
Epstein-Barr Virus Promotes the Production of Inflammatory Cytokines in Gingival Fibroblasts and RANKL-Induced Osteoclast Differentiation in RAW264.7 Cells


 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
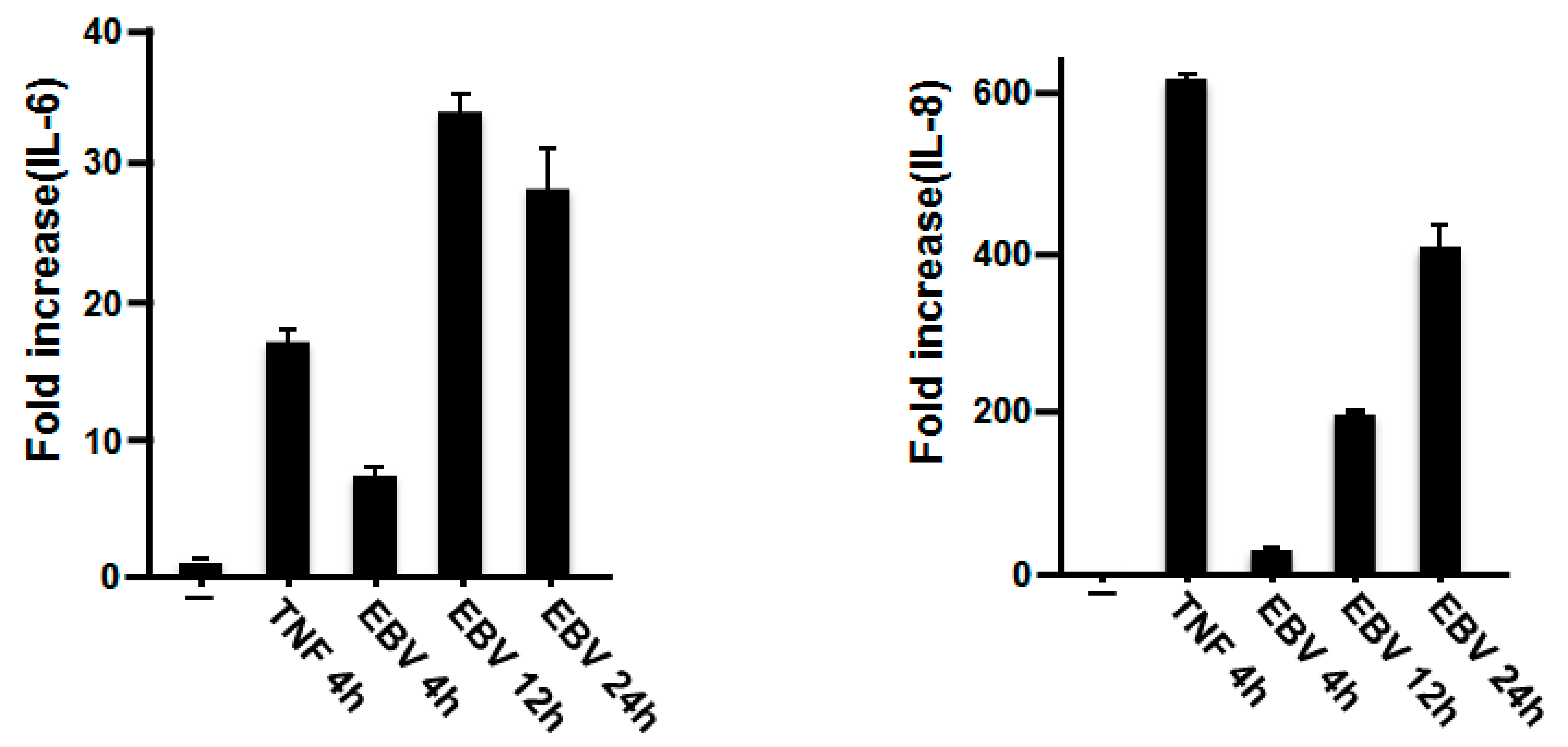
2.1. Effect of EBV on IL-6 and IL-8 mRNA Expression in Gingival Fibroblasts
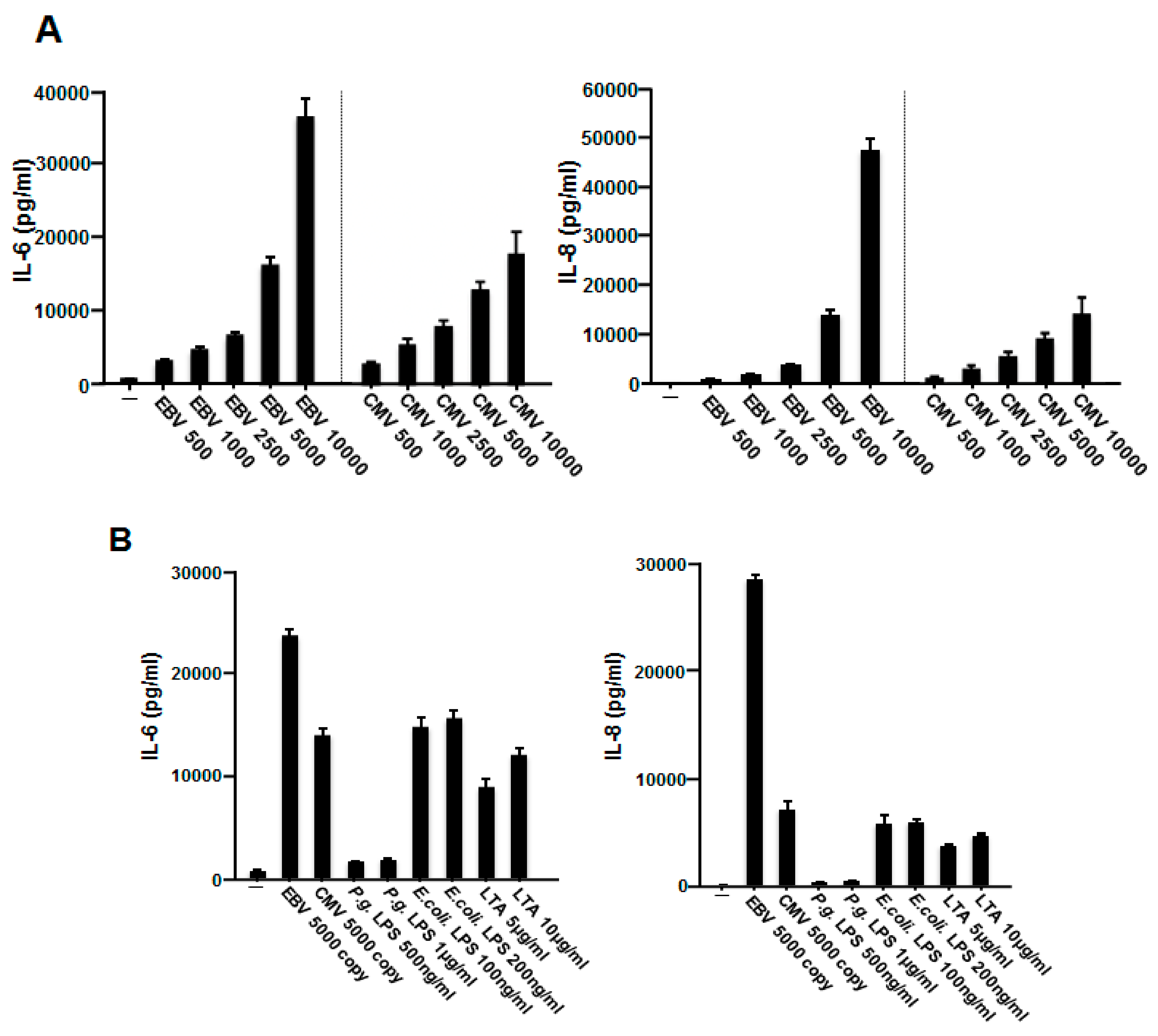
2.2. EBV Promoted the Production of IL-6 and IL-8 in HGFs
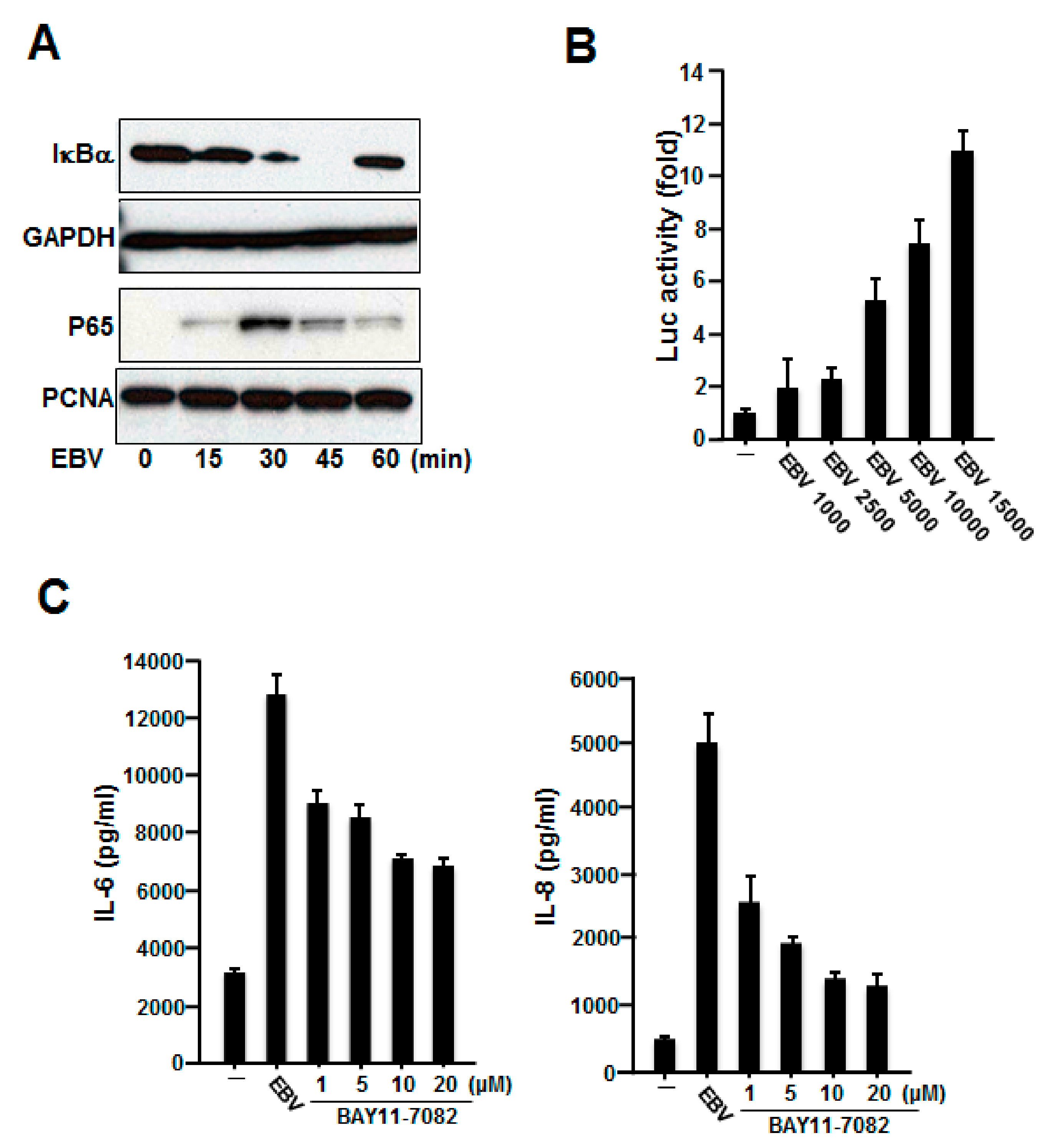
2.3. NF-κB Was Involved in the Production of IL-6 and IL-8 Induced by Inactivated EBV in HGFs
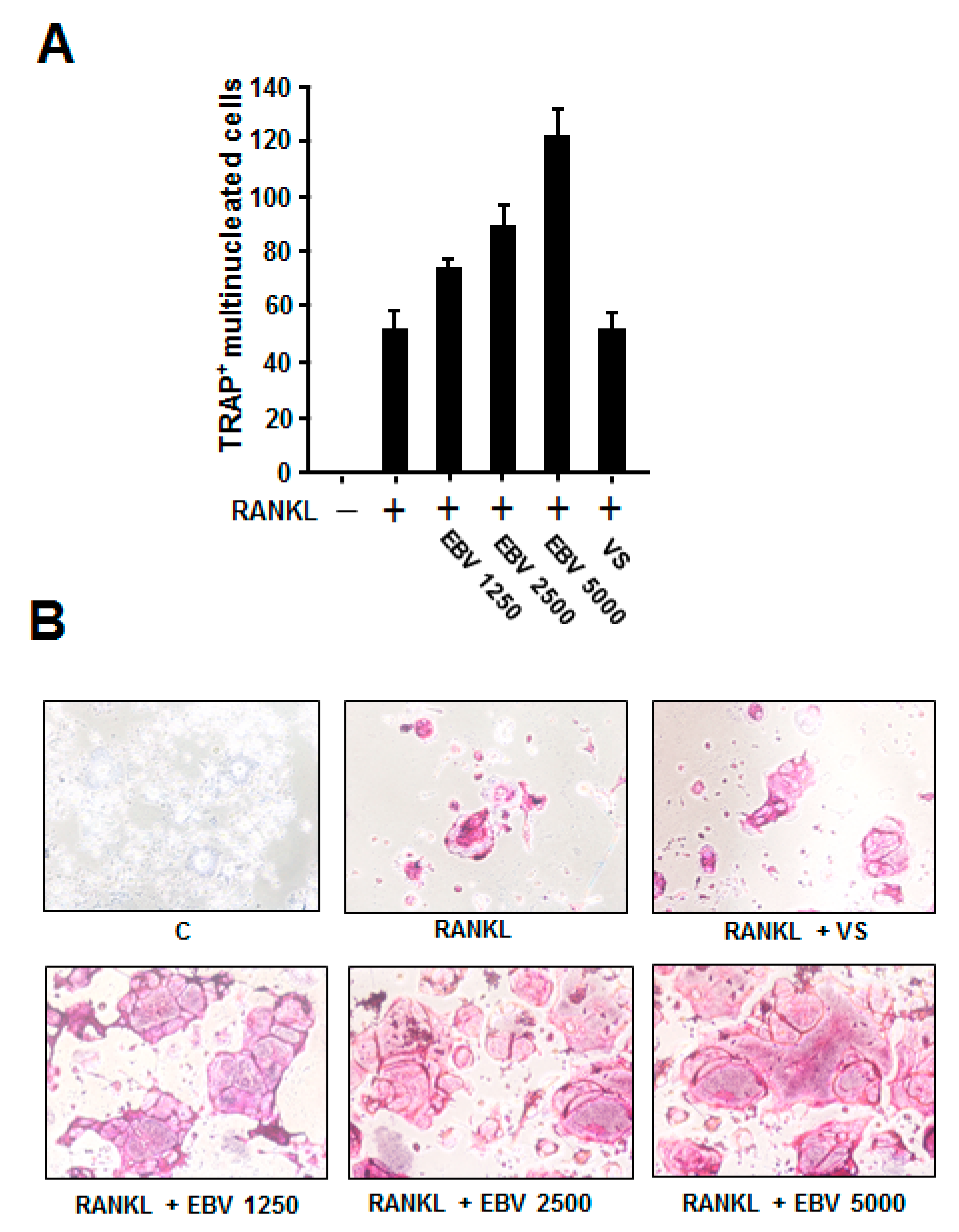
2.4. EBV Promoted RANKL-Induced Osteoclast Differentiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents and Plasmids
4.3. Preparation of mRNA and Real-Time Polymerase Chain Reaction
4.4. Cytokine Measurements
4.5. Preparation of Whole-Cell and Nuclear Extracts
4.6. Western Blot Analysis
4.7. Transfection and Luciferase Assay
4.8. Osteoclast Formation and TRAP Staining
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Holt, S.C.; Ebersole, J.L. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: The “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol. 2000 2005, 38, 72–122. [Google Scholar] [CrossRef] [PubMed]
- Slots, J.; Saygun, I.; Sabeti, M.; Kubar, A. Epstein-Barr virus in oral diseases. J. Periodontal Res. 2006, 41, 235–244. [Google Scholar] [CrossRef]
- Abusleme, L.; Dupuy, A.K.; Dutzan, N.; Silva, N.; Burleson, J.A.; Strausbaugh, L.D.; Gamonal, J.; Diaz, P.I. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7, 1016–1025. [Google Scholar] [CrossRef]
