
Control of Expression of Key Cell Cycle Enzymes Drives Cell Line-Specific Functions of CDK7 in Human PDAC Cells


Abstract
:1. Introduction
2. Results
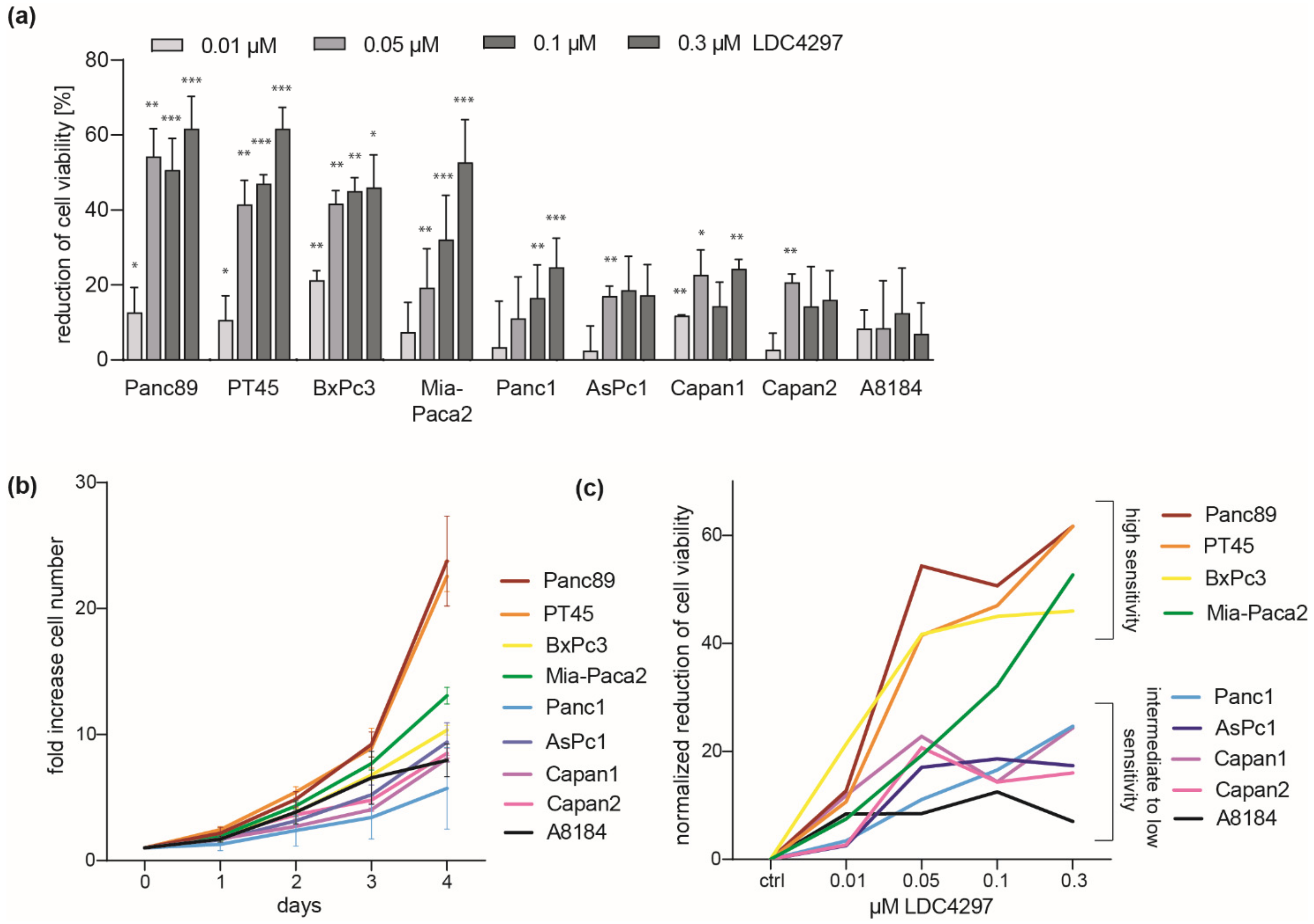
2.1. Broad Response but Selective Sensitivity of PDAC Lines to Inhibition of CDK7
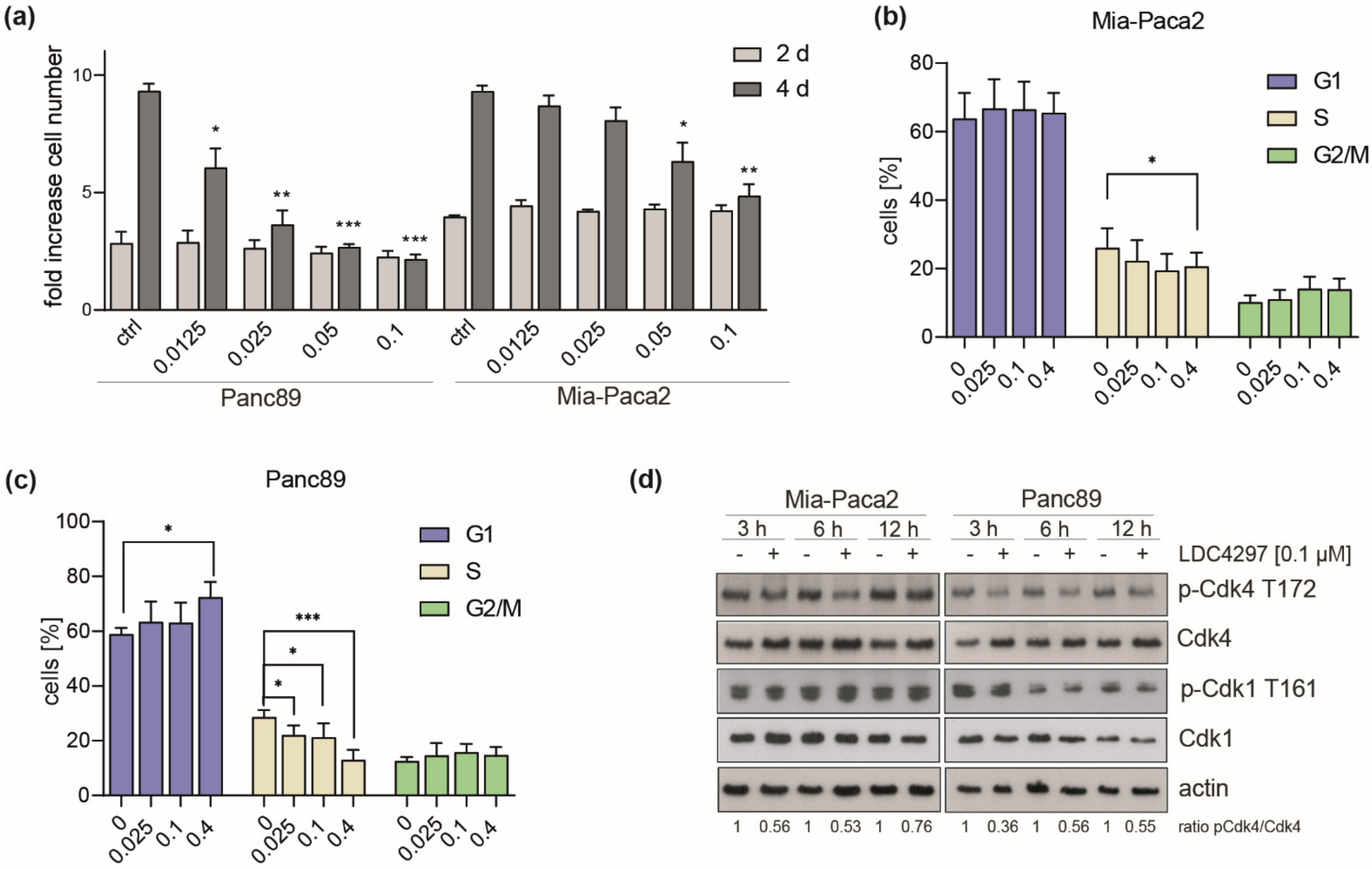
2.2. CDK7 Affects All Cell Cycle Phases with Preference for G1/S in Panc89
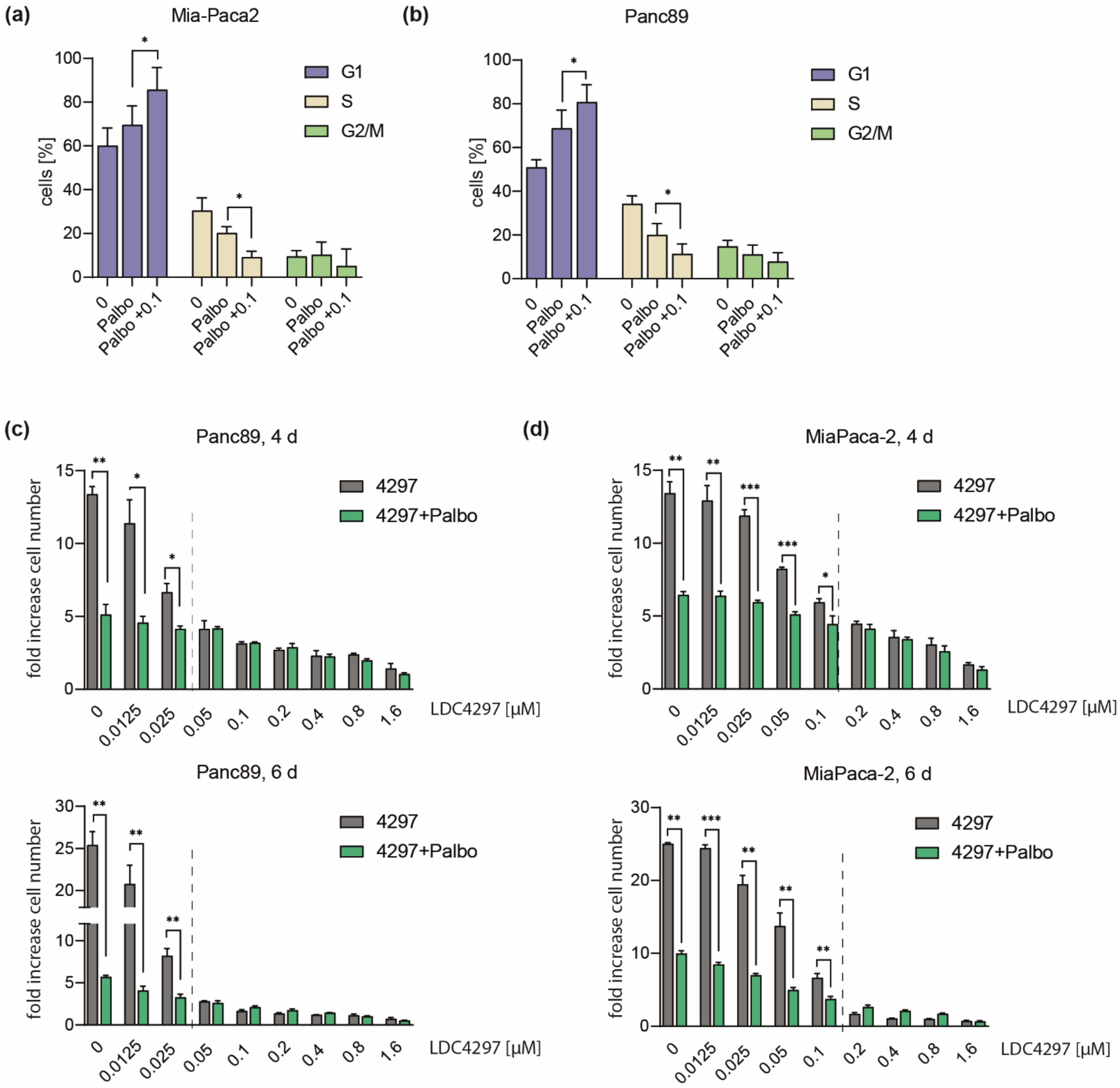
2.3. Evidence for a Concentration-Dependent Crosstalk between CDK7 and CDK4
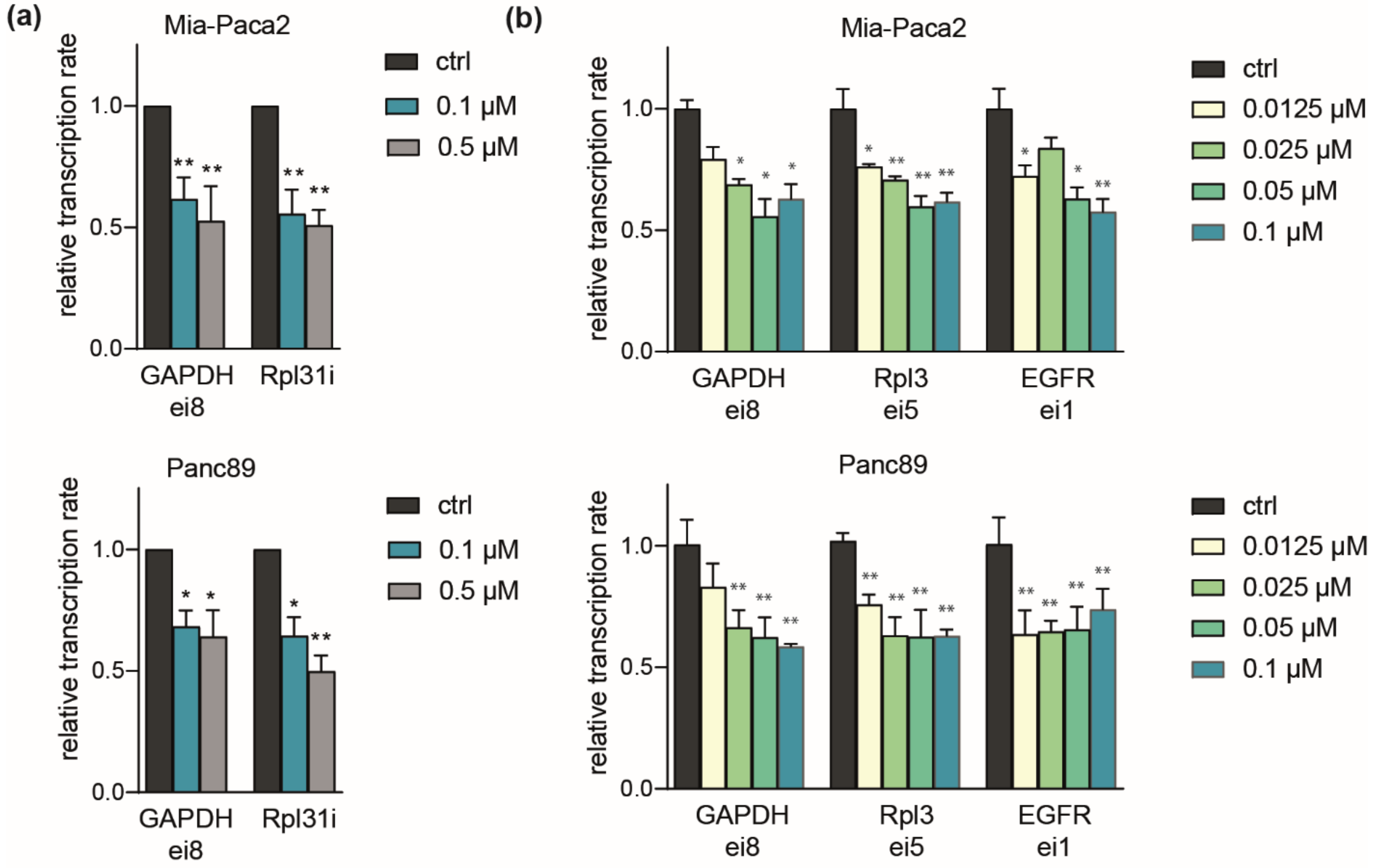
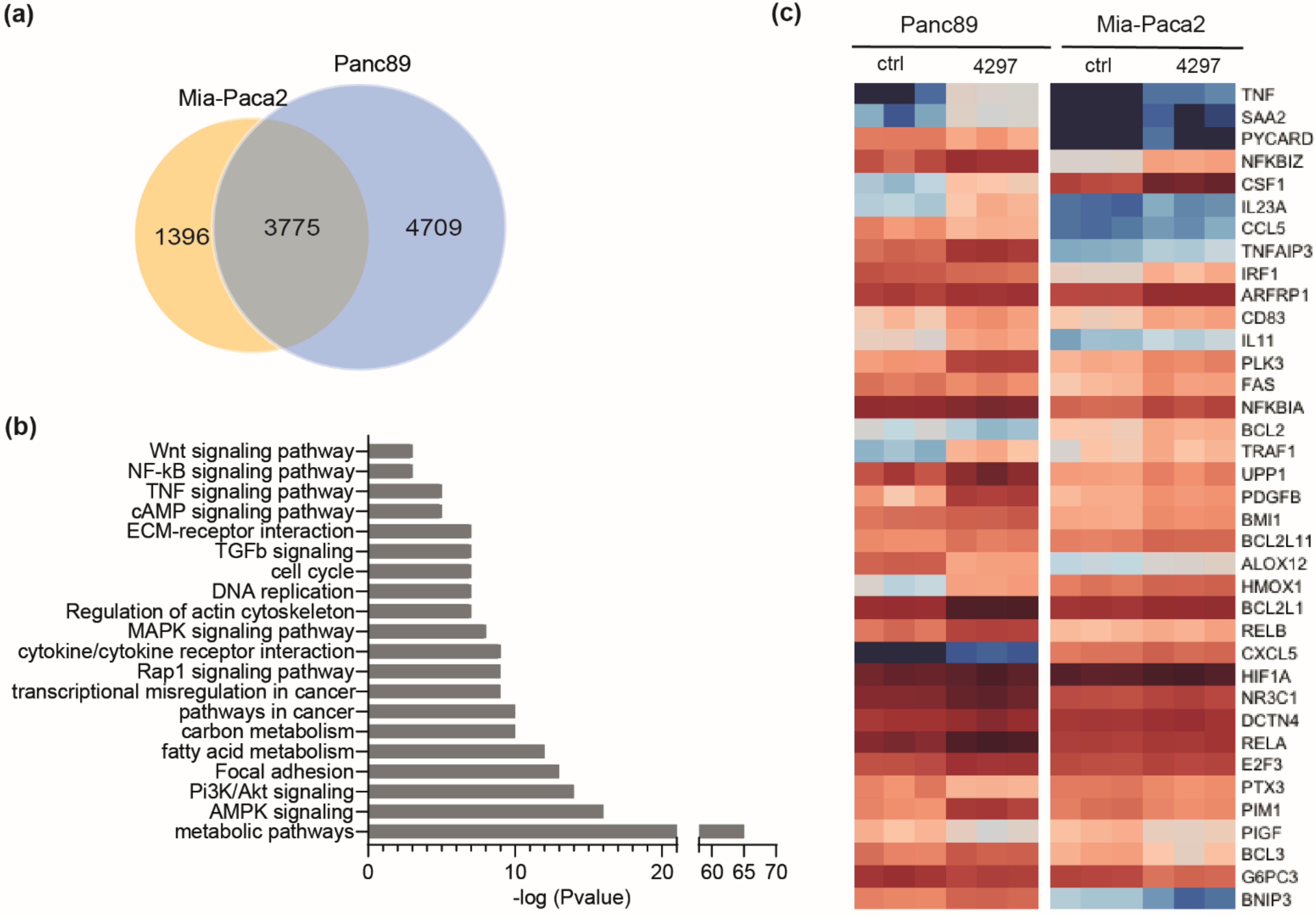
2.4. Comparable Impact of CDK7 Inhibition on Transcription in Mia-Paca2 and Panc89 Cells
2.5. Prolonged Limited Inhibition of CDK7 Leads to Activation of NF-κB Target Genes
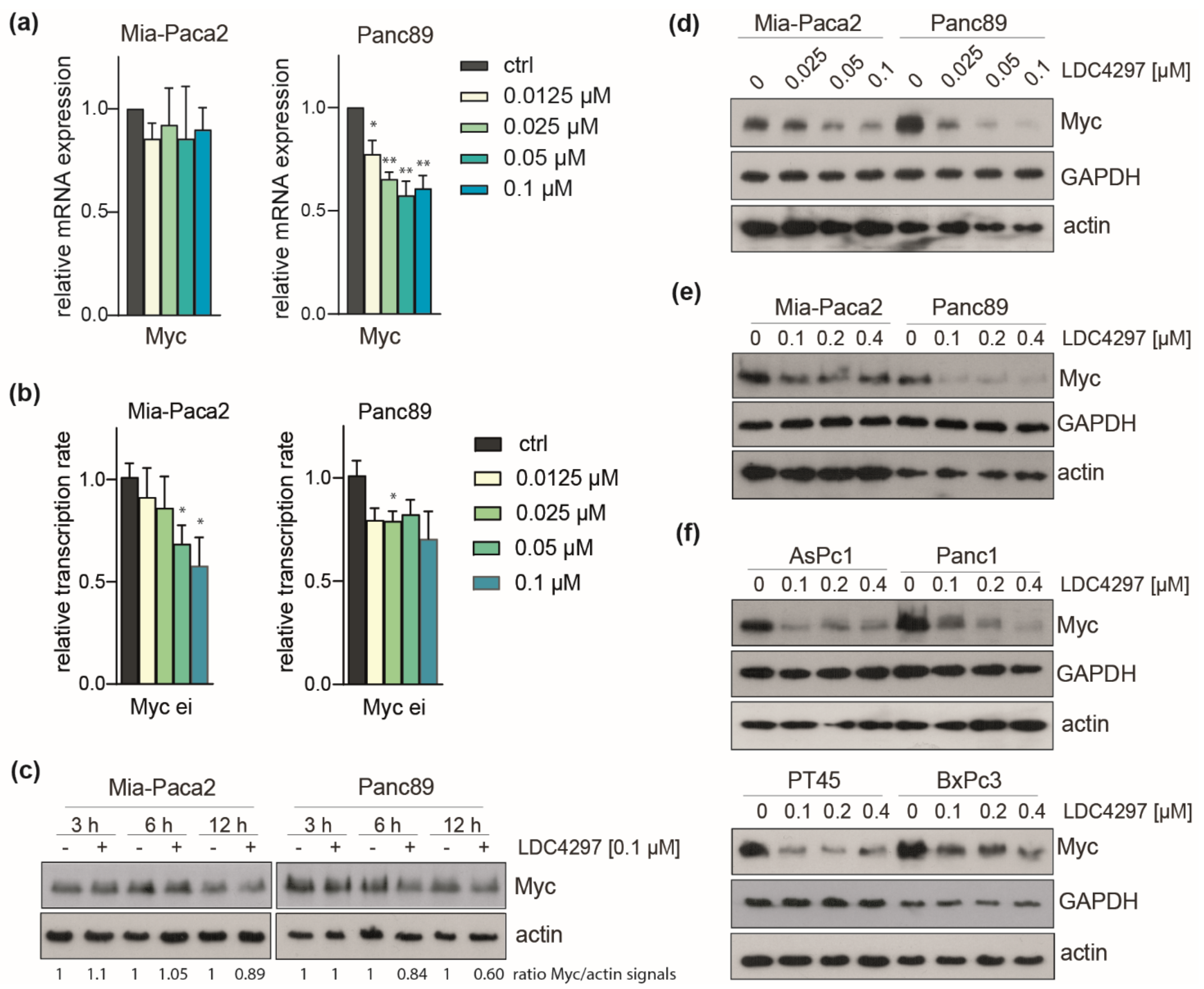
2.6. CDK7 Inhibition Caused Downregulation of Myc in PDAC Lines
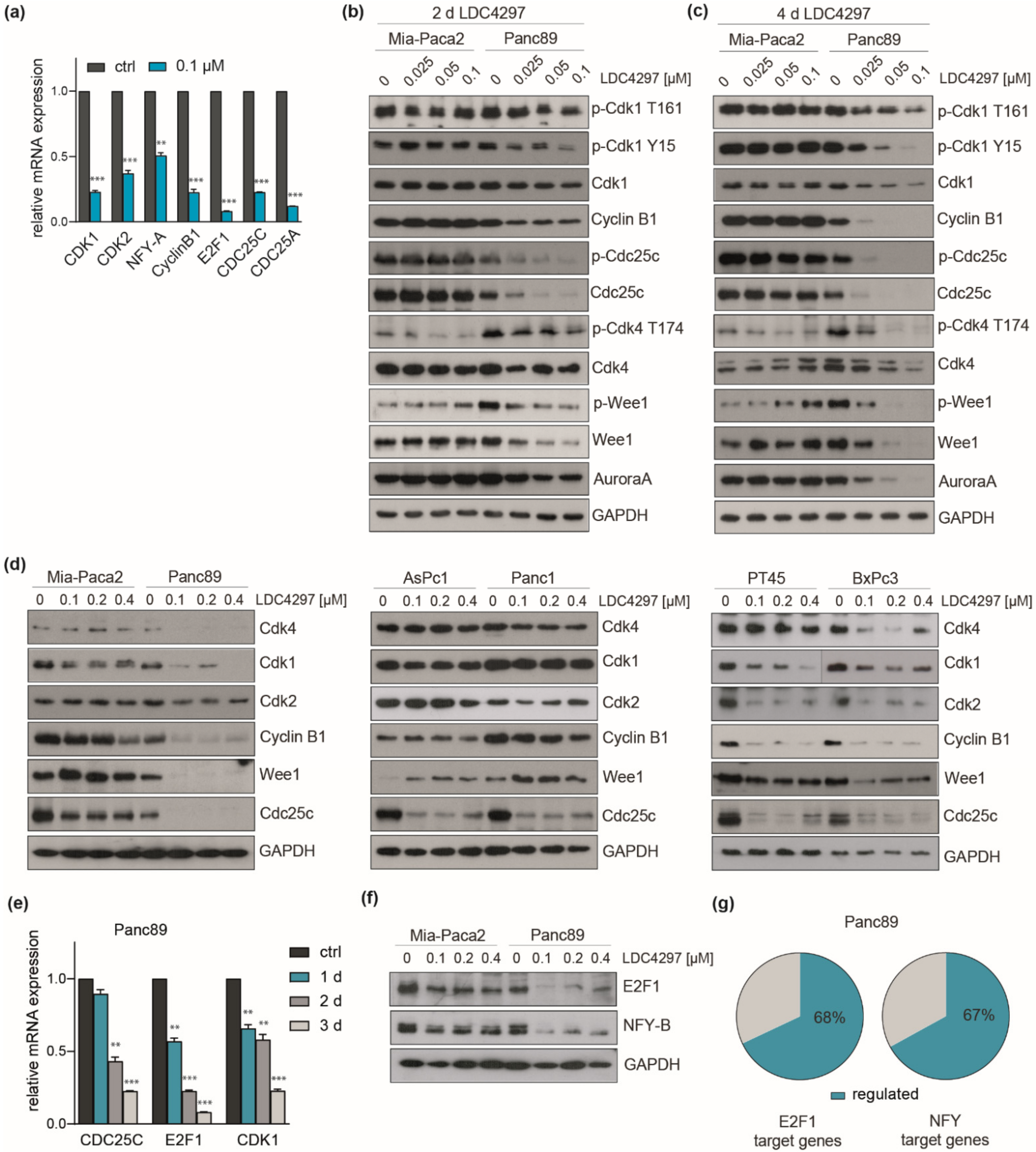
2.7. Evidence for Broad Control of the Expression of Key Cell Cycle Control Genes by CDK7
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Inhibitors
4.2. Cell Viability Assay
4.3. FACS Analyses
4.4. Cell Lysis and Immunoblotting
4.5. RNA-seq Analysis
4.6. RNA Extraction and RT-qPCR
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.P. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J. Cell Sci. 2005, 118, 5171–5180. [Google Scholar] [CrossRef] [Green Version]
- Lolli, G.; Johnson, L.N. CAK—Cyclin-Dependent Activating Kinase: A Key Kinase in Cell Cycle Control and a Target for Drugs? Cell Cycle 2005, 4, 565–570. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, S.; Heidemann, M.; Tietjen, J.R.; Zhang, D.W.; Chapman, R.D.; Eick, D.; Ansari, A.Z. TFIIH Kinase Places Bivalent Marks on the Carboxy-Terminal Domain of RNA Polymerase II. Mol. Cell 2009, 34, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Glover-Cutter, K.; Larochelle, S.; Erickson, B.; Zhang, C.; Shokat, K.; Fisher, R.P.; Bentley, D.L. TFIIH-Associated Cdk7 Kinase Functions in Phosphorylation of C-Terminal Domain Ser7 Residues, Promoter-Proximal Pausing, and Termination by RNA Polymerase II. Mol. Cell. Biol. 2009, 29, 5455–5464. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.H.; Jin, Y.; Struhl, K. TFIIH Phosphorylation of the Pol II CTD Stimulates Mediator Dissociation from the Preinitiation Complex and Promoter Escape. Mol. Cell 2014, 54, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sanso, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 2012, 19, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilson, K.A.; Guo, J.; Turek, M.E.; Brogie, J.E.; Delaney, E.; Luse, D.S.; Price, D.H. