
Pharmacological Dissection of the Crosstalk between NaV and CaV Channels in GH3b6 Cells

 , , ,
, , ,  , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results
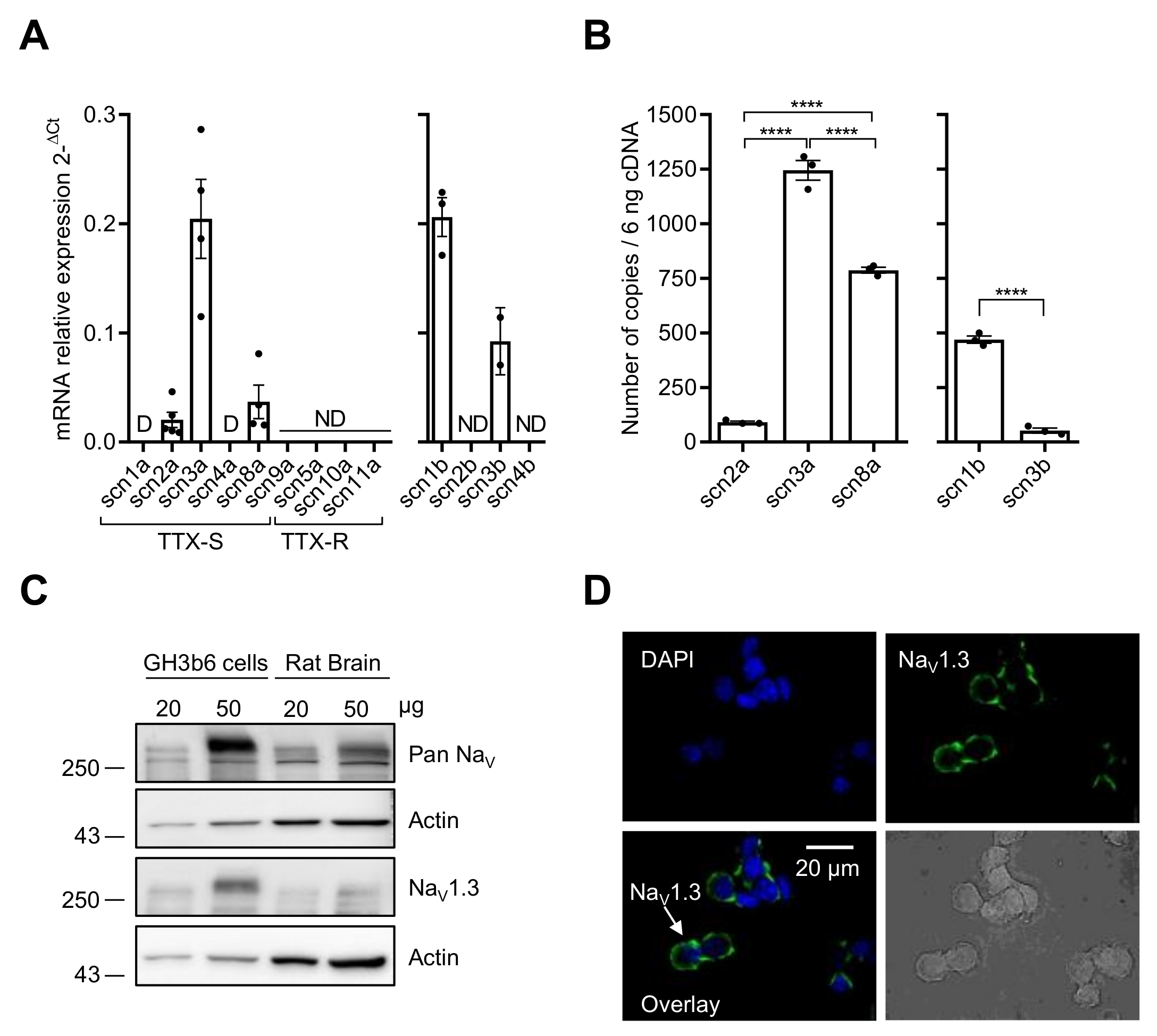
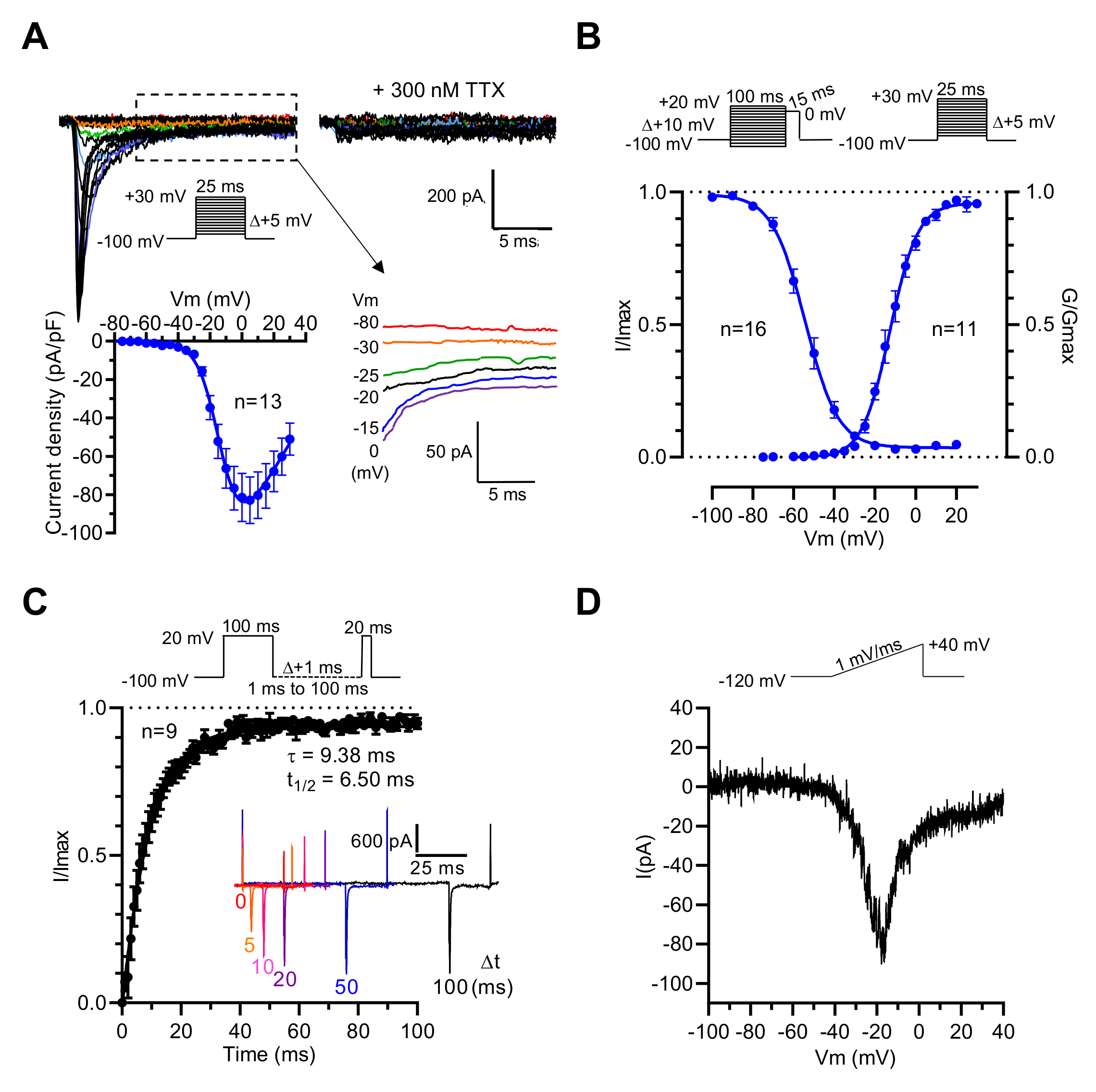
2.1. GH3b6 Cells Mainly Express the NaV1.3 Channel Subtype
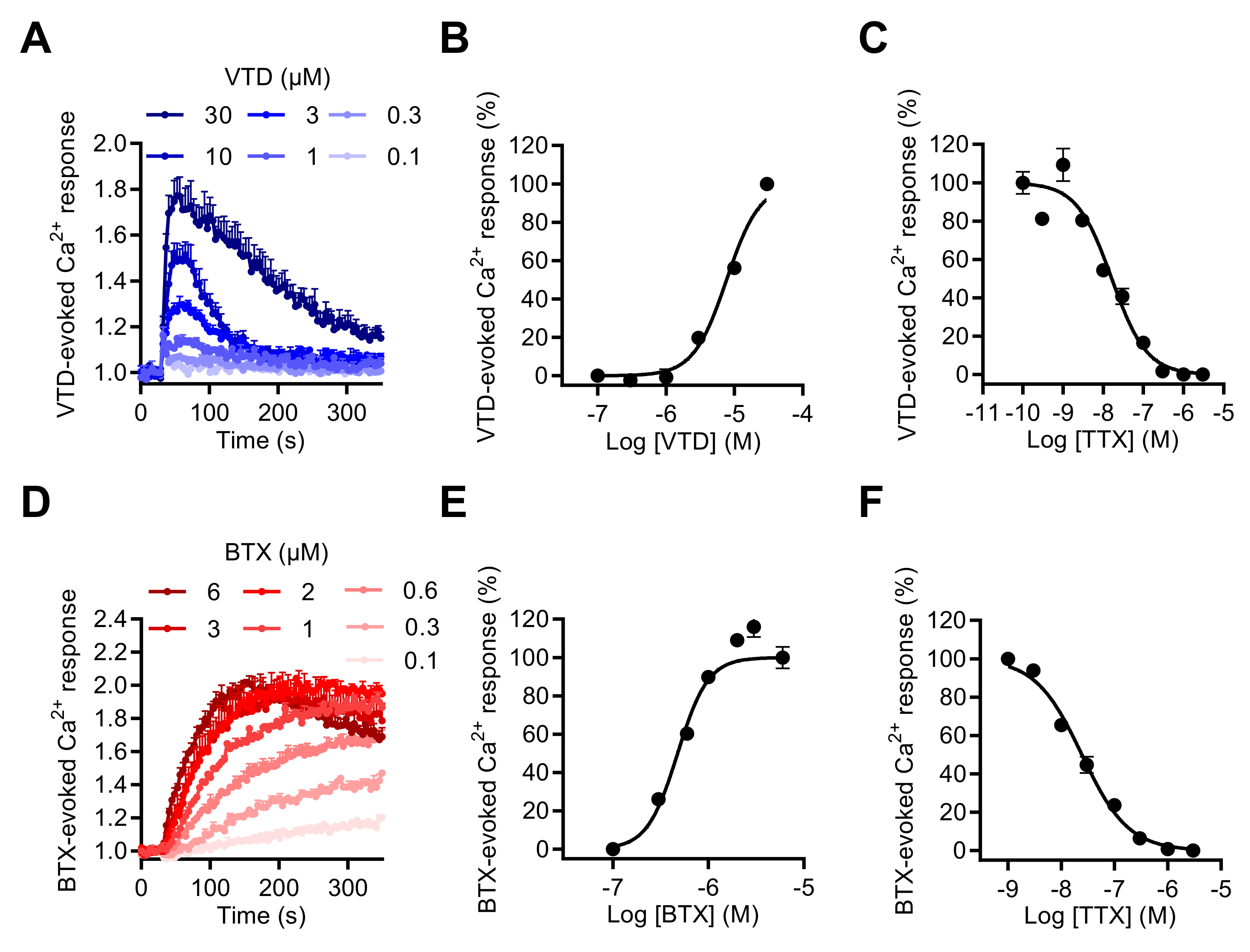
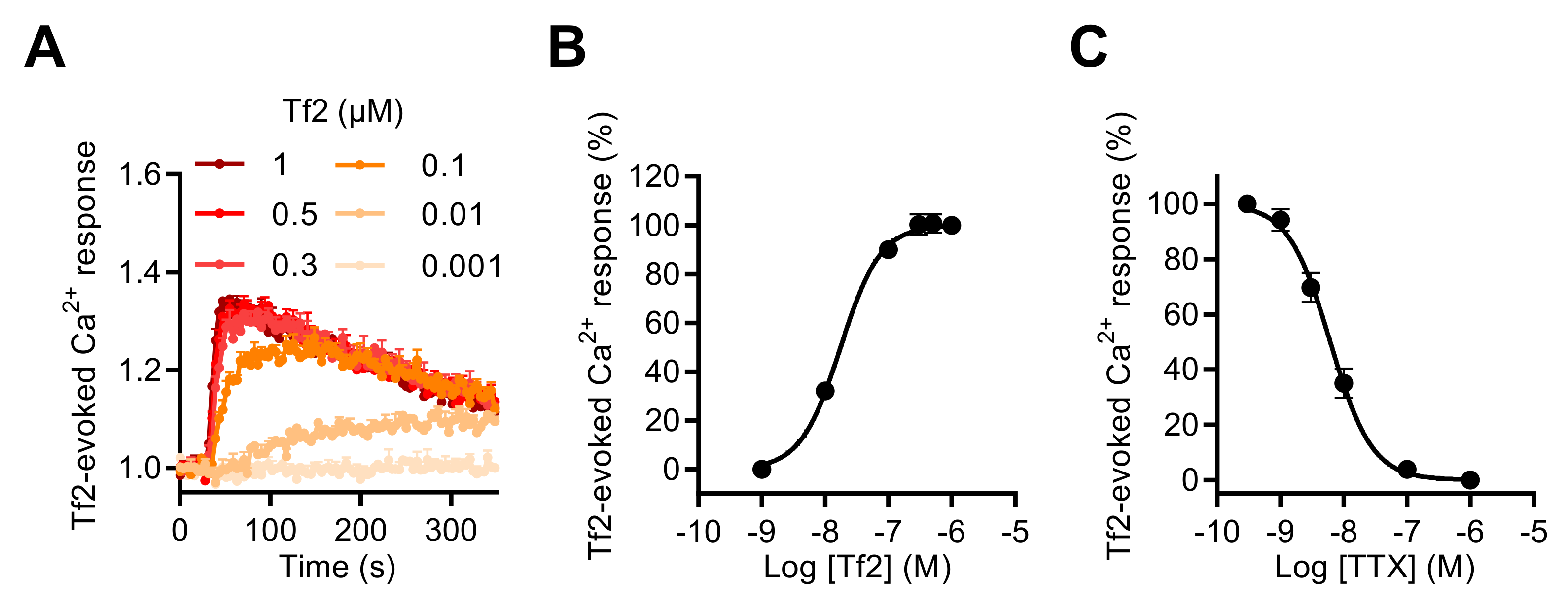
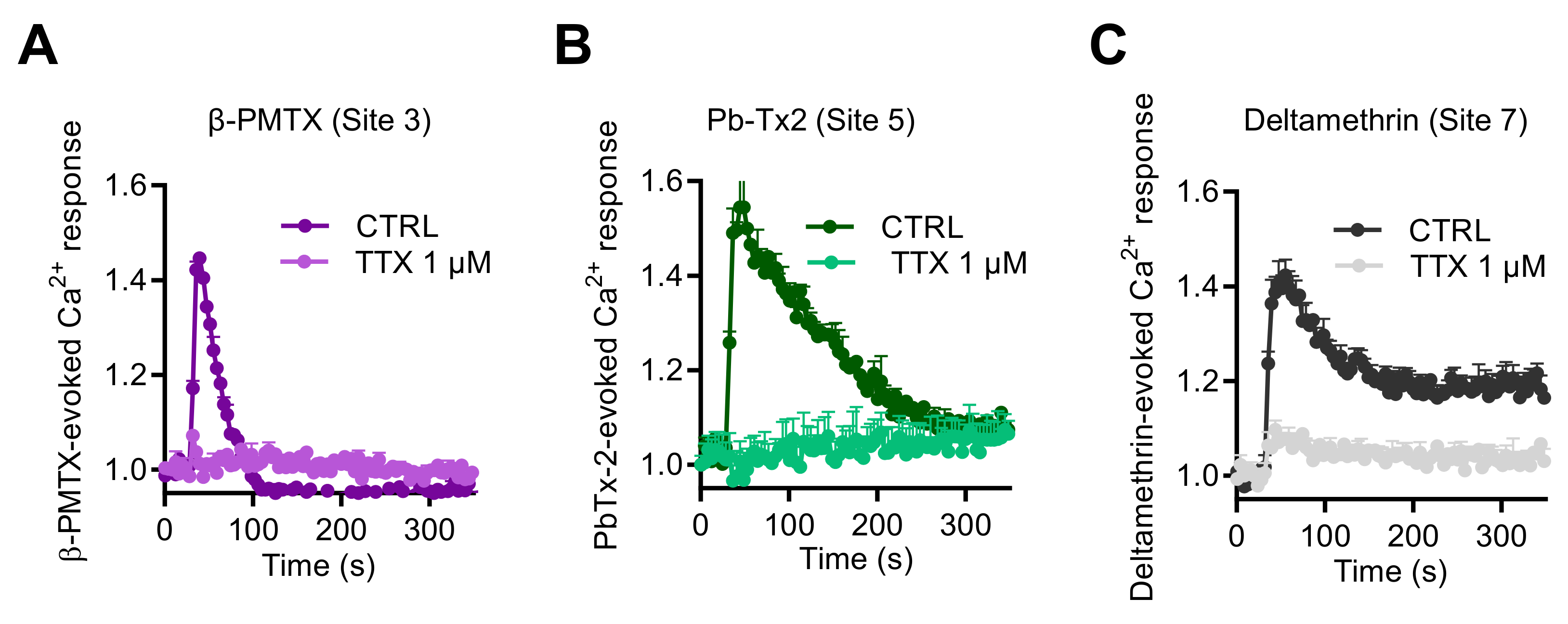
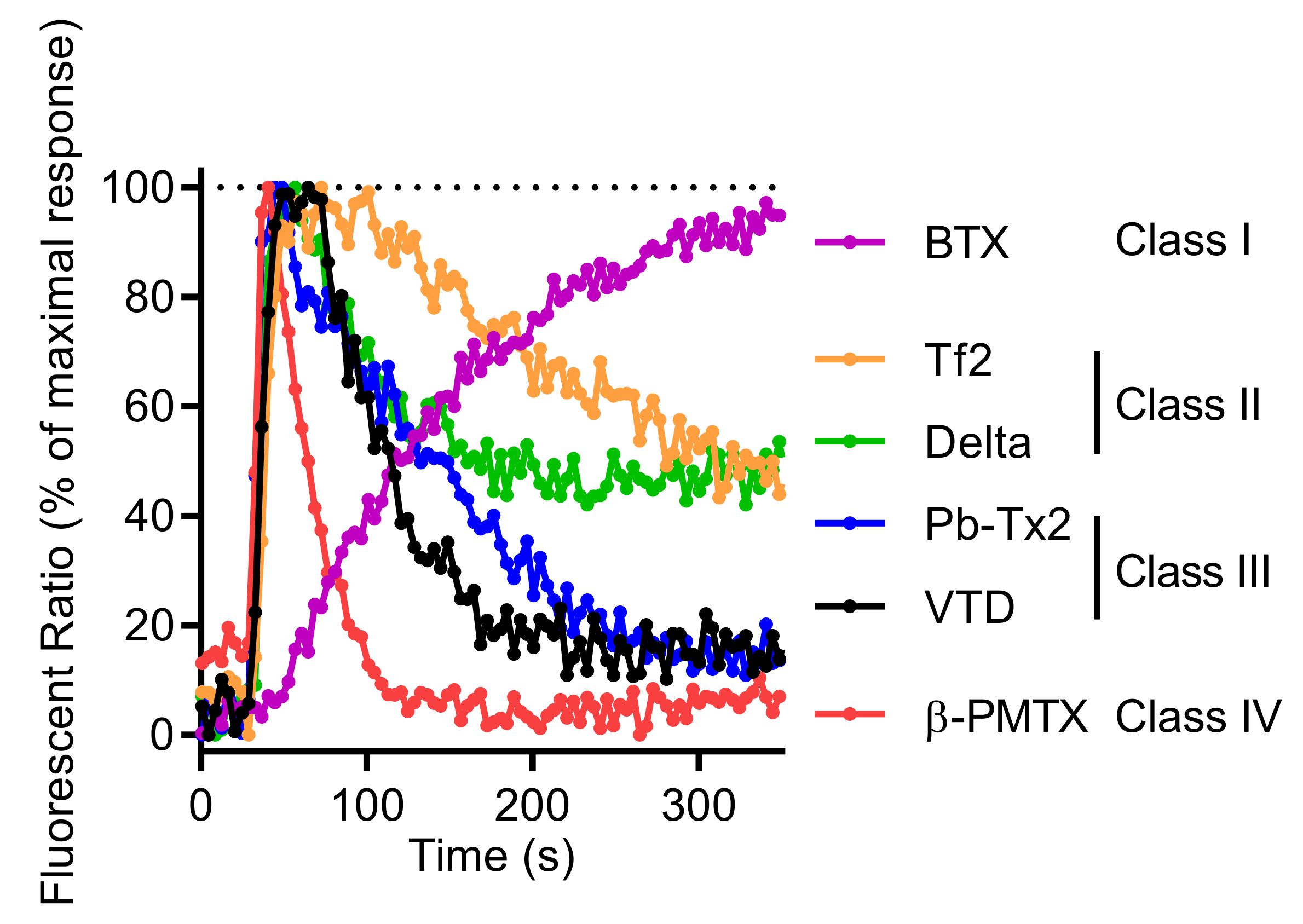
2.2. Activation of NaV Channels by Various Neurotoxins Triggers the Increase of Intracellular Ca2+ in GH3b6 Cells
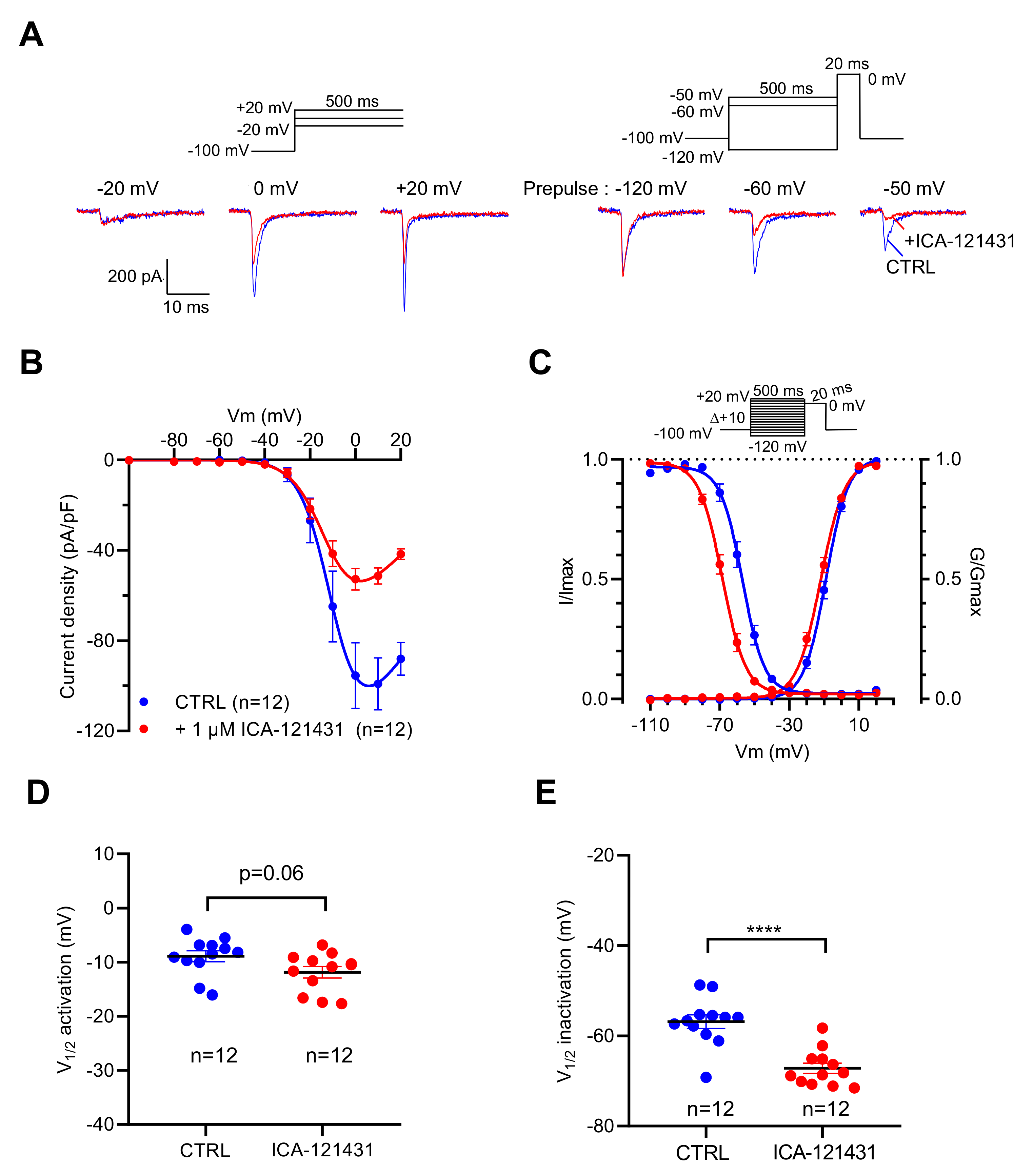
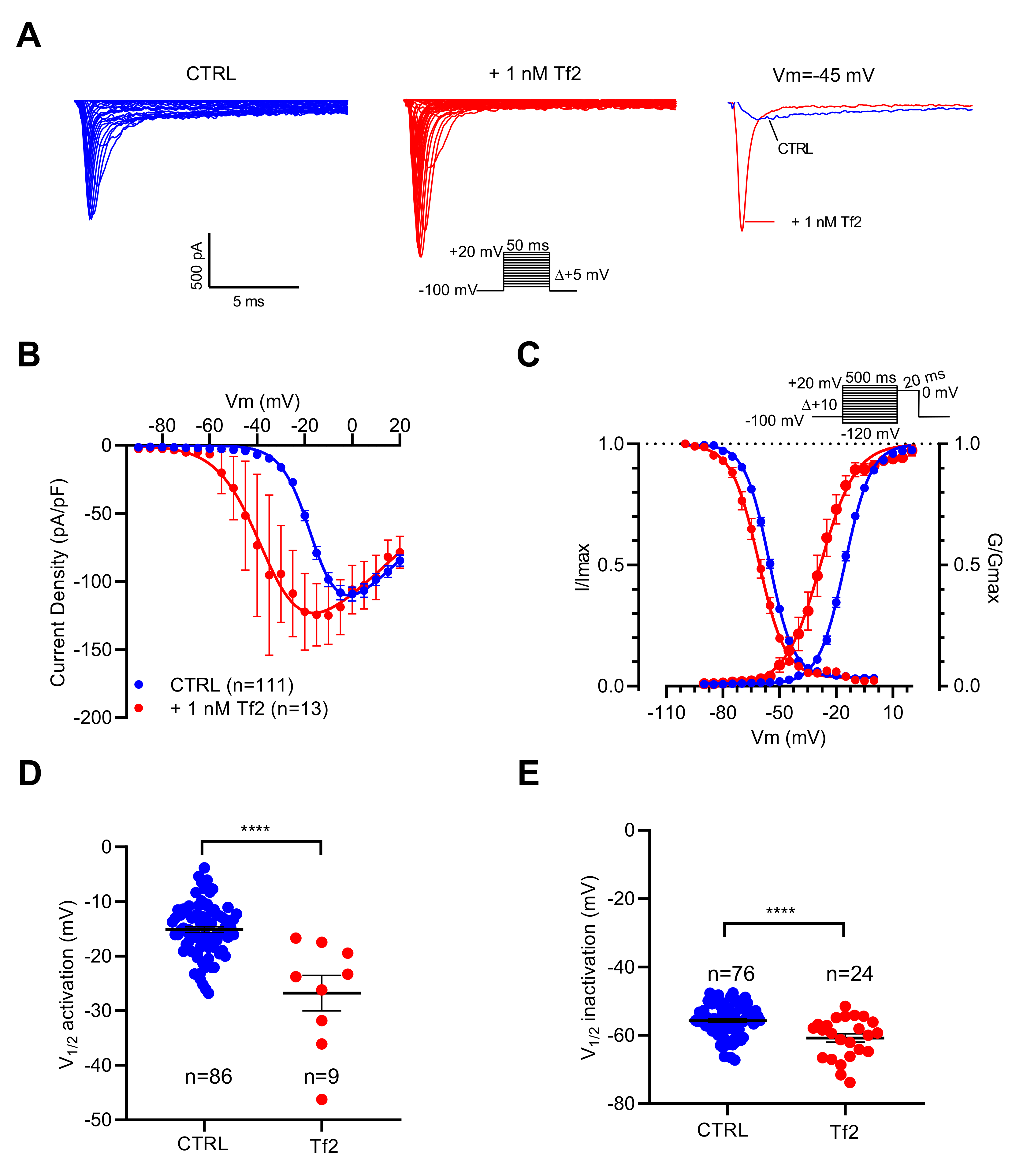
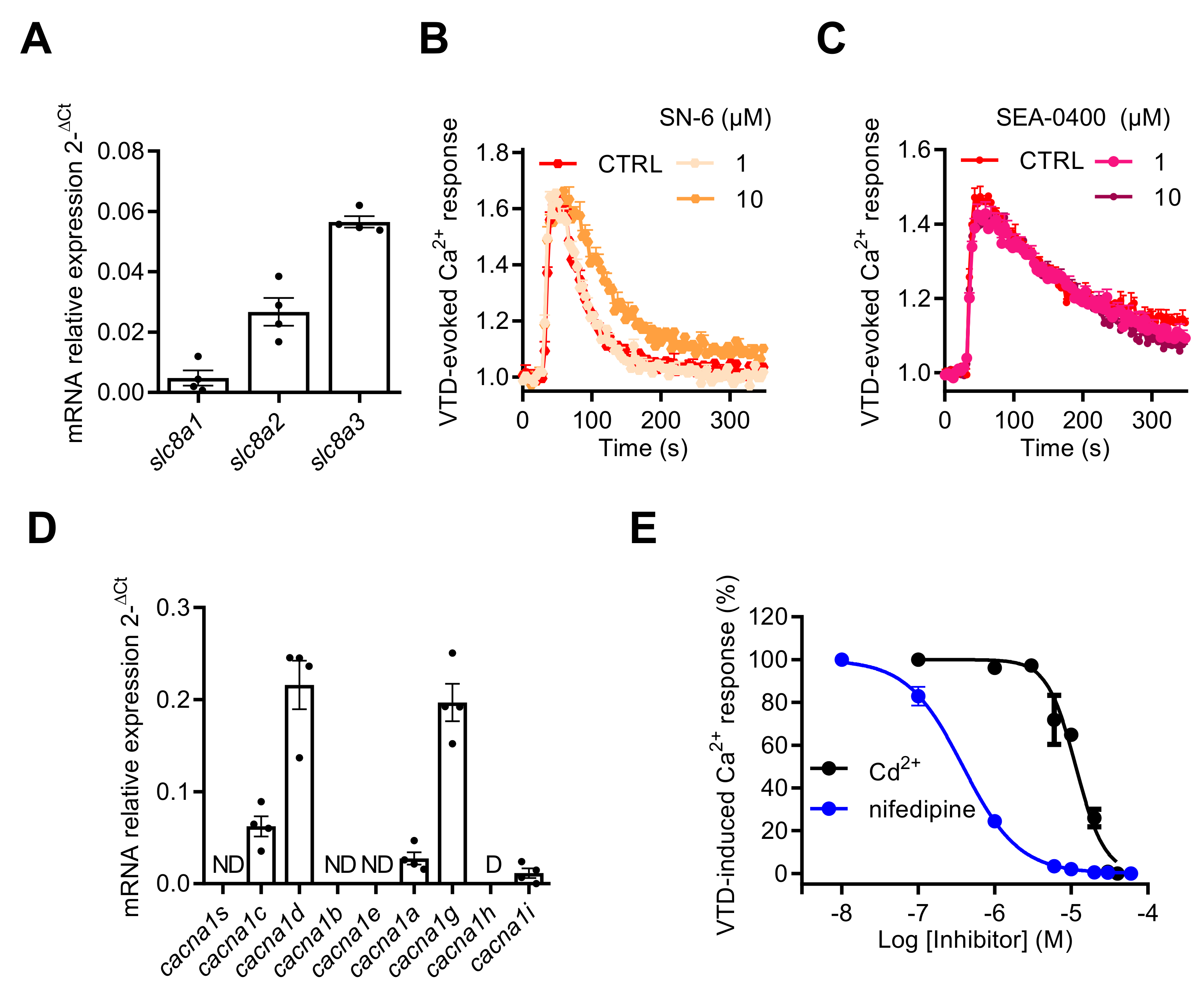
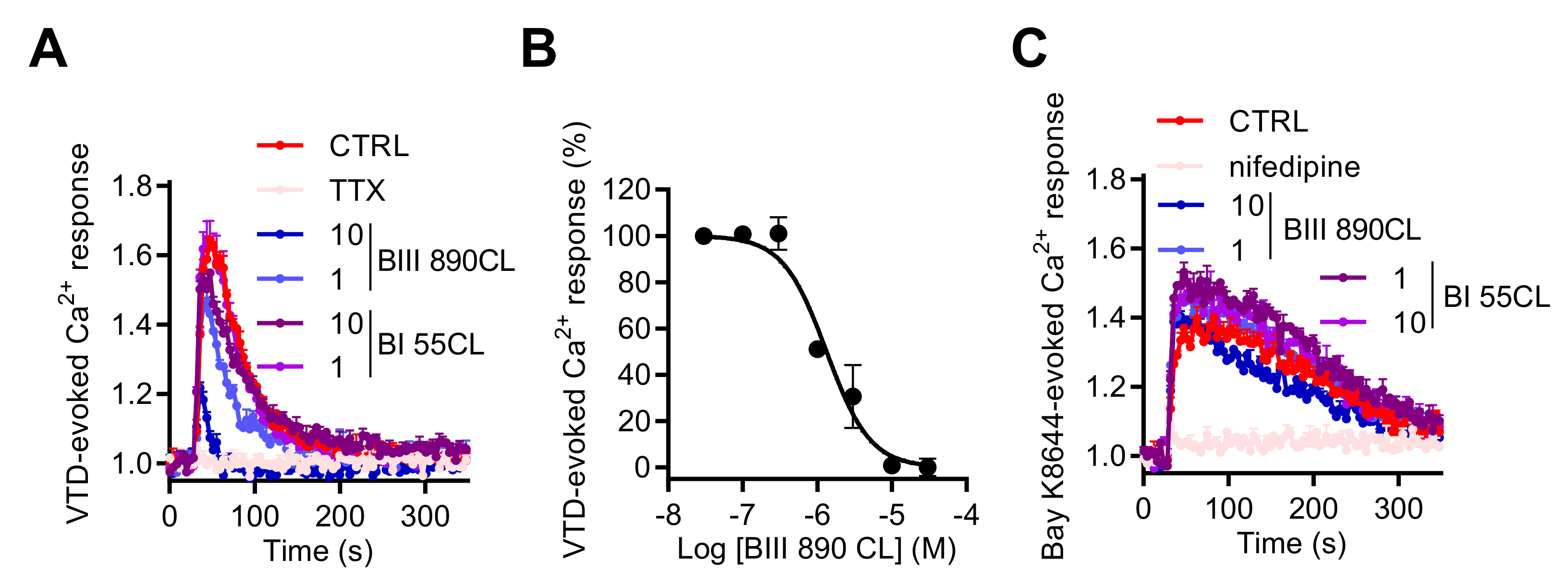
2.3. NaV Channel Activation by Neurotoxins Triggers Ca2+ Influx Mediated by L-Type CaV Channels (LTCC)
2.4. GH3b6 Cell-Based Assay Using Fura-2 Offers a Convenient Model to Characterize NaV Channel Modulators
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Line
4.3. Quantitative Real-Time PCR
4.4. Immunoblot
4.5. Immunofluorescence Staining
4.6. Measurement of Intracellular Ca2+
4.7. Manual Patch-Clamp Experiments
4.8. High-Throughput Electrophysiology
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | area under curve |
| BTX | batrachotoxin |
| INa | Na+ current |
| PbTx | brevetoxin |
| β-PMTX | β-pompidilotoxin |
| CaV channel | voltage-gated Ca2+ channel |
| [Ca2+]i | intracellular Ca2+ concentration |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| GUSB | beta-glucuronidase |
| LTCC | L-type voltage-gated Ca2+ channel |
| NaV channel | voltage-gated Na+ channel |
| NCX | Na+/Ca2+ exchanger |
| TTX | tetrodotoxin |
| TTX-S | sensitive to tetrodotoxin |
| TTX-R | resistant to tetrodotoxin; |
| VTD | veratridine |
References
- Hille, B. Ion Channels of Excitable Membranes; Sinauer: Sunderland, MA, USA, 2001; p. 507. [Google Scholar]
- De Lera Ruiz, M.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef]
- Mattei, C.; Legros, C. The Voltage-Gated Sodium Channel: A Major Target of Marine Neurotoxins. Toxicon 2014, 91, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L. Evolution of Voltage-Gated Na+ Channels. J. Exp. Biol. 2002, 205, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Chahine, M.; O’Leary, M.E. Regulatory Role of Voltage-Gated Na Channel β Subunits in Sensory Neurons. Front. Pharmacol. 2011, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, S.; Ong, S.T.; Chandy, K.G. Contributions of Natural Products to Ion Channel Pharmacology. Nat. Prod. Rep. 2020, 37, 703–716. [Google Scholar] [CrossRef]