- Ledder, R.G.; Gilbert, P.; Huws, S.A.; Aarons, L.; Ashley, M.P.; Hull, P.S.; McBain, A.J. Molecular analysis of the subgingival microbiota in health and disease. J. Appl. Environ. Microbiol. 2007, 73, 516–523. [Google Scholar] [CrossRef]
- Riep, B.; Edesi-Neuss, L.; Claessen, F.; Skarabis, H.; Ehmke, B.; Flemmig, T.F.; Bernimoulin, J.P.; Göbel, U.B.; Moter, A. Are putative periodontal pathogens reliable diagnostic markers? J. Clin. Microbiol. 2009, 47, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Ogata, Y. How dose Epstein-Barr virus contribute to chronic periodontitis? Int. J. Mol. Sci. 2020, 21, 1940. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Inoue, H.; Tamura, M.; Cueno, M.E.; Inoue, H.; Takeichi, O.; Kusama, K.; Saito, I.; Ochiai, K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie 2012, 94, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.; Nowzari, H.; Slots, J. Herpesviruses in periodontal pocket and gingival tissue specimens. Oral Microbiol. Immunol. 2000, 15, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhu, C.; Li, F.; Xu, W.; Tao, D.; Feng, X. Putative periodontopathic bacteria and herpesviruses in pregnant women: A case–control study. Sci. Rep. 2016, 6, 27796. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Herpesviral-bacterial interactions in periodontal diseases. Periodontology 2000, 52, 117–140. [Google Scholar] [CrossRef] [PubMed]