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol. Cell 2015, 59, 576–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coin, F.; Egly, J.-M. Revisiting the Function of CDK7 in Transcription by Virtue of a Recently Described TFIIH Kinase Inhibitor. Mol. Cell 2015, 59, 513–514. [Google Scholar] [CrossRef] [Green Version]
- Rochette-Egly, C.; Adam, S.; Rossignol, M.; Egly, J.-M.; Chambon, P. Stimulation of RARα Activation Function AF-1 through Binding to the General Transcription Factor TFIIH and Phosphorylation by CDK7. Cell 1997, 90, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Keriel, A.; Stary, A.; Sarasin, A.; Rochette-Egly, C.; Egly, J.-M. XPD Mutations Prevent TFIIH-Dependent Transactivation by Nuclear Receptors and Phosphorylation of RARα. Cell 2002, 109, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Bastien, J.; Adam-Stitah, S.; Riedl, T.; Egly, J.M.; Chambon, P.; Rochette-Egly, C. TFIIH Interacts with the Retinoic Acid Receptor Gamma and Phosphorylates Its AF-1-Activating Domain through Cdk7. J. Biol. Chem. 2000, 275, 21896–21904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chymkowitch, P.; Le May, N.; Charneau, P.; Compe, E.; Egly, J.-M. The Phosphorylation of the Androgen Receptor by TFIIH Directs the Ubiquitin/Proteasome Process. EMBO J. 2011, 30, 468–479. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.K.; Duan, H.O.; Chang, C. From Androgen Receptor to the General Transcription Factor TFIIH: Identification of cdk Activating Kinase (CAK) as an Androgen Receptor NH2-Terminal Associated Coactivator. J. Biol. Chem. 2000, 275, 9308–9313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Riedl, T.; Washbrook, E.; Pace, P.E.; Coombes, R.C.; Egly, J.-M.; Ali, S. Activation of Estrogen Receptor α by S118 Phosphorylation Involves a Ligand-Dependent Interaction with TFIIH and Participation of CDK7. Mol. Cell 2000, 6, 127–137. [Google Scholar] [CrossRef]
- Chen, D.; Washbrook, E.; Sarwar, N.; Bates, G.J.; Pace, P.E.; Thirunuvakkarasu, V.; Taylor, J.; Epstein, R.J.; Fuller-Pace, F.V.; Egly, J.-M.; et al. Phosphorylation of Human Estrogen Receptor Alpha at Serine 118 by Two Distinct Signal Transduction Pathways Revealed by Phosphorylation-Specific Antisera. Oncogene 2002, 21, 4921–4931. [Google Scholar] [CrossRef] [Green Version]