- England, S.; de Groot, M.J. Subtype-Selective Targeting of Voltage-Gated Sodium Channels. Br. J. Pharmacol. 2009, 158, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Berman, F.W.; Murray, T.F. Brevetoxin-Induced Autocrine Excitotoxicity Is Associated with Manifold Routes of Ca2+ Influx. J. Neurochem. 2000, 74, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dravid, S.M.; Baden, D.G.; Murray, T.F. Brevetoxin Activation of Voltage-Gated Sodium Channels Regulates Ca Dynamics and ERK1/2 Phosphorylation in Murine Neocortical Neurons. J. Neurochem. 2004, 89, 739–749. [Google Scholar] [CrossRef] [Green Version]
- Fekete, Á.; Franklin, L.; Ikemoto, T.; Rózsa, B.; Lendvai, B.; Vizi, E.S.; Zelles, T. Mechanism of the Persistent Sodium Current Activator Veratridine-evoked Ca2+ Elevation: Implication for Epilepsy. J. Neurochem. 2009, 111, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Li, W.I.; Berman, F.W.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. Antillatoxin Is a Marine Cyanobacterial Toxin That Potently Activates Voltage-Gated Sodium Channels. Proc. Natl. Acad. Sci. USA 2001, 98, 7599–7604. [Google Scholar] [CrossRef] [Green Version]
- Meder, W.; Fink, K.; Zentner, J.; Göthert, M. Calcium Channels Involved in K+- and Veratridine-Induced Increase of Cytosolic Calcium Concentration in Human Cerebral Cortical Synaptosomes. J. Pharmacol. Exp. Ther. 1999, 290, 1126–1131. [Google Scholar] [PubMed]
- George, J.; Baden, D.G.; Gerwick, W.H.; Murray, T.F. Bidirectional Influence of Sodium Channel Activation on NMDA Receptor-Dependent Cerebrocortical Neuron Structural Plasticity. Proc. Natl. Acad. Sci. USA 2012, 109, 19840–19845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopper, K.L.; Adorante, J.S. Regulation of Intracellular Calcium in N1E-115 Neuroblastoma Cells: The Role of Na+/Ca2+ Exchange. Am. J. Physiol. Cell Physiol. 2002, 282, C1000–C1008. [Google Scholar] [CrossRef] [Green Version]
- LePage, K.T.; Goeger, D.; Yokokawa, F.; Asano, T.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. The Neurotoxic Lipopeptide Kalkitoxin Interacts with Voltage-Sensitive Sodium Channels in Cerebellar Granule Neurons. Toxicol. Lett. 2005, 158, 133–139. [Google Scholar] [CrossRef]