- Saygun, I.; Kubar, A.; Özdemir, A.; Slots, J. Periodontitis lesions are a source of salivary cytomegalovirus and Epstein-Barr virus. J. Periodontal Res. 2005, 40, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, A.; Sakellari, D.; Papa, A.; Antoniadis, A. Real-time polymerase chain reaction quantification of Epstein-Barr virus in chronic periodontitis patients. J. Periodontal Res. 2005, 40, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Imai, K.; Ochiai, K.; Ogata, Y. Higher prevalence of Epstein-Barr virus DNA in deeper periodontal pockets of chronic periodontitis in Japanese patients. PLoS ONE 2013, 8, e71990. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Imai, K.; Ochiai, K.; Ogata, Y. Prevalence and quantitative analysis of Epstein-Barr virus DNA and Porphyromonas gingivalis associated with Japanese chronic periodontitis patients. Clin. Oral Investig. 2015, 19, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Prêcheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV infection is common in gingival epithelial cells of the periodontium and worsens during chronic periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [PubMed]
- Sunde, P.T.; Olsen, I.; Enersen, M.; Grinde, B. Patient with severe periodontitis and subgingival Epstein-Barr virus treated with antiviral therapy. J. Clin. Virol. 2008, 42, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef]
- Mundy, G.R. Osteoporosis and inflammation. Nutr. Rev. 2007, 65 Pt 2, S147–S151. [Google Scholar] [CrossRef] [PubMed]