- Ko, L.J.; Shieh, S.Y.; Chen, X.; Jayaraman, L.; Tamai, K.; Taya, Y.; Prives, C.; Pan, Z.Q. P53 Is Phosphorylated by CDK7-Cyclin H in a P36MAT1-Dependent Manner. Mol. Cell. Biol. 1997, 17, 7220–7229. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Fisher, R.P.; Bailey, P.; Levine, A.J. The CDK7-CycH-P36 Complex of Transcription Factor IIH Phosphorylates P53, Enhancing Its Sequence-Specific DNA Binding Activity in Vitro. Mol. Cell. Biol. 1997, 17, 5923–5934. [Google Scholar] [CrossRef] [Green Version]
- Rimel, J.K.; Poss, Z.C.; Erickson, B.; Maas, Z.L.; Ebmeier, C.C.; Johnson, J.L.; Decker, T.-M.; Yaron, T.M.; Bradley, M.J.; Hamman, K.B.; et al. Selective Inhibition of CDK7 Reveals High-Confidence Targets and New Models for TFIIH Function in Transcription. Genes Dev. 2020, 34, 1452–1473. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, L.; Jiang, G.; Chen, Z.; Li, J.; An, P.; Chen, L.; Du, J.; Wang, H. Targeting CDK7 Increases the Stability of Snail to Promote the Dissemination of Colorectal Cancer. Cell Death Differ. 2019, 26, 1442–1452. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Li, M.; Zhang, X.; Huang, H.; Huang, J.; Ke, J.; Ding, H.; Xiao, J.; Shan, X.; Liu, Q.; et al. Upregulation of CDK7 in Gastric Cancer Cell Promotes Tumor Cell Proliferation and Predicts Poor Prognosis. Exp. Mol. Pathol. 2016, 100, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.-G.; Sun, Y.-W.; et al. THZ1 Reveals CDK7-Dependent Transcriptional Addictions in Pancreatic Cancer. Oncogene 2019, 38, 3932–3945. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting Transcription Regulation in Cancer with a Covalent CDK7 Inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Dang, L.; Zheng, X.; Lu, Y.; Lu, Y.; Ji, R.; Zhang, T.; Ruan, X.; Zhi, J.; Hou, X.; et al. Targeting Super-Enhancer-Driven Oncogenic Transcription by CDK7 Inhibition in Anaplastic Thyroid Carcinoma. Thyroid 2019, 29, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.Y.; Kim, H.R.; Lee, J.Y.; Park, S.; Hwang, J.A.; Kim, W.S.; Yoon, S.; Choi, C.-M.; Rho, J.K.; Lee, J.C. CDK7 Inhibition as a Promising Therapeutic Strategy for Lung Squamous Cell Carcinomas with a SOX2 Amplification. Cell. Oncol. 2019, 42, 449–458. [Google Scholar] [CrossRef]
- Zhong, S.; Zhang, Y.; Yin, X.; Di, W. CDK7 Inhibitor Suppresses Tumor Progression through Blocking the Cell Cycle at the G2/M Phase and Inhibiting Transcriptional Activity in Cervical Cancer. OncoTargets Ther. 2019, 12, 2137–2147. [Google Scholar] [CrossRef] [Green Version]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-Driven Cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, C.L.; Kwiatkowski, N.; Abraham, B.J.; Carretero, J.; Al-Shahrour, F.; Zhang, T.; Chipumuro, E.; Herter-Sprie, G.S.; Akbay, E.A.; Altabef, A.; et al. Targeting Transcriptional Addictions in Small Cell Lung Cancer with a Covalent CDK7 Inhibitor. Cancer Cell 2014, 26, 909–922. