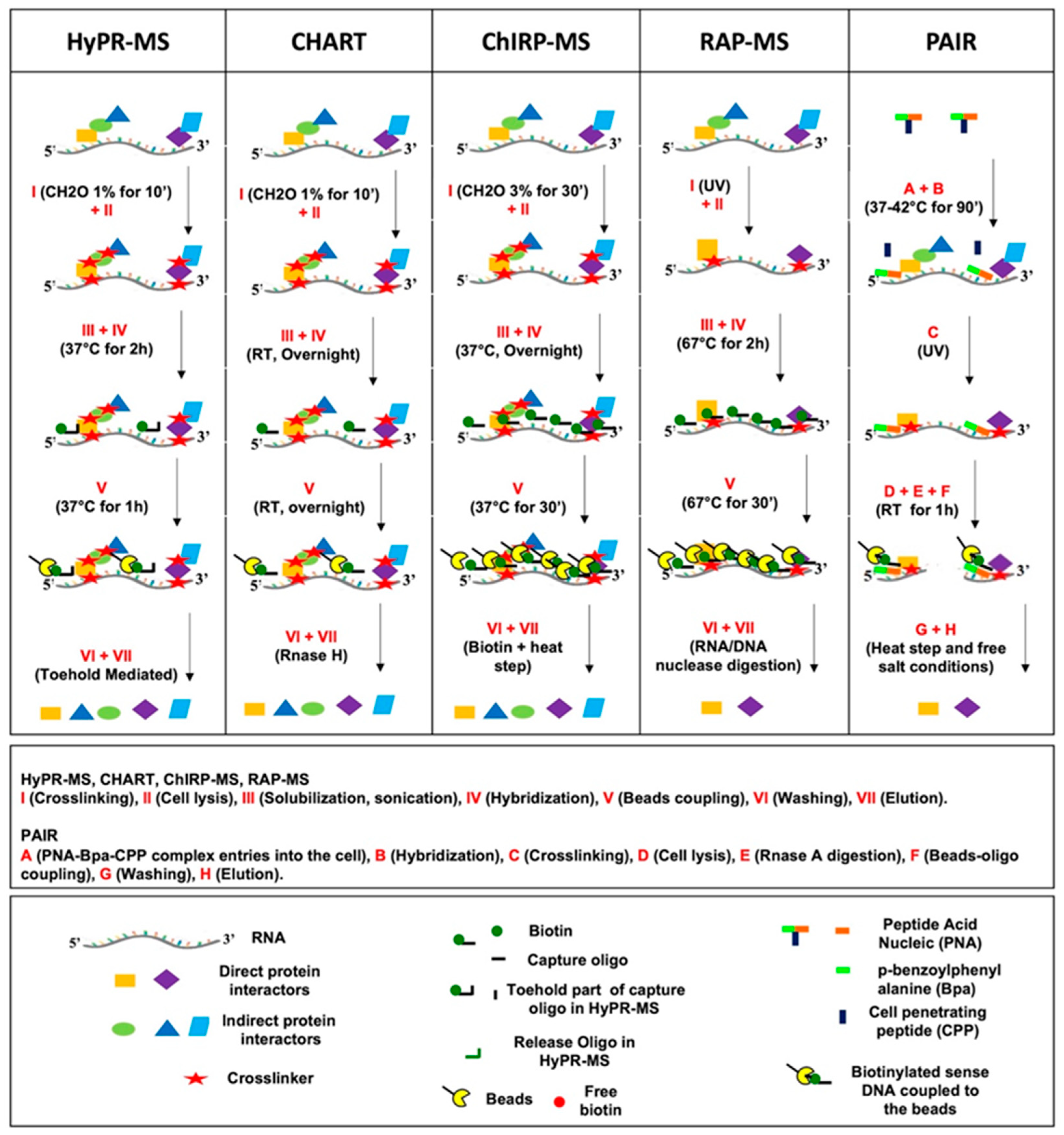{kind=link}
| PROCEDURE | CHART-MS [37,38] | ChIRP-MS [39] | RAP-MS [40] | PAIR [41] | HyPR-MS [42,43,44,45,46] |
|---|---|---|---|---|---|
| Type, cell amount used and target RNA studied with copy number per cell (cpc) estimation | MCF-7 and BJ cells (108 cells)
| HeLa S3 cells (1–5 × 108 cells)
(1–5 × 108 cells)
| - Wild-type V6.5 ES cells (2–8 × 108 cells)
| Cortical cells (1–5 × 104) - ank mRNA | HIV-1 infected Jurkat cells (5–7.5 × 107 cells)
(1 × 108 cells)
(1 × 108 cells)
|
| Controls | Lysate | Xist lncRNA capture:
| - Xist lncRNA capture:
|
|
|
| Crosslinking | 1% formaldehyde for 10 min on cells and 3% formaldehyde for 30 min on nuclei | 3% formaldehyde for 30 min | UV irradiation | UV irradiation of Bpa, a photoactivatable compound linked to capture probe system | 1% formaldehyde for 10 min (0.25% formaldehyde for 10 min in HIV1 splice variant study) |
| Solubilization | Sonication; 3 kbp is median size of chromatin fragments | Sonication; 100–500 bp is the length of chromatin fragments | Sonication and DNAse digestion;100–300 bp is the size of chromatin fragments length | Sonication; About 6 kbp is the median size of chromatin fragments | |
| Probes features | 25 nt DNA biotinylated at 3′ Tm 55–65 °C | 20 nt DNA biotinylated at 3′ | 90 nt DNA biotinylated at 3′ | PNA coupled to a CPP and to a Bpa Biotinylated sense DNA (antisense to PNA) | Hybridization part of 20–30 nt DNA biotinylated at 3′ and toehold part of 8 nt DNA Tm 56.8–68 °C |
| Hybridization | 2 COs per target each one used for a single experiment 1.3 M urea, 800 mM NaCl, 33 mM HEPES pH 7.5, 0.33% SDS Overnight at 20 °C | 43 COs per Xist lncRNA (1 probe/100 bp of RNA length) 15% formamide, 750 mM NaCl, 1% SDS, 50 mM Tris-Cl pH 7.0 Overnight at 37 °C | 142 COs per Xist lncRNA (probes span the entire length of the target RNA) 4 M urea, 500 m MLiCl, 10 mM Tris pH 7.5, 0.2% SDS 67 °C for 2 h | 3 COs (PNA) per target each one used for a single experiment