- Roberge, C.J.; McColl, S.R.; Larochelle, B.; Gosselin, J. Granulocyte-macrophage colony-stimulating factor enhances EBV-induced synthesis of chemotactic factors in human neutrophils. J. Immunol. 1998, 160, 2442–2448. [Google Scholar] [PubMed]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J Virol. 2007, 81, 8016–8024. [Google Scholar] [CrossRef] [PubMed]
- Roberge, C.J.; Larochelle, B.; Rola-Pleszczynski, M.; Gosselin, J. Epstein-Barr virus induces GM-CSF synthesis by monocytes: Effect on EBV-induced IL-1 and IL-1 receptor antagonist production in neutrophils. Virology 1997, 238, 344–352. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roberge, C.J.; Poubelle, P.E.; Beaulieu, A.D.; Heitz, D.; Gosselin, J. The IL-1 and IL-1 receptor antagonist (IL-1Ra) response of human neutrophils to EBV stimulation. Preponderance of IL-Ra detection. J. Immunol. 1996, 156, 4884–4891. [Google Scholar] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N. Interleukin-8: An expanding universe beyond neutrophil chemotaxis and activation. Int. J. Hematol. 2000, 72, 391–398. [Google Scholar]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Bickel, M. The role of interleukin-8 in inflammation and mechanisms of regulation. J. Periodontol. 1993, 64, 456–460. [Google Scholar]
- Hwang, Y.S.; Lee, S.K.; Park, K.K.; Chung, W.Y. Secretion of IL-6 and IL-8 from lysophosphatidic acid-stimulated oral squamous cell carcinoma promotes osteoclastogenesis and bone resorption. Oral Oncol. 2012, 48, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Ho, Y.P.; Chen, C.C. Levels of interleukin-1 beta and interleukin-8 in gingival crevicular fluids in adult periodontitis. J. Periodontol. 