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Kwiatkowski, N.P.; Zhang, T.; Nabet, B.; Xu, M.; Liang, Y.; Quan, C.; Wang, J.; Hao, M.; Palakurthi, S.; et al. Targeting MYC Dependency in Ovarian Cancer through Inhibition of CDK7 and CDK12/13. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 Inhibitors as Anticancer Drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Diab, S.; Yu, M.; Wang, S. CDK7 Inhibitors in Cancer Therapy: The Sweet Smell of Success? J. Med. Chem. 2020, 63, 7458–7474. [Google Scholar] [CrossRef] [PubMed]
- Kelso, T.W.R.; Baumgart, K.; Eickhoff, J.; Albert, T.; Antrecht, C.; Lemcke, S.; Klebl, B.; Meisterernst, M. Cyclin-Dependent Kinase 7 Controls MRNA Synthesis by Affecting Stability of Preinitiation Complexes, Leading to Altered Gene Expression, Cell Cycle Progression, and Survival of Tumor Cells. Mol. Cell. Biol. 2014, 34, 3675–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutterer, C.; Eickhoff, J.; Milbradt, J.; Korn, K.; Zeitträger, I.; Bahsi, H.; Wagner, S.; Zischinsky, G.; Wolf, A.; Degenhart, C.; et al. A Novel CDK7 Inhibitor of the Pyrazolotriazine Class Exerts Broad-Spectrum Antiviral Activity at Nanomolar Concentrations. Antimicrob. Agents Chemother. 2015, 59, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Bardeesy, N.; DePinho, R.A. Pancreatic Cancer Biology and Genetics. Nat. Rev. Cancer 2002, 2, 897–909. [Google Scholar] [CrossRef]
- Liu, J.; Ji, S.; Liang, C.; Qin, Y.; Jin, K.; Liang, D.; Xu, W.; Shi, S.; Zhang, B.; Liu, L.; et al. Critical Role of Oncogenic KRAS in Pancreatic Cancer (Review). Mol. Med. Rep. 2016, 13, 4943–4949. [Google Scholar] [CrossRef] [Green Version]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a Tumor Suppressor Gene Product, P19Arf, Interacts with MDM2 and Neutralizes MDM2’s Inhibition of P53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent Somatic Mutations and Homozygous Deletions of the P16 (MTS1) Gene in Pancreatic Adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/P16 Tumor-Suppressive Pathway in Virtually All Pancreatic Carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar] [PubMed]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant P53 Drives Metastasis and Overcomes Growth Arrest/Senescence in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a Candidate Tumor Suppressor Gene at Human Chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.A.; Freitas, J.P.; Mazher Hussain, S.; Glazer, E.S. TGF-β Inhibitors in Metastatic Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Cancer 2019, 50, 207–213. [Google Scholar] [CrossRef]