| 2–3 COs per target per experiment 375 mM LiCl, 50 mM Tris pH 7.5, 1% LiDS 37 °C for 2 h |
| Beads coupling | Overnight, RT | 37 °C for 30 min | 67 °C for 30 min | 1 h at RT (Biotinylated sense DNA coupled to the beads) | 37 °C for 1 h |
| Washing | 250 mM NaCl, 0.22% SDS, 10 mM Hepes pH 7.5 Five washing cycles at 20 °C (RT) | 300 mM NaCl and 30 mM Sodium citrate (2X SSC), 0.5% SDS Five washing cycles at 37 °C, each one of 5 min | 4 M urea, 500 mM LiCl, 10 mM Tris pH 7.5, 0.2% SDS Sic washing cycles at 67 °C for 5 min | 25 mM Hepes, pH 7.4, 0.1% Triton X-100, 300 mM NaCl Two washing steps at RT | 375 mM LiCl, 50 mM Tris pH 7.5, 0.2% LiDS, 0.2% Triton X-100 37 °C for 15 min |
| Elution | RNase-H digestion | Biotin-elution at RT for 20 min and at 65 °C for 10 min | Benzonase nonspecific RNA/DNA nuclease digestion for 2 h at 37 °C | 50 °C for 20 min in salt-free buffer | Toehold mediated release (RT for 30 min) |
| CHART-MS | ChIRP-MS | RAP-MS | PAIR | HyPR-MS | ||
|---|---|---|---|---|---|---|
| SETTING-UP | Cost saving | • | •• | •• | • | ••• |
| Time saving | • | ••• | ••• | • | •• | |
| Technical accessibility | ••• | •• | •• | ••• | •• | |
| Characterization level | •• | ••• | ••• | •• | •• |
| CHART-MS | ChIRP-MS | RAP-MS | PAIR | HyPR-MS | ||
|---|---|---|---|---|---|---|
| PROCEDURE | Cost saving | •• | •• | •• | •• | ••• |
| Time saving | •• | •• | •• | •• | ••• | |
| Versatility | •• | • | • | •• | ••• | |
| Purification Efficiency | • MALAT, NEAT lncRNAS (1–10%) * | ••• XIST lncRNA (>60%) * | ••• XIST lncRNA (>60%) * | •• UFL HIV-1 RNA (35%) * US, PS, CS HIV-1 RNA (>70%) ** MALAT lncRNA (8%) * NEAT lncRNA (20%) * NORAD lncRNA (28%) * C-Myc mRNA (30%) * | ||
| Purification Specificity | 58S RNA enrichment using NEAT and MALAT COs (<1%) ## NEAT, MALAT DNA enrichment (<1%) | No GAPDH detected using XIST Cos ## | 18S RNA enrichment using XIST COs (~1%) ## | UFL HIV-1 RNA (60-fold) # US, PS, CS HIV-1 RNA (10-fold comparing one HIV splice variant to the other two or at least 200-fold relative to cellular RNAs) #
|
