
Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo



Abstract
:1. Introduction
2. Results
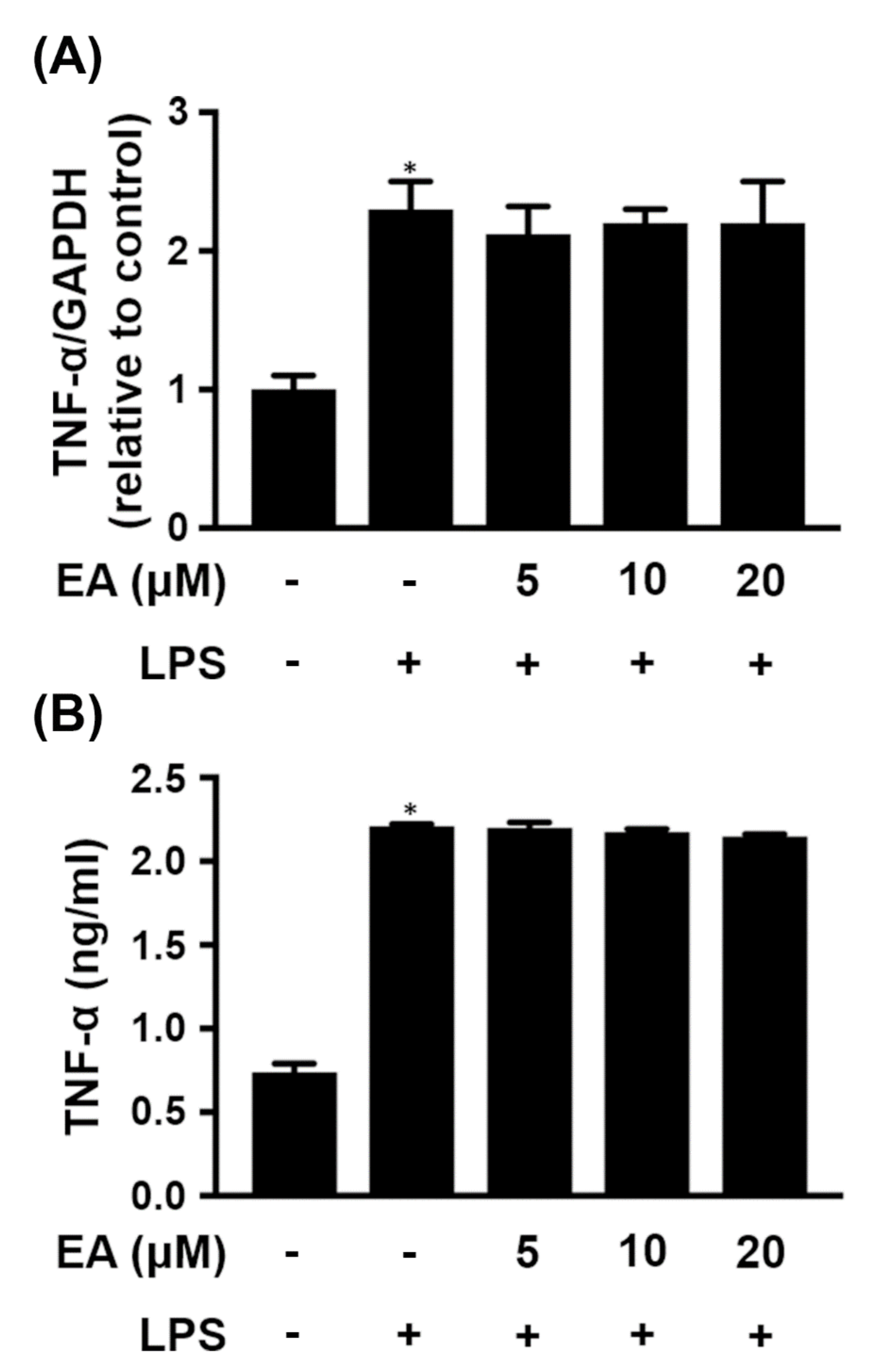
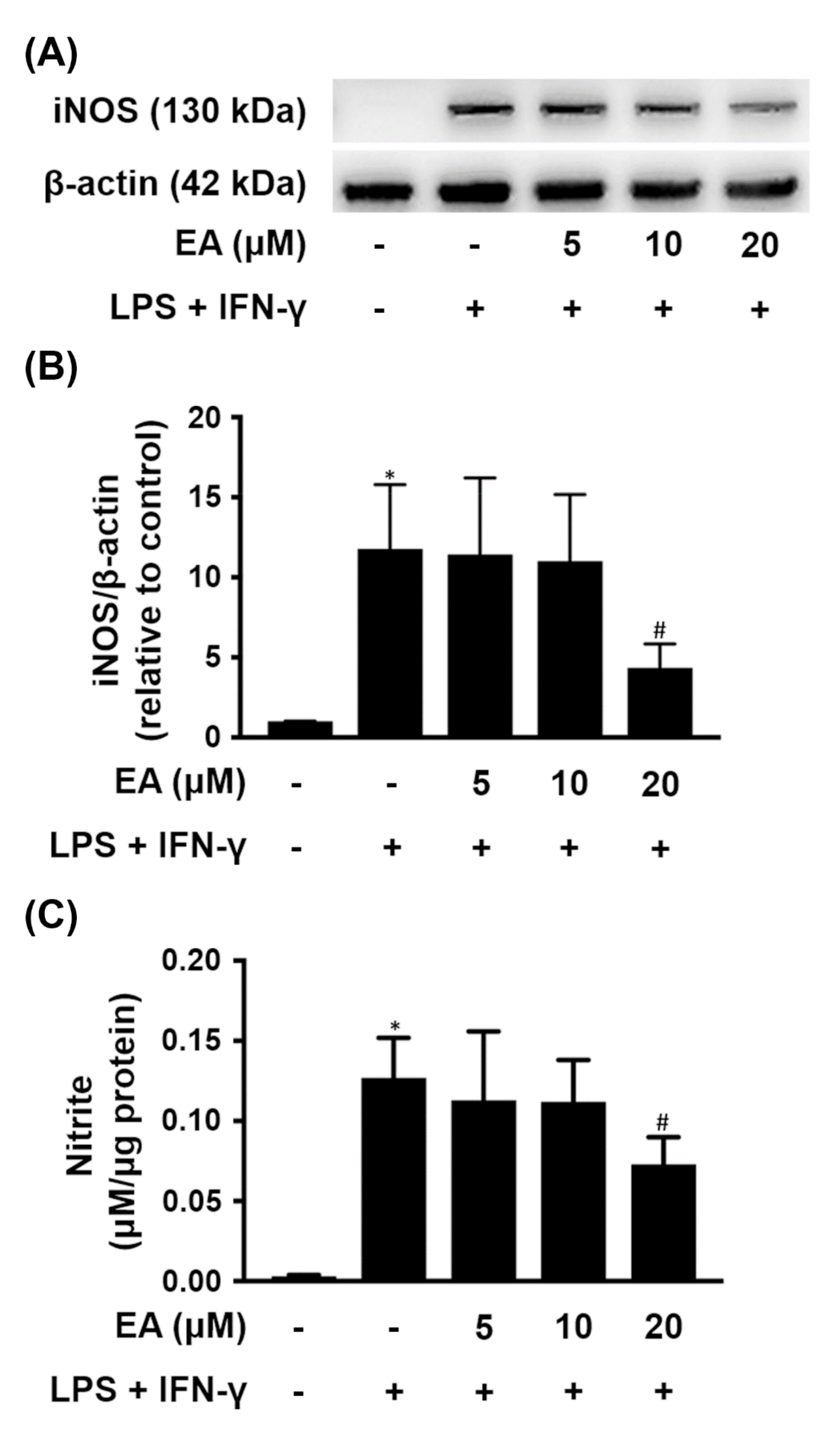
2.1. EA Selectively Modulates the Expression of Proinflammatory Genes in Glia
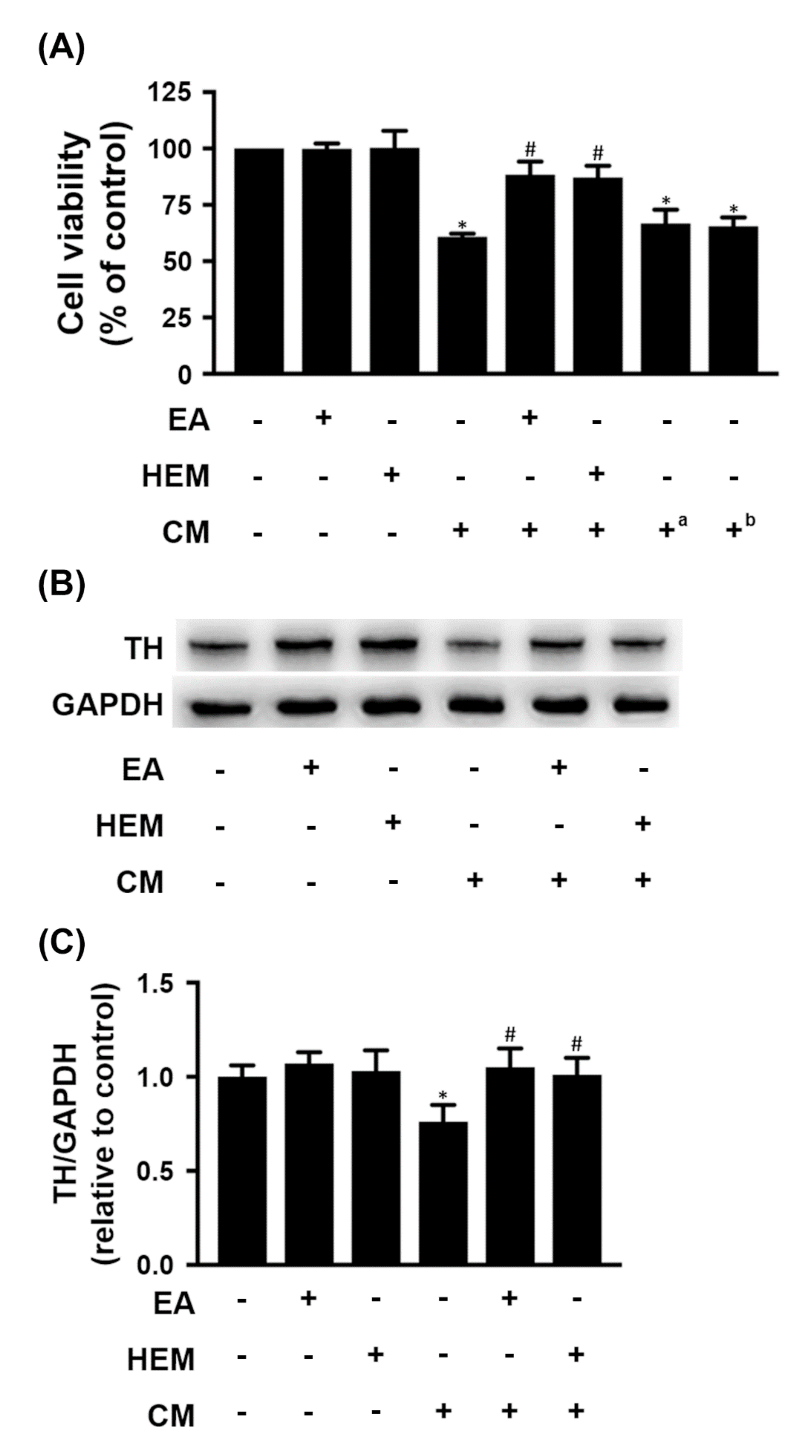
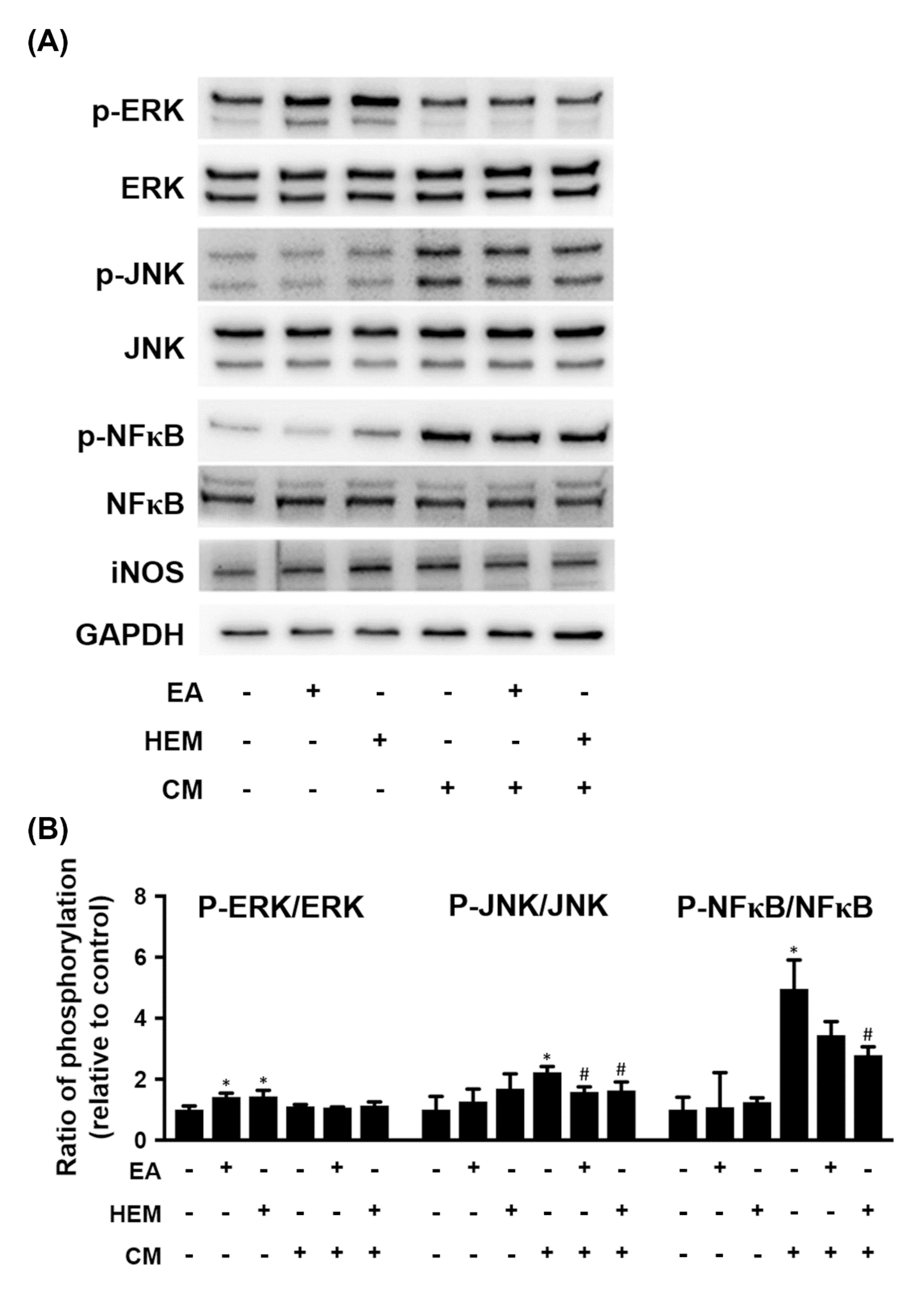
2.2. HEM/EA Ameliorates Microglia-Induced Neurotoxicity
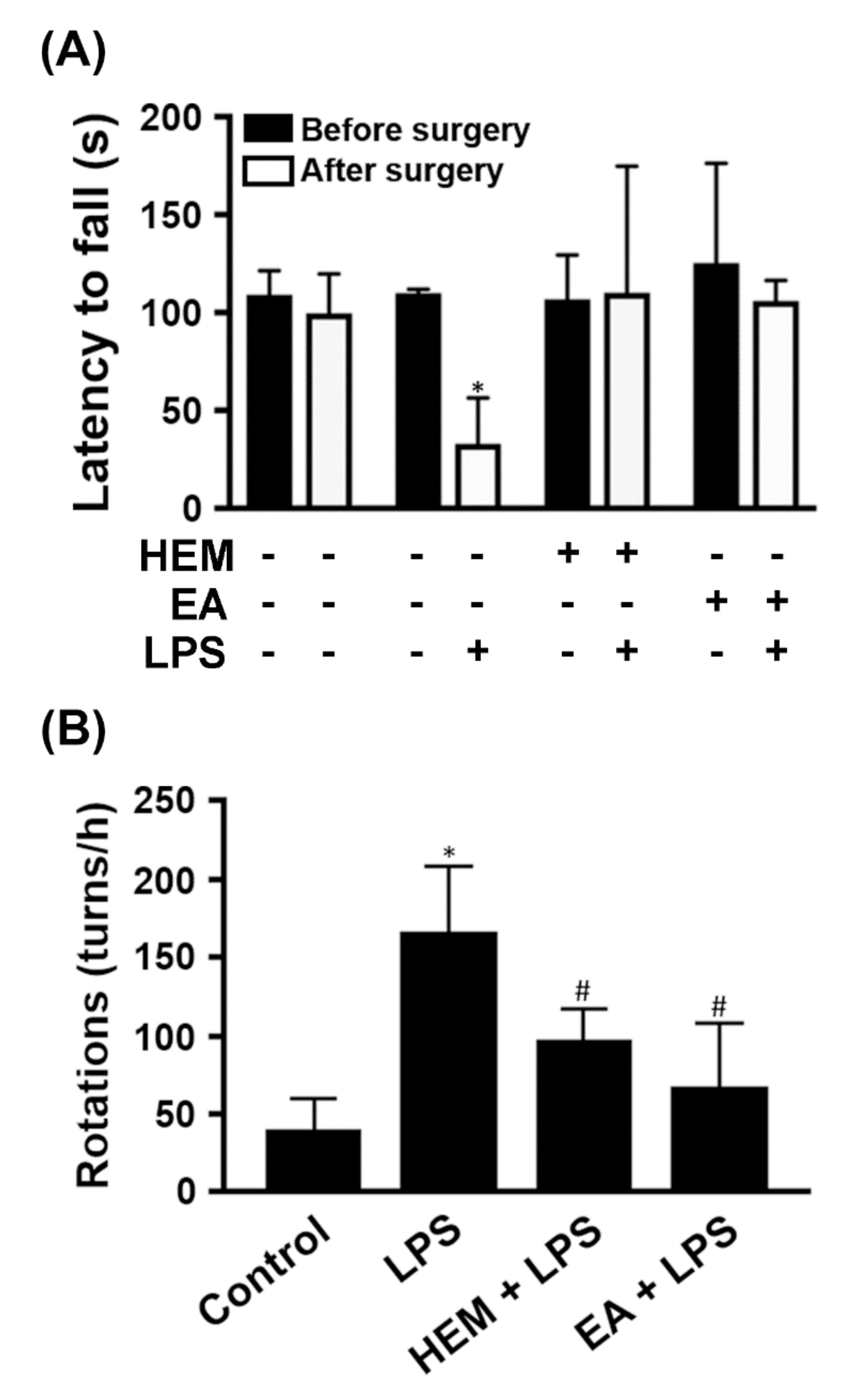
2.3. HEM/EA Alleviates Motor Dysfunction in an LPS-Induced PD Rat Model
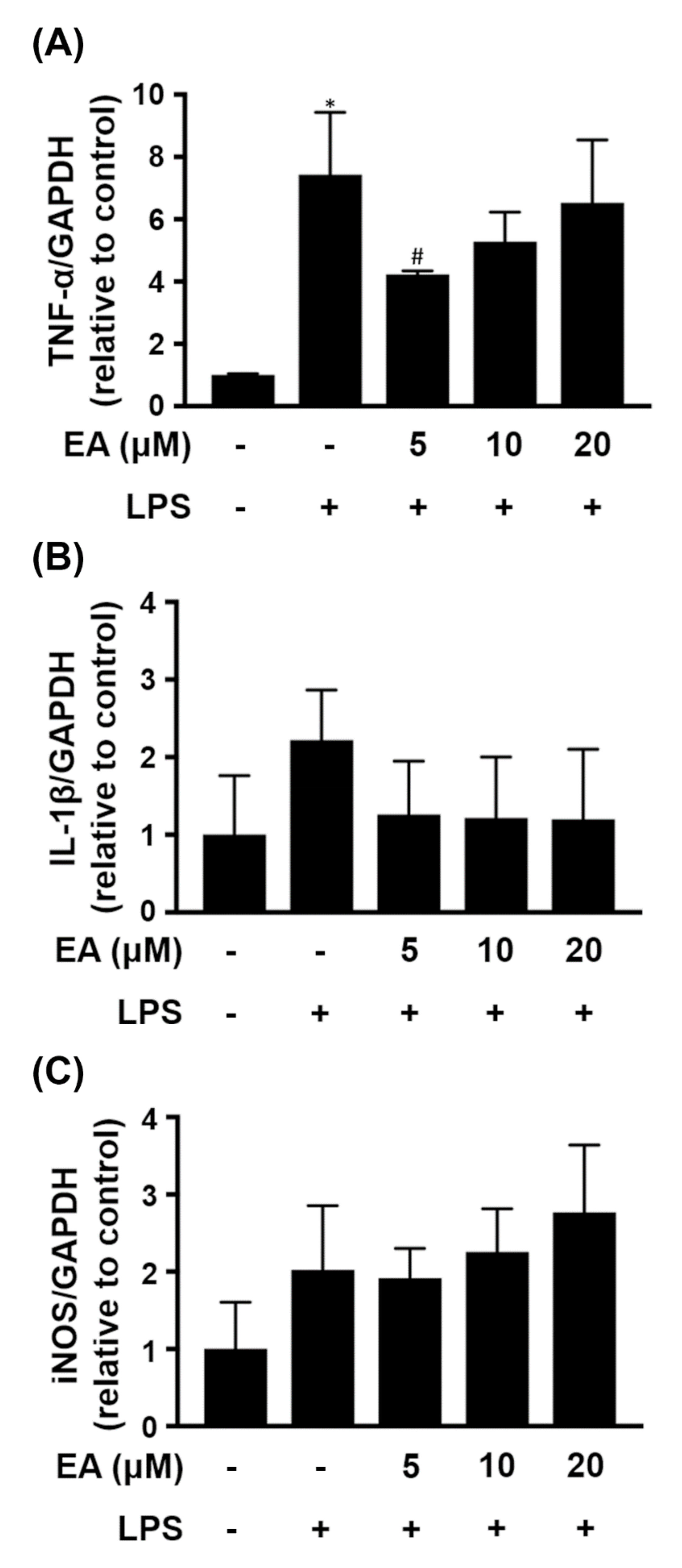
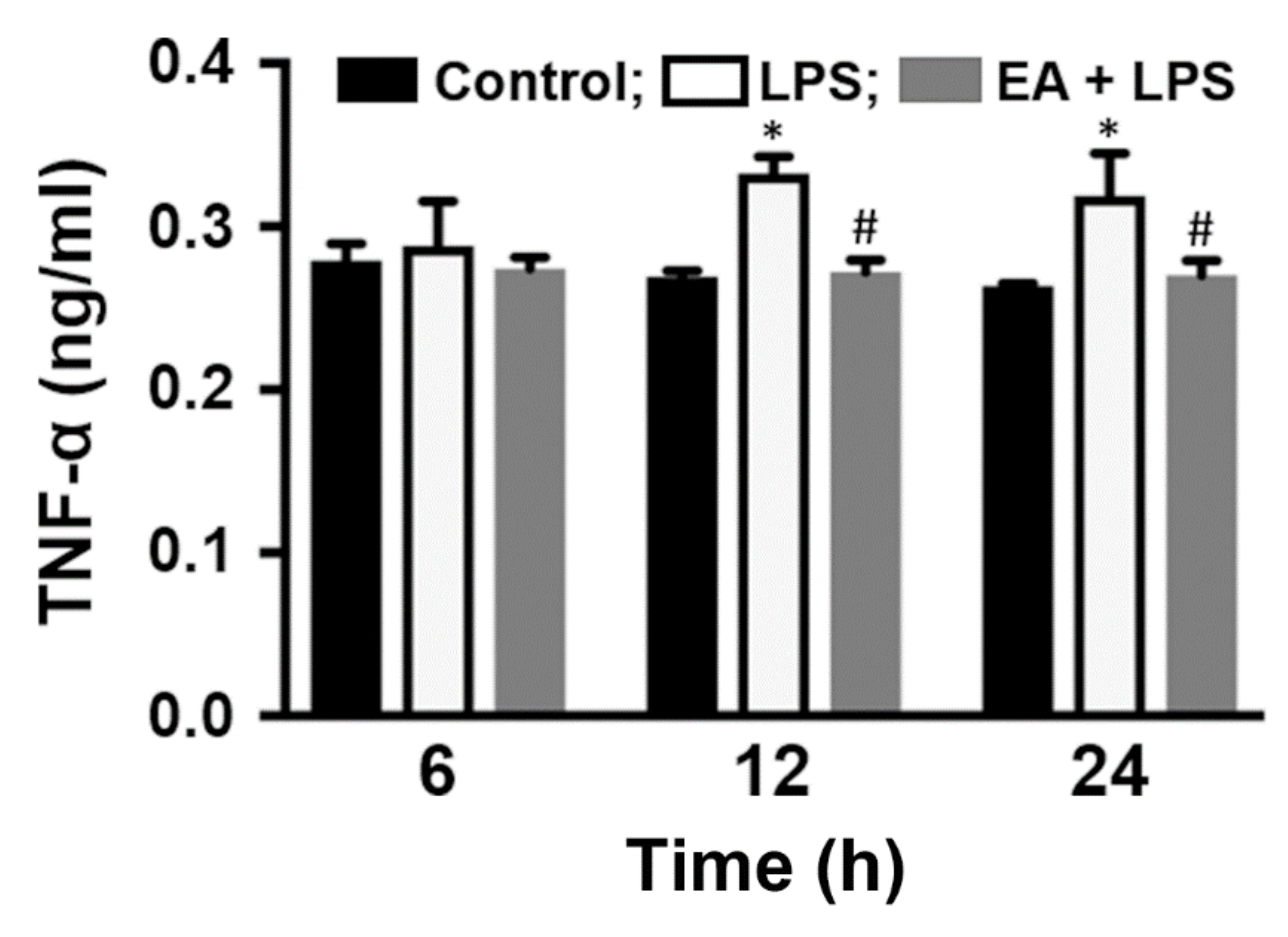
2.4. HEM/EA Downregulates Proinflammatory Mediators in the Midbrain of an LPS-Induced PD Rat Model
3. Discussion
3.1. EA Modulates LPS-Induced Expression of Proinflammatory Factors in Glial Cells
3.2. EA Protects Dopaminergic Cells against Activated Microglia-Induced Neuronal Cell Death
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. Animal Preparation and Surgery
4.4. Real-Time Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
4.5. Locomotor Activity Measurements
4.6. Rotational Behavior Assay
4.7. Western Blot Analysis
4.8. Nitric Oxide Release
4.9. TNF-α Assessment
4.10. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Afridi, R.; Kim, J.H.; Rahman, M.H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron-Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef]