- Ahmed, S.; Bradshaw, A.-D.; Gera, S.; Dewan, M.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, M.L.; Muñoz-Bellvís, L.; Orfao, A. Genomic Heterogeneity of Pancreatic Ductal Adenocarcinoma and Its Clinical Impact. Cancers 2021, 13, 4451. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M.M.; Merrick, K.A.; Larochelle, S.; Hirschi, A.; Zhang, C.; Shokat, K.M.; Rubin, S.M.; Fisher, R.P. A Cdk7-Cdk4 T-Loop Phosphorylation Cascade Promotes G1 Progression. Mol. Cell 2013, 50, 250. [Google Scholar] [CrossRef] [Green Version]
- Takaki, T.; Echalier, A.; Brown, N.R.; Hunt, T.; Endicott, J.A.; Noble, M.E.M. The Structure of CDK4/Cyclin D3 Has Implications for Models of CDK Activation. Proc. Natl. Acad. Sci. USA 2009, 106, 4171–4176. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, C.; Paul, S.; Dandawate, P.; Gunewardena, S.S.; Subramaniam, D.; West, C.; Anant, S.; Dhar, A. Super-Enhancers: Novel Target for Pancreatic Ductal Adenocarcinoma. Oncotarget 2019, 10, 1554–1571. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; You, X.; Dai, C.; Xu, Q.; Li, F.; Wang, L.; Huang, X.; Wang, J.; Li, S.; Gao, Z.; et al. Targeting Super-Enhancers via Nanoparticle-Facilitated BRD4 and CDK7 Inhibitors Synergistically Suppresses Pancreatic Ductal Adenocarcinoma. Adv. Sci. 2020, 7, 1902926. [Google Scholar] [CrossRef] [Green Version]
- Gurtner, A.; Manni, I.; Piaggio, G. NF-Y in Cancer: Impact on Cell Transformation of a Gene Essential for Proliferation. Biochim. Biophys. Acta-Gene Regul. Mech. 2017, 1860, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiao, X.; Chen, H.; Chen, Z.; Hu, K.; Yin, D. Transcription Factor NFYA Promotes G1/S Cell Cycle Transition and Cell Proliferation by Transactivating Cyclin D1 and CDK4 in Clear Cell Renal Cell Carcinoma. Am. J. Cancer Res. 2020, 10, 2446–2463. [Google Scholar]
- Chae, H.-D.; Kim, J.; Shin, D.Y. NF-Y Binds to Both G1- and G2-Specific Cyclin Promoters; a Possible Role in Linking CDK2/Cyclin A to CDK1/Cyclin B. BMB Rep. 2011, 44, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Z.; Lin, M.; Li, C.; Liu, H.; Gong, C. A Comprehensive Review of the Roles of E2F1 in Colon Cancer. Am. J. Cancer Res. 2020, 10, 757–768. [Google Scholar]