1995, 66, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Söder, B.; Corbet, E.F. Interleukin-8 and granulocyte elastase in gingival crevicular fluid in relation to periodontopathogens in untreated adult periodontitis. J. Periodontol. 2000, 71, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, F.Y.; Berker, E.; Akkuş, S.; Bulut, S. Relationship between interleukin-6 levels in gingival crevicular fluid and periodontal status in patients with rheumatoid arthritis and adult periodontitis. J. Periodontol. 2000, 71, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, L.; Sergi, R.; Caselli, G.; Boraschi, D.; Locati, M.; Sozzani, S.; Bertini, R. Interleukin-1β primes interleukin-8-stimulated chemotaxis and elastase release in human neutrophils via its type I receptor. Eur. Cytokine Netw. 1997, 8, 173–178. [Google Scholar] [PubMed]
- Reinhardt, R.A.; Masada, M.P.; Kaldahl, W.B.; DuBois, L.M.; Kornman, K.S.; Choi, J.I.; Kalkwarf, K.L.; Allison, A.C. Gingival fluid IL-1 and IL-6 levels in refractory periodontitis. J. Clin. Periodontol. 1993, 20, 225–231. [Google Scholar] [CrossRef]
- Tanner, J.E.; Alfieri, C.; Chatila, T.A.; Diaz-Mitoma, F. Induction of interleukin-6 after stimulation of human B-cell CD21 by Epstein-Barr virus glycoproteins gp350 and gp220. J. Virol. 1996, 70, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, I.; Shima, K.; Saito, I.; Kiyoshima, T.; Matsuo, K.; Ozeki, S.; Ohishi, M.; Sakai, H. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma. J. Pathol. 1999, 189, 34–39. [Google Scholar] [CrossRef]
