
PAR4-Mediated PI3K/Akt and RhoA/ROCK Signaling Pathways Are Essential for Thrombin-Induced Morphological Changes in MEG-01 Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
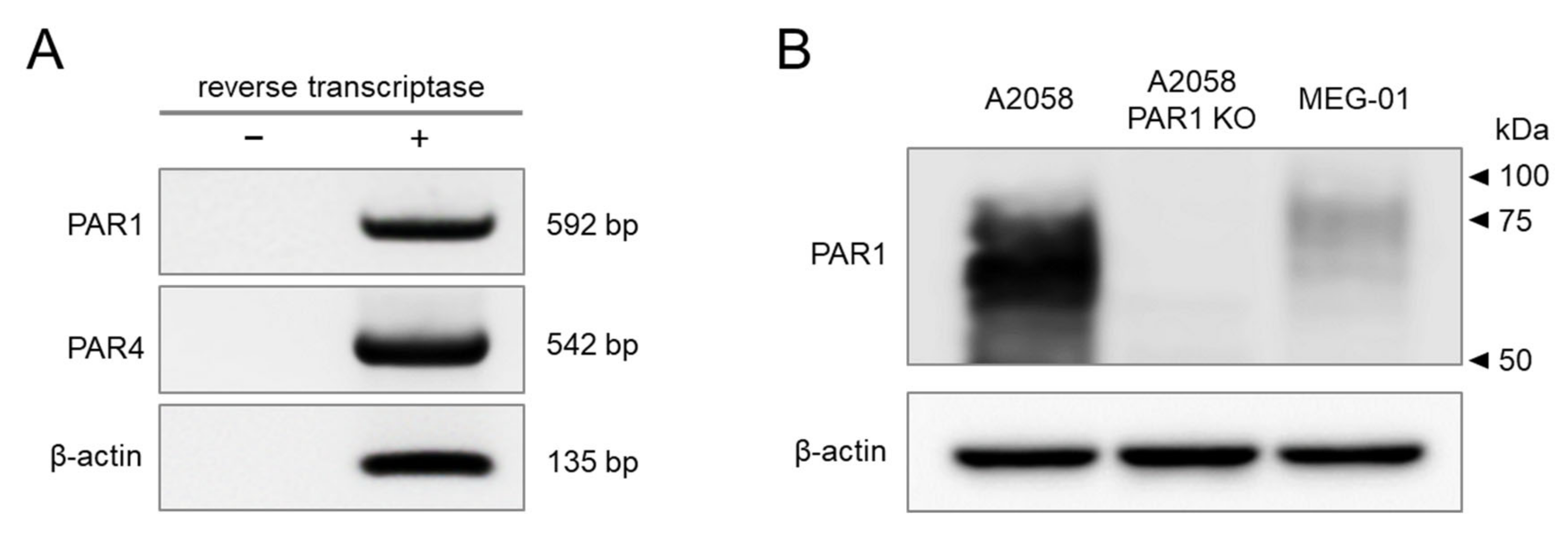
2.1. PAR1 and PAR4 Are Endogenously Expressed in MEG-01 Cells