- Franco, R.; Fernandez-Suarez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.H.; Oyarzabal, E.A.; Sung, Y.F.; Chu, C.H.; Wang, Q.; Chen, S.L.; Lu, R.B.; Hong, J.S. Microglial regulation of immunological and neuroprotective functions of astroglia. Glia 2015, 63, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Moehle, M.S.; West, A.B. M1 and M2 immune activation in Parkinson’s Disease: Foe and ally? Neuroscience 2015, 302, 59–73. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Kim, C.; Lee, S.J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxid. Med. Cell Longev. 2010, 3, 283–287. [Google Scholar] [CrossRef]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate infammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Ros, C.M.; Annese, V.; Gómez, A.; Ros-Bernal, F.; Aguado-Year, D.; Martínez-Pagán, M.E.; de Pablos, V.; Fernandez-Villalba, E.; Herrero, M.T. IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis. 2011, 2, e142. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [Green Version]
- Tufekci, K.U.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in Parkinson’s disease. Adv. Protein Chem. Struct. Biol. 2012, 88, 69–132. [Google Scholar] [PubMed]
- Khan, M.A.; Tania, M.; Liu, R.; Rahman, M.M. Hericium erinaceus: An edible mushroom with medicinal values. J. Complement. Integr. Med. 2013, 10, 253–258. [Google Scholar] [CrossRef]