- DeGregori, J.; Kowalik, T.; Nevins, J.R. Cellular Targets for Activation by the E2F1 Transcription Factor Include DNA Synthesis- and G1/S-Regulatory Genes. Mol. Cell. Biol. 1995, 15, 4215–4224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachter, M.M.; Fisher, R.P. The CDK-Activating Kinase Cdk7: Taking Yes for an Answer. Cell Cycle 2013, 12, 3239–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, R.P. The CDK Network: Linking Cycles of Cell Division and Gene Expression. Genes Cancer 2012, 3, 731–738. [Google Scholar] [CrossRef] [Green Version]
- Kent, L.N.; Leone, G. The Broken Cycle: E2F Dysfunction in Cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Li, B.-B.; Wang, B.; Zhu, C.-M.; Tang, D.; Pang, J.; Zhao, J.; Sun, C.-H.; Qiu, M.-J.; Qian, Z.-R. Cyclin-Dependent Kinase 7 Inhibitor THZ1 in Cancer Therapy. Chronic Dis. Transl. Med. 2019, 5, 155–169. [Google Scholar] [CrossRef]
- Hausmann, S.; Kong, B.; Michalski, C.; Erkan, M.; Friess, H. The Role of Inflammation in Pancreatic Cancer. Adv. Exp. Med. Biol. 2014, 816, 129–151. [Google Scholar] [CrossRef]
- Wirth, M.; Mahboobi, S.; Krämer, O.H.; Schneider, G. Concepts to Target MYC in Pancreatic Cancer. Mol. Cancer Ther. 2016, 15, 1792–1798. [Google Scholar] [CrossRef] [Green Version]
- Hessmann, E.; Schneider, G.; Ellenrieder, V.; Siveke, J.T. MYC in Pancreatic Cancer: Novel Mechanistic Insights and Their Translation into Therapeutic Strategies. Oncogene 2016, 35, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Salghetti, S.E.; Kim, S.Y.; Tansey, W.P. Destruction of Myc by Ubiquitin-Mediated Proteolysis: Cancer-Associated and Transforming Mutations Stabilize Myc. EMBO J. 1999, 18, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, A.S.; Sears, R.C. MYC Degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef] [PubMed]
- Ganuza, M.; Sáiz-Ladera, C.; Cañamero, M.; Gómez, G.; Schneider, R.; Blasco, M.A.; Pisano, D.; Paramio, J.M.; Santamaría, D.; Barbacid, M. Genetic Inactivation of Cdk7 Leads to Cell Cycle Arrest and Induces Premature Aging Due to Adult Stem Cell Exhaustion. EMBO J. 2012, 31, 2498–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, C.M.; Liang, Y.; Leggett, A.; Park, W.D.; Li, L.; Mills, C.E.; Elsarrag, S.Z.; Ficarro, S.B.; Zhang, T.; Düster, R.; et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem. Biol. 2019, 26, 792–803. [Google Scholar] [CrossRef]