- Vetter, I.; Mozar, C.A.; Durek, T.; Wingerd, J.S.; Alewood, P.F.; Christie, M.J.; Lewis, R.J. Characterisation of Na(v) Types Endogenously Expressed in Human SH-SY5Y Neuroblastoma Cells. Biochem. Pharmacol. 2012, 83, 1562–1571. [Google Scholar] [CrossRef]
- Fletcher, P.A.; Sherman, A.; Stojilkovic, S.S. Common and Diverse Elements of Ion Channels and Receptors Underlying Electrical Activity in Endocrine Pituitary Cells. Mol. Cell. Endocrinol. 2018, 463, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Trebak, M.; Perocchi, F.; Khananshvili, D.; Sekler, I. Crosslink between Calcium and Sodium Signalling. Exp. Physiol. 2018, 103, 157–169. [Google Scholar] [CrossRef]
- Szabat, M.; Modi, H.; Ramracheya, R.; Girbinger, V.; Chan, F.; Lee, J.T.C.; Piske, M.; Kamal, S.; Carol Yang, Y.H.; Welling, A.; et al. High-Content Screening Identifies a Role for Na+ Channels in Insulin Production. R. Soc. Open Sci. 2015, 2, 150306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandael, D.H.F.; Ottaviani, M.M.; Legros, C.; Lefort, C.; Guérineau, N.C.; Allio, A.; Carabelli, V.; Carbone, E. Reduced Availability of Voltage-Gated Sodium Channels by Depolarization or Blockade by Tetrodotoxin Boosts Burst Firing and Catecholamine Release in Mouse Chromaffin Cells. J. Physiol. 2015, 593, 905–927. [Google Scholar] [CrossRef] [Green Version]
- Stojilkovic, S.S.; Bjelobaba, I.; Zemkova, H. Ion Channels of Pituitary Gonadotrophs and Their Roles in Signaling and Secretion. Front. Endocrinol. 2017, 8, 126. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Chibalina, M.V.; Bengtsson, M.; Groschner, L.N.; Ramracheya, R.; Rorsman, N.J.G.; Leiss, V.; Nassar, M.A.; Welling, A.; Gribble, F.M.; et al. Na+ Current Properties in Islet α- and β-Cells Reflect Cell-Specific Scn3a and Scn9a Expression. J. Physiol. 2014, 592, 4677–4696. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Parrazal, L.; Charles, A. Riluzole Inhibits Spontaneous Ca2+ Signaling in Neuroendocrine Cells by Activation of K+ Channels and Inhibition of Na+ Channels. Br. J. Pharmacol. 2003, 140, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Campos, F.V.; Coronas, F.I.V.; Beirão, P.S.L. Voltage-Dependent Displacement of the Scorpion Toxin Ts3 from Sodium Channels and Its Implication on the Control of Inactivation. Br. J. Pharmacol. 2004, 142, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Kushmerick, C.; Romano-Silva, M.A.; Gomez, M.V.; Prado, M.A. Changes in Ca2+ Channel Expression upon Differentiation of SN56 Cholinergic Cells. Brain Res. 2001, 916, 199–210. [Google Scholar] [CrossRef]