- Friedman, M. Chemistry, Nutrition, and Health-Promoting Properties of Hericium erinaceus (Lion’s Mane) Mushroom Fruiting Bodies and Mycelia and Their Bioactive Compounds. J. Agric. Food Chem. 2015, 63, 7108–7123. [Google Scholar] [CrossRef]
- Kawagishi, H.; Zhuang, C. Compounds for dementia from Hericium erinaceum. Drugs Future 2008, 33, 149–155. [Google Scholar] [CrossRef]
- Tang, H.Y.; Yin, X.; Zhang, C.C.; Jia, Q.; Gao, J.M. Structure Diversity, Synthesis, and Biological Activity of Cyathane Diterpenoids in Higher Fungi. Curr. Med. Chem. 2015, 22, 2375–2391. [Google Scholar] [CrossRef]
- Chiu, C.H.; Chyau, C.C.; Chen, C.C.; Lee, L.Y.; Chen, W.P.; Liu, J.L.; Lin, W.H.; Mong, M.C. Erinacine A-Enriched Hericium erinaceus Mycelium Produces Antidepressant-Like Effects through Modulating BDNF/PI3K/Akt/GSK-3β Signaling in Mice. Int. J. Mol. Sci. 2018, 19, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.F.; Chen, J.H.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; Lu, C.C.; Lee, K.C.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; et al. Protective effects of Hericium erinaceus mycelium and its isolated erinacine A against ischemia-injury-induced neuronal cell death via the inhibition of iNOS/p38 MAPK and nitrotyrosine. Int. J. Mol. Sci. 2014, 15, 15073–15089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzeng, T.T.; Chen, C.C.; Chen, C.C.; Tsay, H.J.; Lee, L.Y.; Chen, W.P.; Shen, C.C.; Shiao, Y.J. The Cyanthin Diterpenoid and Sesterterpene Constituents of Hericium erinaceus Mycelium Ameliorate Alzheimer’s Disease-Related Pathologies in APP/PS1 Transgenic Mice. Int. J. Mol. Sci. 2018, 19, 598. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, T.T.; Chen, C.C.; Lee, L.Y.; Chen, W.P.; Lu, C.K.; Shen, C.C.; Huang, F.C.Y.; Chen, C.C.; Shiao, Y.J. Erinacine A-enriched Hericium erinaceus mycelium ameliorates Alzheimer’s disease-related pathologies in APPswe/PS1dE9 transgenic mice. J. Biomed. Sci. 2016, 23, 49. [Google Scholar]
- Lee, K.F.; Tung, S.Y.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; Huang, C.Y.; Lee, K.C.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; et al. Post-Treatment with Erinacine A, a Derived Diterpenoid of H. erinaceus, Attenuates Neurotoxicity in MPTP Model of Parkinson’s Disease. Antioxidants 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef]
- White, R.B.; Thomas, M.G. Moving Beyond Tyrosine Hydroxylase to Define Dopaminergic Neurons for Use in Cell Replacement Therapies for Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2012, 11, 340–349. [Google Scholar] [CrossRef]
- Hoban, D.B.; Connaughton, E.; Connaughton, C.; Hogan, G.; Thornton, C.; Mulcahy, P.; Moloney, T.C.; Dowd, E. Further characterisation of the LPS model of Parkinson’s disease: A comparison of intra-nigral and intra-striatal lipopolysaccharide administration on motor function, microgliosis and nigrostriatal neurodegeneration in the rat. Brain Behav. Immun. 