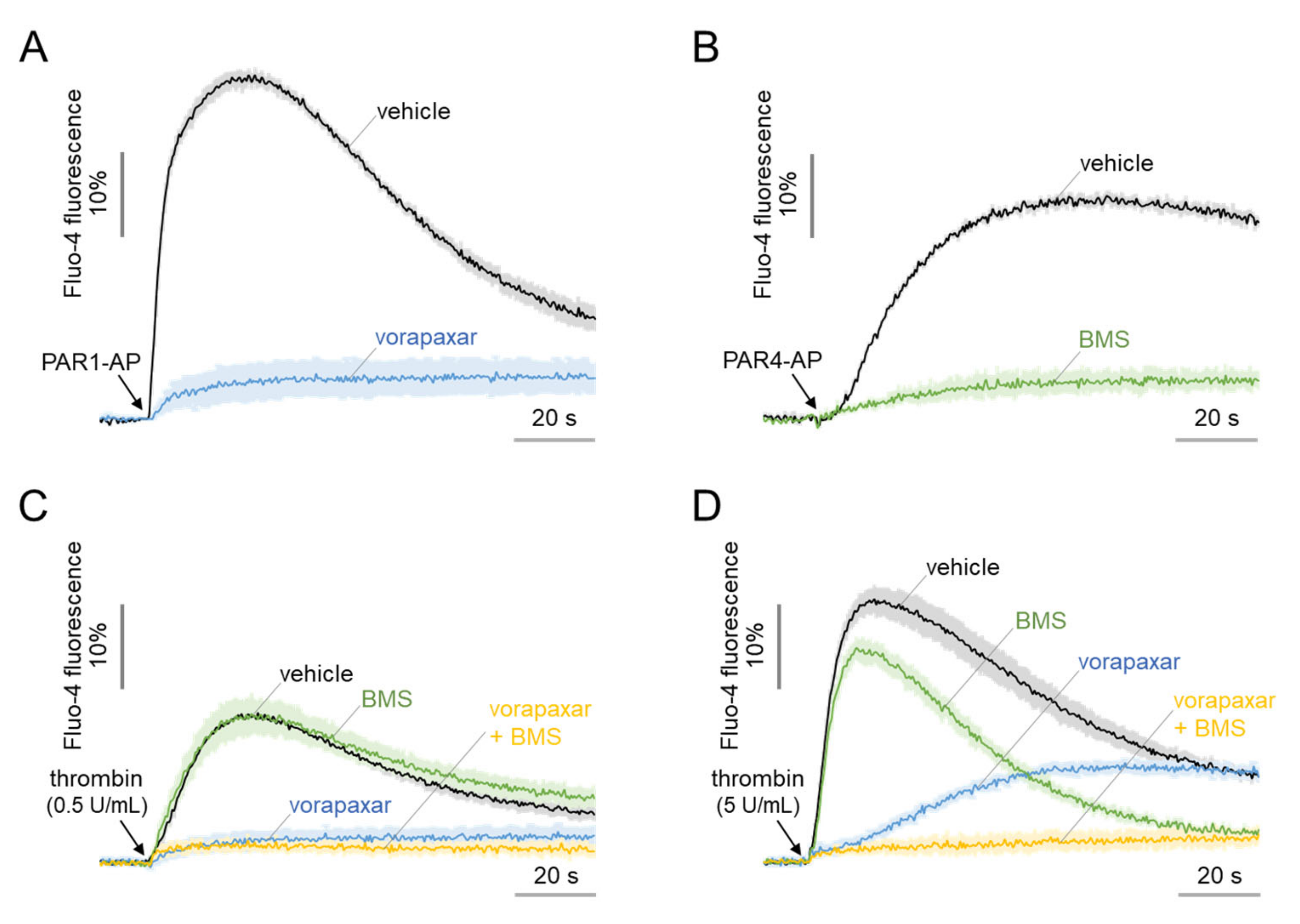
2.2. Functional Expression of PAR1 and PAR4 in MEG-01 Cells
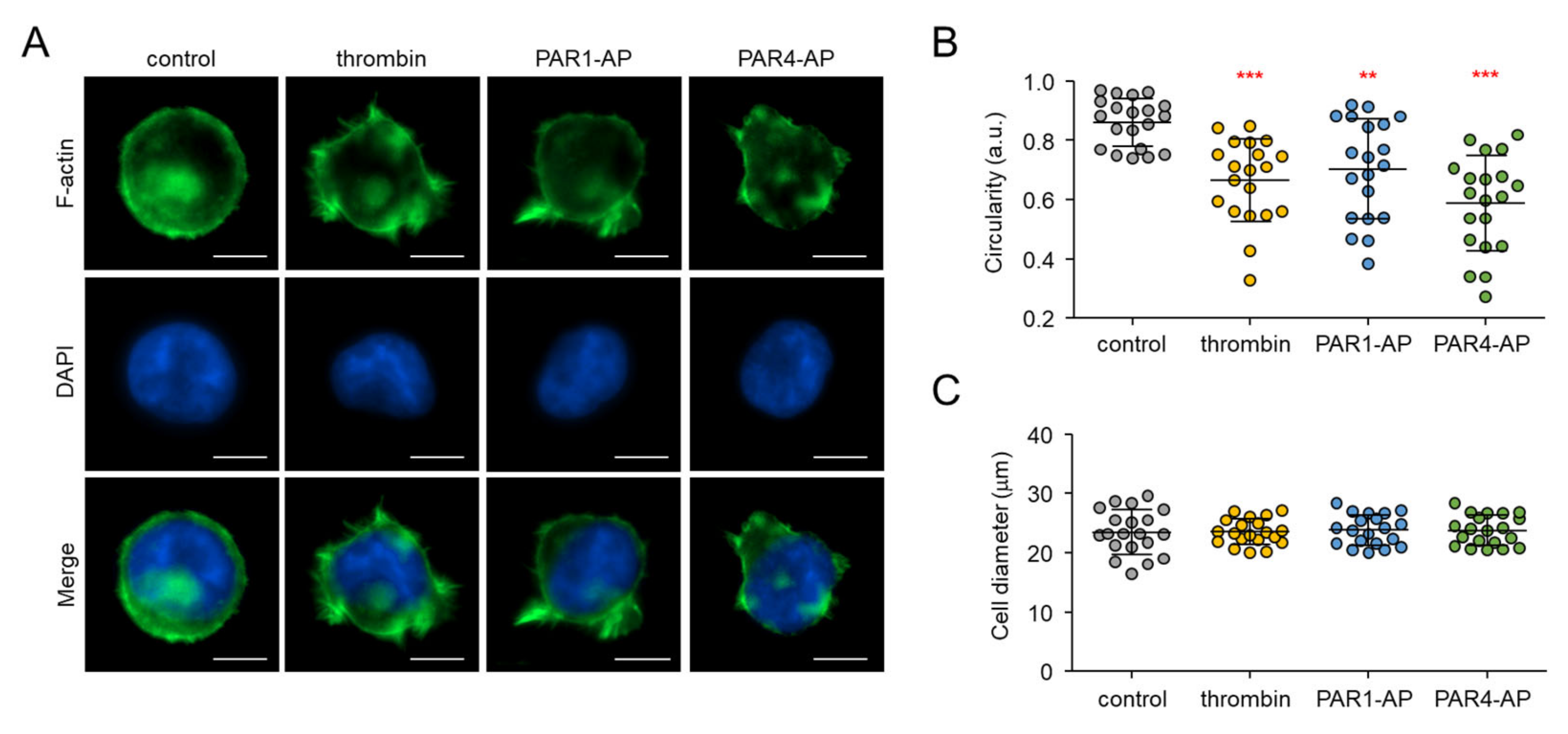