- Dubinsky, J.M.; Oxford, G.S. Ionic Currents in Two Strains of Rat Anterior Pituitary Tumor Cells. J. Gen. Physiol. 1984, 83, 309–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matteson, D.R.; Armstrong, C.M. Na and Ca Channels in a Transformed Line of Anterior Pituitary Cells. J. Gen. Physiol. 1984, 83, 371–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushmerick, C.; Kalapothakis, E.; Beirão, P.S.; Penaforte, C.L.; Prado, V.F.; Cruz, J.S.; Diniz, C.R.; Cordeiro, M.N.; Gomez, M.V.; Romano-Silva, M.A.; et al. Phoneutria Nigriventer Toxin Tx3-1 Blocks A-Type K+ Currents Controlling Ca2+ Oscillation Frequency in GH3 Cells. J. Neurochem. 1999, 72, 1472–1481. [Google Scholar] [CrossRef] [PubMed]
- Theile, J.W.; Cummins, T.R. Recent Developments Regarding Voltage-Gated Sodium Channel Blockers for the Treatment of Inherited and Acquired Neuropathic Pain Syndromes. Front. Pharmacol. 2011, 2, 54. [Google Scholar] [CrossRef] [Green Version]
- Garrison, C.E.; Guan, W.; Kato, M.; Tamsett, T.; Patel, T.; Sun, Y.; Pathak, T.P. Structure-Activity Relationship Evaluation of Wasp Toxin β-PMTX Leads to Analogs with Superior Activity for Human Neuronal Sodium Channels. ACS Med. Chem. Lett. 2020, 11, 353–357. [Google Scholar] [CrossRef]
- Israel, M.R.; Dash, T.S.; Bothe, S.N.; Robinson, S.D.; Deuis, J.R.; Craik, D.J.; Lampert, A.; Vetter, I.; Durek, T. Characterization of Synthetic Tf2 as a NaV1.3 Selective Pharmacological Probe. Biomedicines 2020, 8, 155. [Google Scholar] [CrossRef]
- McCormack, K.; Santos, S.; Chapman, M.L.; Krafte, D.S.; Marron, B.E.; West, C.W.; Krambis, M.J.; Antonio, B.M.; Zellmer, S.G.; Printzenhoff, D.; et al. Voltage Sensor Interaction Site for Selective Small Molecule Inhibitors of Voltage-Gated Sodium Channels. Proc. Natl. Acad. Sci. USA 2013, 110, E2724–E2732. [Google Scholar] [CrossRef] [Green Version]
- Camargos, T.S.; Bosmans, F.; Rego, S.C.; Mourão, C.B.F.; Schwartz, E.F. The Scorpion Toxin Tf2 from Tityus Fasciolatus Promotes Nav1.3 Opening. PLoS ONE 2015, 10, e0128578. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; Maejima, H.; Yamaoka, K.; Konno, K.; Kawai, N.; Shimizu, E.; Yokote, S.; Nakayama, H.; Seyama, I. Novel Wasp Toxin Discriminates between Neuronal and Cardiac Sodium Channels. Mol. Pharmacol. 2001, 59, 1457–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavon, E.; Stevens, M.; Zaharenko, A.J.; Konno, K.; Tytgat, J.; Wanke, E. Voltage-Gated Sodium Channel Isoform-Specific Effects of Pompilidotoxins. FEBS J. 2010, 277, 918–930. [Google Scholar] [CrossRef]