2013, 27, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Creese, I.; Burt, D.R.; Snyder, S.H. Dopamine receptor binding enhancement accompanies lesion-induced behavioral supersensitivity. Science 1977, 197, 596–598. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.J.; Xiao, L.J.; Zhong, Z.Y.; Wang, L.M.; Li, Z.; Pan, X.P.; Liu, Z.L. Astaxanthin acts via LRP-1 to inhibit inflammation and reverse lipopolysaccharide-induced M1/M2 polarization of microglial cells. Oncotarget 2017, 8, 69370–69385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Yang, H.; Chen, Z.; Hu, X.; Wu, T.; Liu, W. Long Noncoding RNA X-Inactive-Specific Transcript Promotes the Secretion of Inflammatory Cytokines in LPS Stimulated Astrocyte Cell Via Sponging miR-29c-3p and Regulating Nuclear Factor of Activated T cell 5 Expression. Front. Endocrinol. 2021, 12, 573143. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.J.; Castano, A.; Venero, J.L.; Cano, J.; Machado, A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol. Dis. 2000, 7, 429–447. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, X.; Wu, J.; Li, J.; Zhang, L.; Xiong, T.; Tang, J.; Qu, Y.; Mu, D. Involvement of the JNK/FOXO3a/Bim Pathway in Neuronal Apoptosis after Hypoxic-Ischemic Brain Damage in Neonatal Rats. PLoS ONE 2015, 10, e0132998. [Google Scholar] [CrossRef] [Green Version]