| CHART-MS | ChIRP-MS | RAP-MS | PAIR | HyPR-MS | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| STUDIED RNA (lncRNA, snRNA, rRNA, mRNA, HIV-1 RNA) | Malat001 | Neat | U1 | U2 | Xist | U1 | 18S | Xist | ANK | US full-length HIV-1 | US, PS, CS HIV-1 | Malat-001 | Neat | Norad | c-Myc | |
| IDENTIFIED PROTEINS | 69 | 71 | 418 | 370 | 81 | 9 | 105 | 10 | 13 | 189 | 926 (CI) 212 (DI) | 127 | 94 | 415 | 229 | |
| V A L I D A T I O N | Previous known interactors | 9/9 | 92 | 117/926 49/212 | 52 | 24 | 33 | 25 | ||||||||
| Interacting proteins ‘functionally’ linked to the related RNA | ~42 | ~41 | 143 | 143 | 13 | 98 | 9 | 13 | 89 | 633/926 131/212 | ~117 | ~66 | 145 | 209 | ||
| Direct validation | 2/69–2/71 (WB) | 1 (IP) | 3 (IB) | 8 (IP) | 2 (IP) | 8 (siRNA KD and FM) | 84 (DI) (siRNA KD) 15 (DI) (siRNA KD and IB) | 2/127–2/94–2/415 (IP) | 2 (IP) | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spiniello, M.; Scalf, M.; Casamassimi, A.; Abbondanza, C.; Smith, L.M. Towards an Ideal In Cell Hybridization-Based Strategy to Discover Protein Interactomes of Selected RNA Molecules. Int. J. Mol. Sci. 2022, 23, 942. https://doi.org/10.3390/ijms23020942
Spiniello M, Scalf M, Casamassimi A, Abbondanza C, Smith LM. Towards an Ideal In Cell Hybridization-Based Strategy to Discover Protein Interactomes of Selected RNA Molecules. International Journal of Molecular Sciences. 2022; 23(2):942. https://doi.org/10.3390/ijms23020942
Chicago/Turabian StyleSpiniello, Michele, Mark Scalf, Amelia Casamassimi, Ciro Abbondanza, and Lloyd M. Smith. 2022. "Towards an Ideal In Cell Hybridization-Based Strategy to Discover Protein Interactomes of Selected RNA Molecules" International Journal of Molecular Sciences 23, no. 2: 942. https://doi.org/10.3390/ijms23020942
APA StyleSpiniello, M., Scalf, M., Casamassimi, A., Abbondanza, C., & Smith, L. M. (2022). Towards an Ideal In Cell Hybridization-Based Strategy to Discover Protein Interactomes of Selected RNA Molecules. International Journal of Molecular Sciences, 23(2), 942. https://doi.org/10.3390/ijms23020942