- White, R.J.; Reynolds, I.J. Mitochondria and Na+/Ca2+ Exchange Buffer Glutamate-Induced Calcium Loads in Cultured Cortical Neurons. J. Neurosci. 1995, 15, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Nishimaru, K.; Aikawa, T.; Hirayama, W.; Tanaka, Y.; Shigenobu, K. Effect of SEA0400, a Novel Inhibitor of Sodium-Calcium Exchanger, on Myocardial Ionic Currents. Br. J. Pharmacol. 2002, 135, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, T.; Inoue, Y.; Ito, K.; Sakaue, T.; Kita, S.; Katsuragi, T. The Exchanger Inhibitory Peptide Region-Dependent Inhibition of Na+/Ca2+ Exchange by SN-6 [2-[4-(4-Nitrobenzyloxy)Benzyl]Thiazolidine-4-Carboxylic Acid Ethyl Ester], a Novel Benzyloxyphenyl Derivative. Mol. Pharmacol. 2004, 66, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Kita, S.; Uehara, A.; Imanaga, I.; Matsuda, T.; Baba, A.; Katsuragi, T. Molecular Determinants of Na+/Ca2+ Exchange (NCX1) Inhibition by SEA0400. J. Biol. Chem. 2004, 279, 7544–7553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassmeier, G.; Hauber, M.; Wulfsen, I.; Weinsberg, F.; Bauer, C.K.; Schwarz, J.R. Ca2+ Channels in Clonal Rat Anterior Pituitary Cells (GH3/B6). Pflug. Arch. 2001, 442, 577–587. [Google Scholar]
- Safa, P.; Boulter, J.; Hales, T.G. Functional Properties of Cav1.3 (Alpha1D) L-Type Ca2+ Channel Splice Variants Expressed by Rat Brain and Neuroendocrine GH3 Cells. J. Biol. Chem. 2001, 276, 38727–38737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, A.J.; Grauert, M.; Pschorn, U.; Bechtel, W.D.; Bartmann-Lindholm, C.; Qu, Y.; Scheuer, T.; Catterall, W.A.; Weiser, T. Potent Blockade of Sodium Channels and Protection of Brain Tissue from Ischemia by BIII 890 CL. Proc. Natl. Acad. Sci. USA 2000, 97, 4944–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, L.V.; Daniels, Z.; Hick, C.; Elsegood, K.; Bowden, S.; Szestak, T.; Burley, J.R.; Southan, A.; Cronk, D.; James, I.F. Analysis of Human Nav1.8 Expressed in SH-SY5Y Neuroblastoma Cells. Eur. J. Pharmacol. 2005, 528, 52–58. [Google Scholar] [CrossRef]
- Laird, J.M.A.; Carter, A.J.; Grauert, M.; Cervero, F. Analgesic Activity of a Novel Use-Dependent Sodium Channel Blocker, Crobenetine, in Mono-Arthritic Rats: Sodium Channel Blockers in Arthritic Rats. Br. J. Pharmacol. 2001, 134, 1742–1748. [Google Scholar] [CrossRef] [Green Version]