- Chen, H.-D.; Huang, C.-S.; Xu, Q.-C.; Li, F.; Huang, X.-T.; Wang, J.-Q.; Li, S.-J.; Zhao, W.; Yin, X.-Y. Therapeutic Targeting of CDK7 Suppresses Tumor Progression in Intrahepatic Cholangiocarcinoma. Int. J. Biol. Sci. 2020, 16, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, J.; Yue, M.; Cai, X.; Wang, T.; Wu, C.; Su, H.; Wang, Y.; Han, M.; Zhang, Y.; et al. Direct Phosphorylation and Stabilization of MYC by Aurora B Kinase Promote T-Cell Leukemogenesis. Cancer Cell 2020, 37, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Nevins, J.R.; Shats, I.; Chi, J.-T. E2F1-Mediated Induction of NFYB Attenuates Apoptosis via Joint Regulation of a Pro-Survival Transcriptional Program. PLoS ONE 2015, 10, e0127951. [Google Scholar] [CrossRef]
- Dolfini, D.; Andrioletti, V.; Mantovani, R. Overexpression and Alternative Splicing of NF-YA in Breast Cancer. Sci. Rep. 2019, 9, 12955. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Zhang, M.; Wang, Y.; Wang, Y. NF-YC in Glioma Cell Proliferation and Tumor Growth and Its Role as an Independent Predictor of Patient Survival. Neurosci. Lett. 2016, 631, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kottorou, A.E.; Antonacopoulou, A.G.; Dimitrakopoulos, F.-I.D.; Tsamandas, A.C.; Scopa, C.D.; Petsas, T.; Kalofonos, H.P. Altered Expression of NFY-C and RORA in Colorectal Adenocarcinomas. Acta Histochem. 2012, 114, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, E.; Hahn, F.; Bertzbach, L.D.; Seyler, L.; Wangen, C.; Müller, R.; Tannig, P.; Grau, B.; Baumann, M.; Zent, E.; et al. In Vivo Proof-of-Concept for Two Experimental Antiviral Drugs, Both Directed to Cellular Targets, Using a Murine Cytomegalovirus Model. Antivir. Res. 2019, 161, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Panc89 | MP2 |
|---|---|---|
| Log2FC | Log2FC | |
| CDKN1A (p21) | 2.00 | 0.66 |
| CDKN1B (p27) | −0.59 | n.a. |
| CDKN1C (p57) | 1.38 | 0.15 |
| CDKN2A (p16) | 1.11 | n.e. |
| CDKN2B (p15) | 2.78 | n.e. |
| CDKN2C (p18) | −2.43 | 0.60 |
| CDK1 | −1.57 | −0.60 |
| CDK2 | −1.02 | −0.53 |
| CDK4 | −1.12 | n.a. |
| CDK7 | 0.74 | n.a. |
| CCNA1 | −0.90 | n.e. |
| CCNA2 | −1.85 | −0.62 |
| CCNB1 | −1.51 | −0.30 |
| CCNB2 | −1.15 | −0.44 |
| CCND1 | 0.62 | 0.50 |
| CCND3 | 0.53 | n.a. |
| CDC25A | −1.19 | n.a. |
| CDC25B | −1.46 | −0.26 |
| CDC25C | −2.05 | −0.58 |
| CHEK1 | −0.95 | −0.52 |
| CHEK2 | −1.81 | −1.20 |
| WEE1 | −1.45 | 0.53 |
| E2F1 | −1.90 | −0.83 |
| E2F2 | −1.52 | −1.09 |
| E2F4 | −0.30 | −0.41 |
| E2F5 | −0.55 | n.a. |
| E2F7 | −0.59 | n.a. |
| E2F8 | −1.71 | n.a. |
| NFYA | −0.65 | n.a. |
| NFYB | −0.99 | n.a. |
| NFYC | 0.59 | 0.19 |
| RBL1 | −1.48 | −0.75 |
| RBL2 | −0.69 | −0.31 |
| TRAP1 | −1.50 | −0.68 |
| AURKA | −1.83 | −0.43 |
| AURKB | −1.70 | −0.43 |
| AURKC | 2.70 | 1.31 |
| Primer | Sequences |
|---|---|