- Son, E.W.; Mo, S.J.; Rhee, D.K.; Pyo, S. Inhibition of ICAM-1 expression by garlic component, allicin, in gamma-irradiated human vascular endothelial cells via downregulation of the JNK signaling pathway. Int. Immunopharmacol. 2006, 6, 1788–1795. [Google Scholar] [CrossRef]
- Flood, P.M.; Qian, L.; Peterson, L.J.; Zhang, F.; Shi, J.S.; Gao, H.M.; Hong, J.S. Transcriptional Factor NF-κB as a Target for Therapy in Parkinson’s Disease. Parkinsons Dis. 2011, 2011, 216298. [Google Scholar] [CrossRef] [Green Version]
- Shimbo, M.; Kawagishi, H.; Yokogoshi, H. Erinacine A increases catecholamine and nerve growth factor content in the central nervous system of rats. Nutr. Res. 2005, 25, 617–623. [Google Scholar] [CrossRef]
- Lee, L.Y.; Chou, W.; Chen, W.P.; Wang, M.F.; Chen, Y.J.; Chen, C.C.; Tung, K.C. Erinacine A-Enriched Hericium erinaceus Mycelium Delays Progression of Age-Related Cognitive Decline in Senescence Accelerated Mouse Prone 8 (SAMP8) Mice. Nutrients 2021, 13, 3659. [Google Scholar] [CrossRef]
- Jang, H.J.; Kim, J.E.; Jeong, K.H.; Lim, S.C.; Kim, S.Y.; Cho, K.O. The Neuroprotective Effect of Hericium erinaceus Extracts in Mouse Hippocampus after Pilocarpine-Induced Status Epilepticus. Int. J. Mol. Sci. 2019, 20, 859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratto, D.; Corana, F.; Mannucci, B.; Priori, E.C.; Cobelli, F.; Roda, E.; Ferrari, B.; Occhinegro, A.; Di Iorio, C.; De Luca, F.; et al. Hericium erinaceus Improves