- Cusdin, F.S.; Nietlispach, D.; Maman, J.; Dale, T.J.; Powell, A.J.; Clare, J.J.; Jackson, A.P. The Sodium Channel {beta}3-Subunit Induces Multiphasic Gating in NaV1.3 and Affects Fast Inactivation via Distinct Intracellular Regions. J. Biol. Chem. 2010, 285, 33404–33412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, B.S.; Stevens, E.B.; Pinnock, R.D.; Dixon, A.K.; Lee, K. Developmental Expression of the Novel Voltage-Gated Sodium Channel Auxiliary Subunit Beta3, in Rat CNS. J. Physiol. 2001, 534, 763–776. [Google Scholar] [CrossRef]
- Baroni, D.; Moran, O. Molecular Differential Expression of Voltage-Gated Sodium Channel α and β Subunit MRNAs in Five Different Mammalian Cell Lines. J. Bioenerg. Biomembr. 2011, 43, 729–738. [Google Scholar] [CrossRef]
- Vega, A.V.; Espinosa, J.L.; López-Domínguez, A.M.; López-Santiago, L.F.; Navarrete, A.; Cota, G. L-Type Calcium Channel Activation up-Regulates the MRNAs for Two Different Sodium Channel α Subunits (Nav1.2 and Nav1.3) in Rat Pituitary GH3 Cells. Brain Res. 2003, 116, 115–125. [Google Scholar] [CrossRef]
- Cummins, T.R.; Aglieco, F.; Renganathan, M.; Herzog, R.I.; Dib-Hajj, S.D.; Waxman, S.G. Nav1.3 Sodium Channels: Rapid Repriming and Slow Closed-State Inactivation Display Quantitative Differences after Expression in a Mammalian Cell Line and in Spinal Sensory Neurons. J. Neurosci. 2001, 21, 5952–5961. [Google Scholar] [CrossRef] [Green Version]
- Estacion, M.; Waxman, S.G. The Response of Na V 1.3 Sodium Channels to Ramp Stimuli: Multiple Components and Mechanisms. J. Neurophysiol. 2013, 109, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Hobai, I.A.; Bates, J.A.; Howarth, F.C.; Levi, A.J. Inhibition by External Cd2+ of Na/Ca Exchange and L-Type Ca Channel in Rabbit Ventricular Myocytes. Am. J. Physiol. 1997, 272, H2164–H2172. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.B.; Jiang, B.; Pappano, A.J. Comparison of L-Type Calcium Channel Blockade by Nifedipine and/or Cadmium in Guinea Pig Ventricular Myocytes. J. Pharmacol. Exp. Ther. 2000, 294, 562–570. [Google Scholar] [PubMed]
- Meacham, C.; Brodfuehrer, P.; Watkins, J.; Shafer, T. Developmentally-Regulated Sodium Channel Subunits Are Differentially Sensitive to α-Cyano Containing Pyrethroids. Toxicol. Appl. Pharmacol. 2008, 231, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Membrane Potential-Dependent Binding of Scorpion Toxin to the Action Potential Na+ Ionophore. Studies with a Toxin Derivative Prepared by Lactoperoxidase-Catalyzed Iodination. J. Biol. Chem. 1977, 252, 8660–8668. [Google Scholar] [CrossRef]