- Nagasawa, Y.; Takei, M.; Iwata, M.; Nagatsuka, Y.; Tsuzuki, H.; Imai, K.; Imadome, K.I.; Fujiwara, S.; Kitamura, N. Human osteoclastogenesis in Epstein-Barr virus-induced erosive arthritis in humanized NOD/Shi-scid/IL-2Rγnull mice. PLoS ONE 2021, 16, e0249340. [Google Scholar] [CrossRef] [PubMed]
- Koike, R.; Nodomi, K.; Watanabe, N.; Ogata, Y.; Takeichi, O.; Takei, M.; Kaneko, T.; Tonogi, M.; Kotani, A.I.; Imai, K. Butyric acid in saliva of chronic periodontitis patients induces transcription of the EBV lytic switch activator BZLF1: A Pilot study. In Vivo 2020, 34, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Nodomi, K.; Koike, R.; Kato, A.; Takeichi, O.; Kotani, A.I.; Kaneko, T.; Sakagami, H.; Takei, M.; Ogata, Y.; et al. Ebv lmp1 in gingival epithelium potentially contributes to human chronic periodontitis via inducible il8 production. In Vivo 2019, 33, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Hayata, M.; Watanabe, N.; Kamio, N.; Tamura, M.; Nodomi, K.; Tanaka, K.; Iddamalgoda, A.; Tsuda, H.; Ogata, Y.; Sato, S.; et al. Cynaropicrin from Cynara scolymus L. suppresses Porphyromonas gingivalis LPS-induced production of inflammatory cytokines in human gingival fibroblasts and RANKL-induced osteoclast differentiation in RAW264.7 cells. J. Nat. Med. 2019, 73, 114–123. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yokoe, S.; Hasuike, A.; Watanabe, N.; Tanaka, H.; Karahashi, H.; Wakuda, S.; Takeichi, O.; Kawato, T.; Takai, H.; Ogata, Y.; et al. Epstein-Barr Virus Promotes the Production of Inflammatory Cytokines in Gingival Fibroblasts and RANKL-Induced Osteoclast Differentiation in RAW264.7 Cells. Int. J. Mol. Sci. 2022, 23, 809. https://doi.org/10.3390/ijms23020809
Yokoe S, Hasuike A, Watanabe N, Tanaka H, Karahashi H, Wakuda S, Takeichi O, Kawato T, Takai H, Ogata Y, et al. Epstein-Barr Virus Promotes the Production of Inflammatory Cytokines in Gingival Fibroblasts and RANKL-Induced Osteoclast Differentiation in RAW264.7 Cells. International Journal of Molecular Sciences. 2022; 23(2):809. https://doi.org/10.3390/ijms23020809
Chicago/Turabian StyleYokoe, Sho, Akira Hasuike, Norihisa Watanabe, Hideki Tanaka, Hiroyuki Karahashi, Shin Wakuda, Osamu Takeichi, Takayuki Kawato, Hideki Takai, Yorimasa Ogata, and et al. 2022. "Epstein-Barr Virus Promotes the Production of Inflammatory Cytokines in Gingival Fibroblasts and RANKL-Induced Osteoclast Differentiation in RAW264.7 Cells" International Journal of Molecular Sciences 23, no. 2: 809. https://doi.org/10.3390/ijms23020809
APA StyleYokoe, S., Hasuike, A., Watanabe, N., Tanaka, H., Karahashi, H., Wakuda, S., Takeichi, O., Kawato, T., Takai, H., Ogata, Y., Sato, S., & Imai, K. (2022). Epstein-Barr Virus Promotes the Production of Inflammatory Cytokines in Gingival Fibroblasts and RANKL-Induced Osteoclast Differentiation in RAW264.7 Cells. International Journal of Molecular Sciences, 23(2), 809. https://doi.org/10.3390/ijms23020809