| hActin_F | GCTGTGCTGTCCCTGTATGCCTCT |
| hActin_R | CCTCTCAGCTGTGGTGGTGAAGC |
| hBcl-xL_F | AGGAGAACGGCGGCTGGGATA |
| hBcl-xL_R | GAGCCCAGCAGAACCACGCC |
| hCDC25C_1F | TCTACGGAACTCTTCTCATCCAC |
| hCDC25C_1R | TCCAGGAGCAGGTTTAACATTTT |
| hCDC25A_1F | TTCCTCTTTTTACACCCCAGTCA |
| hCDC25A_1R | TCGGTTGTCAAGGTTTGTAGTTC |
| hCDCP1_F | CTGAACTGCGGGGTCTCTATC |
| hCDCP1_R | GTCCCCAGCTTTATGAGAACTG |
| hCDK1_1F | AAACTACAGGTCAAGTGGTAGCC |
| hCDK1_1R | TCCTGCATAAGCACATCCTGA |
| hCDK2_1F | CCAGGAGTTACTTCTATGCCTGA |
| hCDK2_1R | TTCATCCAGGGGAGGTACAAC |
| hCyclinB1_1F | AATAAGGCGAAGATCAACATGGC |
| hCyclinB1_1R | TTTGTTACCAATGTCCCCAAGAG |
| hEGFR_F | AACTGTGAGGTGGTCCTTGG |
| hEGFR_R | TGAGGACATAACCAGCCACC |
| hEGFRei1_F | AGGGCGTCATCAGTTTCTCA |
| hEGFRei1_R | AGTTCTCCTCTCCTGCACCC |
| hEIFD2_F | GCCTTTCGGGTCAAGTCCAA |
| hEIFD2_R | CCTCCTTTCCAGGTACTAACTCA |
| hGAPDHei8_F | GCCCTGACAACTCTTTTCATCT |
| hGAPDHei8_R | TCTCTCTTCCTCTTGTGCTCTTG |
| hMYBL2_F | CCGGAGCAGAGGGATAGCA |
| hMYBL2_R | CAGTGCGGTTAGGGAAGTGG |
| hMYC_F | TTTCGGGTAGTGGAAAACCA |
| hMYC_R | CACCGAGTCGTAGTCGAGGT |
| hMYCei1_F | TAACTCAAGACTGCCTCCCG |
| hMYCei1_R | AAGCTAACGTTGAGGGGCAT |
| hNFY-A_1F | ATGTGGTCAATTCAGGAGGGA |
| hNFY-A_1R | ATTGTTTGGCATTCACGTAGAGA |
| hPRPF3_F | CAGCAGCATTGAACTGTGTGG |
| hPRPF3_R | TCGTCGCTTCTTTACTCCTGAT |
| hRPL3ei5_F | CAAGGGCAAAGGCTACAAAG |
| hRPL3ei5_R | GAATGGTTCTACACTGTCCGATT |
| hRPL31i1_F | TTTGGGATTGAACTGG |
| hRPL31i1_R | CCCTAAGCCTACTTTC |
| hTAF15_F | GATTCTGGAAGTTACGGTCAGTC |
| hTAF15_R | AGCTTTGTGATGCTTGTCCATAG |
| hTNFRSF10D_F | TACCACGACCAGAGACACC |
| hTNFRSF10D_R | CACCCTGTTCTACACGTCCG |
| hTRAF6_F | ATG CGG CCA TAG GTT CTG C |
| hTRAF6_R | TCCTCAAGATGTCTCAGTTCCAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolloch, L.; Kreinest, T.; Meisterernst, M.; Oeckinghaus, A. Control of Expression of Key Cell Cycle Enzymes Drives Cell Line-Specific Functions of CDK7 in Human PDAC Cells. Int. J. Mol. Sci. 2022, 23, 812. https://doi.org/10.3390/ijms23020812
Kolloch L, Kreinest T, Meisterernst M, Oeckinghaus A. Control of Expression of Key Cell Cycle Enzymes Drives Cell Line-Specific Functions of CDK7 in Human PDAC Cells. International Journal of Molecular Sciences. 2022; 23(2):812. https://doi.org/10.3390/ijms23020812
Chicago/Turabian StyleKolloch, Lina, Teresa Kreinest, Michael Meisterernst, and Andrea Oeckinghaus. 2022. "Control of Expression of Key Cell Cycle Enzymes Drives Cell Line-Specific Functions of CDK7 in Human PDAC Cells" International Journal of Molecular Sciences 23, no. 2: 812. https://doi.org/10.3390/ijms23020812