- Weinsberg, F.; Bauer, C.K.; Schwarz, J.R. The Class III Antiarrhythmic Agent E-4031 Selectively Blocks the Inactivating Inward-Rectifying Potassium Current in Rat Anterior Pituitary Tumor Cells (GH3/B6 Cells). Pflug. Arch. 1997, 434, 1–10. [Google Scholar] [CrossRef]
- Hains, B.C.; Waxman, S.G. Sodium Channel Expression and the Molecular Pathophysiology of Pain after SCI. Prog. Brain Res. 2007, 161, 195–203. [Google Scholar] [CrossRef]
- Luiz, A.P.; Wood, J.N. Sodium Channels in Pain and Cancer: New Therapeutic Opportunities. Adv. Pharmacol. 2016, 75, 153–178. [Google Scholar] [CrossRef]
- Su, S.; Shao, J.; Zhao, Q.; Ren, X.; Cai, W.; Li, L.; Bai, Q.; Chen, X.; Xu, B.; Wang, J.; et al. MiR-30b Attenuates Neuropathic Pain by Regulating Voltage-Gated Sodium Channel Nav1.3 in Rats. Front. Mol. Neurosci. 2017, 10, 126. [Google Scholar] [CrossRef]
- Yang, L.; Li, Q.; Liu, X.; Liu, S. Roles of Voltage-Gated Tetrodotoxin-Sensitive Sodium Channels NaV1.3 and NaV1.7 in Diabetes and Painful Diabetic Neuropathy. Int. J. Mol. Sci. 2016, 17, 1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven Novel Modulators of the Analgesic Target NaV 1.7 Uncovered Using a High-Throughput Venom-Based Discovery Approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [CrossRef] [Green Version]
- Tashjian, A.H. Clonal Strains of Hormone-Producing Pituitary Cells. Meth. Enzymol. 1979, 58, 527–535. [Google Scholar]
- Colborn, J.M.; Byrd, B.D.; Koita, O.A.; Krogstad, D.J. Estimation of Copy Number Using SYBR Green: Confounding by AT-Rich DNA and by Variation in Amplicon Length. Am. J. Trop. Med. Hyg. 2008, 79, 887–892. [Google Scholar] [CrossRef]
- Morrison, T.B.; Weis, J.J.; Wittwer, C.T. Quantification of Low-Copy Transcripts by Continuous SYBR Green I Monitoring during Amplification. BioTechniques 1998, 24, 954–958. [Google Scholar]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Hamill, O.P.; Marty, A.; Neher, E.; Sakmann, B.; Sigworth, F.J. Improved Patch-Clamp Techniques for High-Resolution Current Recording from Cells and Cell-Free Membrane Patches. Pflug. Arch.-Eur. J. Physiol. 1981, 391, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Leipold, E.; Hansel, A.; Borges, A.; Heinemann, S.H. Subtype Specificity of Scorpion β-Toxin Tz1 Interaction with Voltage-Gated Sodium Channels Is Determined by the Pore Loop of Domain 3. Mol. Pharmacol. 2006, 70, 340–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leipold, E.; Borges, A.; Heinemann, S.H. Scorpion β-Toxin Interference with NaV Channel Voltage Sensor Gives Rise to Excitatory and Depressant Modes. J. Gen. Physiol. 2012, 139, 305–319. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Voltage-Dependency of Activation | Voltage-Dependency of Inactivation | ||