Recognition Memory and Induces Hippocampal and Cerebellar Neurogenesis in Frail Mice during Aging. Nutrients 2019, 11, 715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, I.C.; Chen, Y.L.; Lee, L.Y.; Chen, W.P.; Tsai, Y.T.; Chen, C.C.; Chen, C.S. Evaluation of the toxicological safety of erinacine A-enriched Hericium erinaceus in a 28-day oral feeding study in Sprague-Dawley rats. Food Chem. Toxicol. 2014, 70, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Lin, Y.C.; Huang, C.C.; Villaflores, O.B.; Wu, T.Y.; Huang, S.M.; Chin, T.Y. Hericium erinaceus Mycelium and Its Isolated Compound, Erinacine A, Ameliorate High-Fat High-Sucrose Diet-Induced Metabolic Dysfunction and Spatial Learning Deficits in Aging Mice. J. Med. Food 2019, 22, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Barro, L.; Tsai, S.T.; Feng, T.W.; Wu, X.Y.; Chao, C.W.; Yu, R.S.; Chin, T.Y.; Hsieh, M.F. Epigallocatechin-3-Gallate-Loaded Liposomes Favor Anti-Inflammation of Microglia Cells and Promote Neuroprotection. Int. J. Mol. Sci. 2021, 22, 3037. [Google Scholar] [CrossRef]
- Lee, S.L.; Tu, S.C.; Hsu, M.Y.; Chin, T.Y. Diosgenin Prevents Microglial Activation and Protects Dopaminergic Neurons from Lipopolysaccharide-Induced Neural Damage In Vitro and In Vivo. Int. J. Mol. Sci. 2021, 22, 10361. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence |
|---|---|
| BDNF | Forward: 5′-GGTCACAGCGGCAGATAAAAAGA-3′ Reverse: 5′-TTCGGCATTGCGAGTTCCA-3′ |