|---|---|---|---|---|
| V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | |
| Control 2 | −12.23 ± 1.34 n = 11 | 6.06 ± 0.20 | −53.58 ± 1.66 n = 16 | 5.26 ± 0.17 |
| Control 3 | −8.92 ± 1.01 n = 12 | 6.06 ± 0.20 | −56.85 ± 1.54 n = 12 | 5.26 ± 0.17 |
| + 1 µM ICA-121431 3 | −11.86 ± 1.05 n = 12 (p = 0.06) | 6.67 ± 0.36 n = 12 (p < 0.05) | −67.19 ± 1.15 n = 12 (p < 0.0001) | 5.18 ± 0.18 (p = 0.72) |
| Control 4 | −15.07 ± 0.51 n = 86 | 5.78 ± 0.14 | −55.69 ± 0.53 n = 76 | 5.86 ± 0.14 |
| +1 nM Tf2 4 | −26.78 ± 3.24 n = 9 (p < 0.0001) | 5.83 ± 0.52 (p = 0.90) | −60.77 ± 1.20 n = 24 (p < 0.0001) | 5.87 ± 0.32 ns, p = 0.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Réthoré, L.; Park, J.; Montnach, J.; Nicolas, S.; Khoury, J.; Le Seac’h, E.; Mabrouk, K.; De Pomyers, H.; Tricoire-Leignel, H.; Mattei, C.; et al. Pharmacological Dissection of the Crosstalk between NaV and CaV Channels in GH3b6 Cells. Int. J. Mol. Sci. 2022, 23, 827. https://doi.org/10.3390/ijms23020827
Réthoré L, Park J, Montnach J, Nicolas S, Khoury J, Le Seac’h E, Mabrouk K, De Pomyers H, Tricoire-Leignel H, Mattei C, et al. Pharmacological Dissection of the Crosstalk between NaV and CaV Channels in GH3b6 Cells. International Journal of Molecular Sciences. 2022; 23(2):827. https://doi.org/10.3390/ijms23020827
Chicago/Turabian StyleRéthoré, Léa, Joohee Park, Jérôme Montnach, Sébastien Nicolas, Joseph Khoury, Elodie Le Seac’h, Kamel Mabrouk, Harold De Pomyers, Hélène Tricoire-Leignel, César Mattei, and et al. 2022. "Pharmacological Dissection of the Crosstalk between NaV and CaV Channels in GH3b6 Cells" International Journal of Molecular Sciences 23, no. 2: 827. https://doi.org/10.3390/ijms23020827
APA StyleRéthoré, L., Park, J., Montnach, J., Nicolas, S., Khoury, J., Le Seac’h, E., Mabrouk, K., De Pomyers, H., Tricoire-Leignel, H., Mattei, C., Henrion, D., Fajloun, Z., De Waard, M., Legendre, C., & Legros, C. (2022). Pharmacological Dissection of the Crosstalk between NaV and CaV Channels in GH3b6 Cells. International Journal of Molecular Sciences, 23(2), 827. https://doi.org/10.3390/ijms23020827