| GAPDH | Forward: 5′-GCAAGAGAGAGGCCCTCA-3′ Reverse: 5′-TGTGAGGGAGATGCTCAGTG-5′ |
| IL-1β | Forward: 5′-GAGCACCTTCTTTTCCTTCATCT-3′ Reverse: 5′-GATATTCTGTCCATTGAGGTGGA-3′ |
| iNOS | Forward: 5′-GGTATGAAATGGCAAATCGG-3′ Reverse: 5′-CGGACCATCTCCTGCATT-3′ |
| TNF-α | Forward: 5′-CTCACACTCAGATCATCTTCT-3′ Reverse: 5′-GGTATGAAATGGCAAATCGG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-L.; Hsu, J.-Y.; Chen, T.-C.; Huang, C.-C.; Wu, T.-Y.; Chin, T.-Y. Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 810. https://doi.org/10.3390/ijms23020810
Lee S-L, Hsu J-Y, Chen T-C, Huang C-C, Wu T-Y, Chin T-Y. Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo. International Journal of Molecular Sciences. 2022; 23(2):810. https://doi.org/10.3390/ijms23020810
Chicago/Turabian StyleLee, Shou-Lun, Jing-Ya Hsu, Ting-Chun Chen, Chun-Chih Huang, Tzong-Yuan Wu, and Ting-Yu Chin. 2022. "Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo" International Journal of Molecular Sciences 23, no. 2: 810. https://doi.org/10.3390/ijms23020810
APA StyleLee, S.-L., Hsu, J.-Y., Chen, T.-C., Huang, C.-C., Wu, T.-Y., & Chin, T.-Y. (2022). Erinacine A Prevents Lipopolysaccharide-Mediated Glial Cell Activation to Protect Dopaminergic Neurons against Inflammatory Factor-Induced Cell Death In Vitro and In Vivo. International Journal of Molecular Sciences, 23(2), 810. https://doi.org/10.3390/ijms23020810