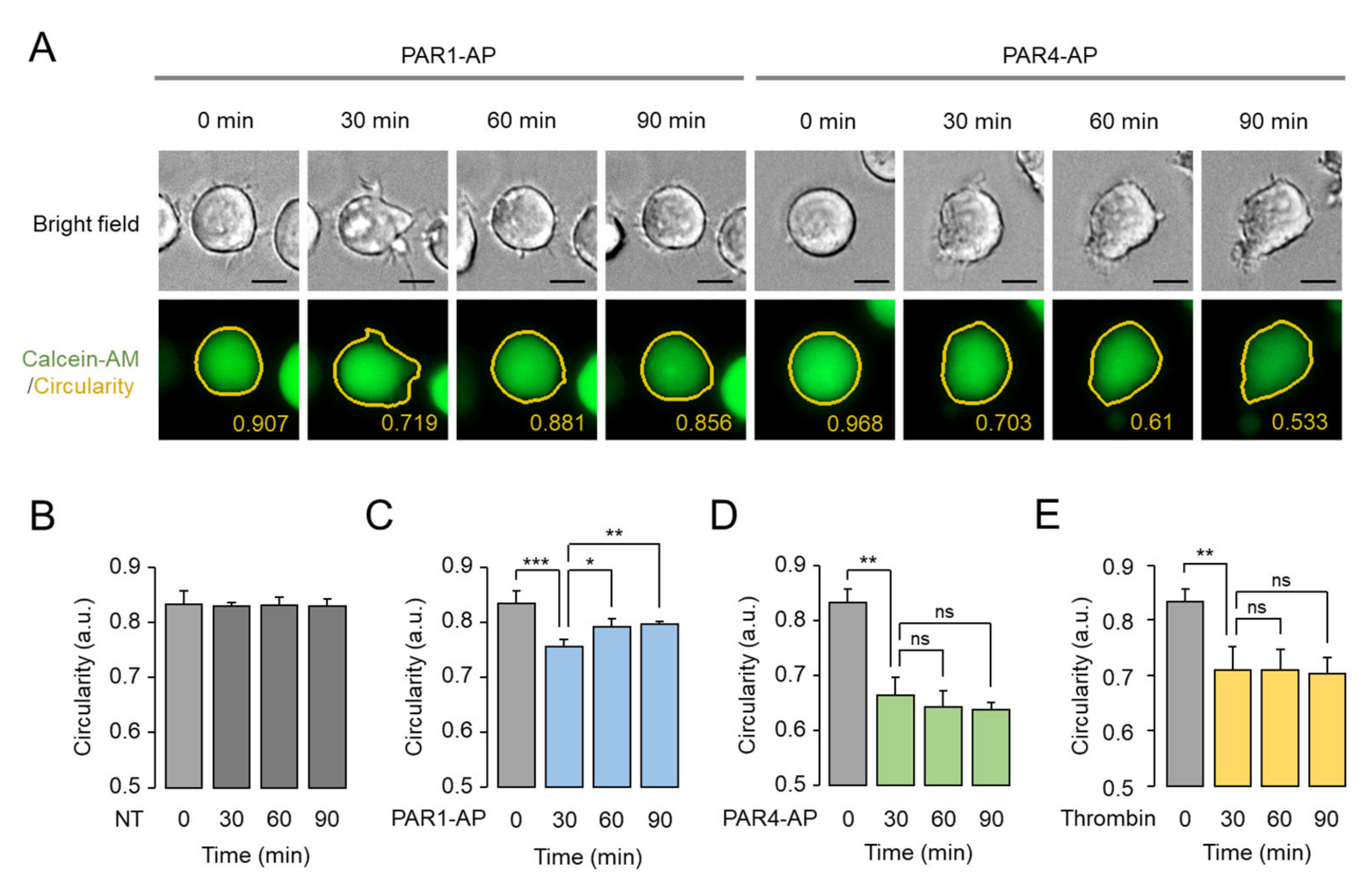
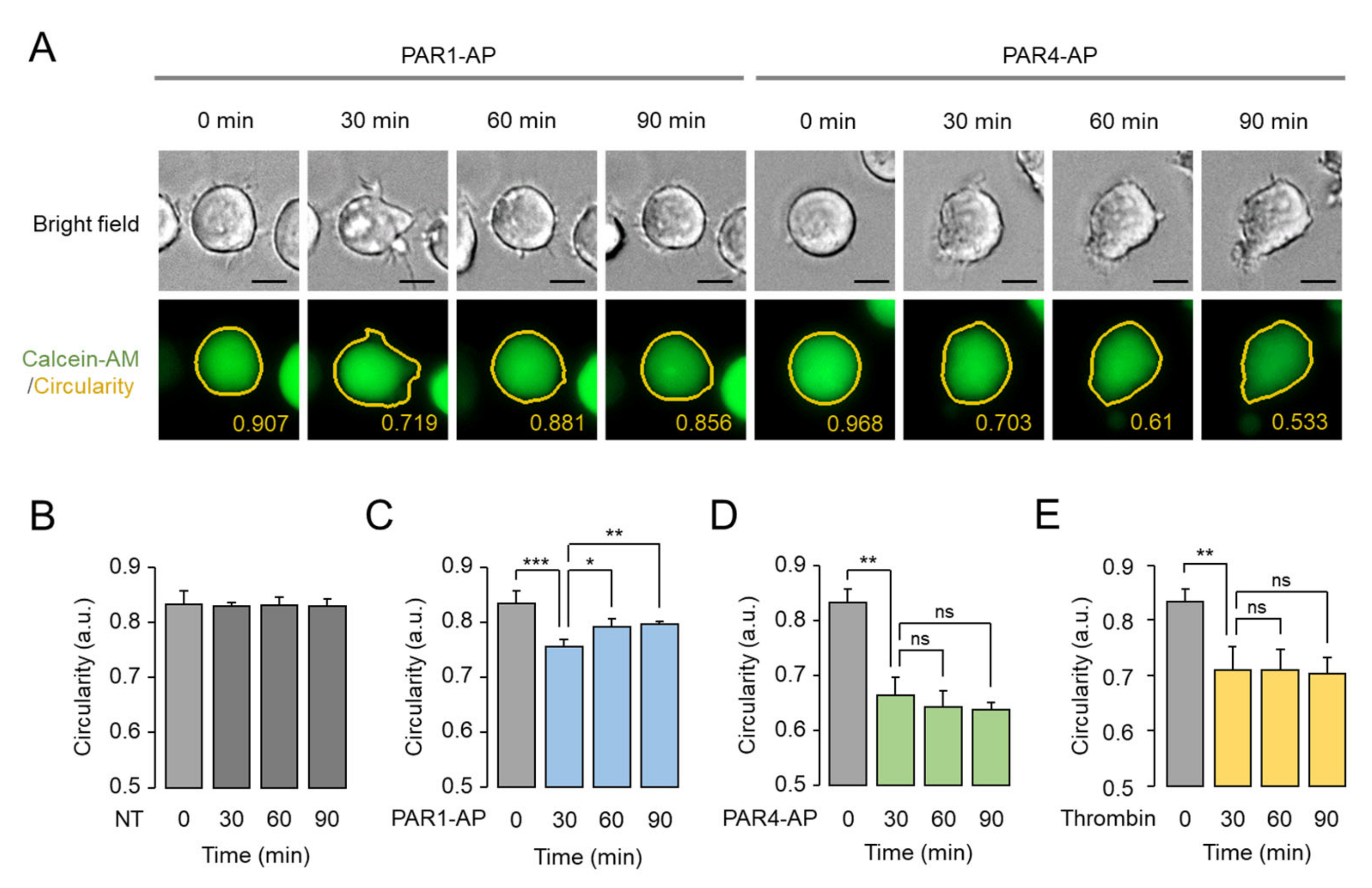
2.3. PAR1- and PAR4-Mediated Morphological Changes in MEG-01 Cells
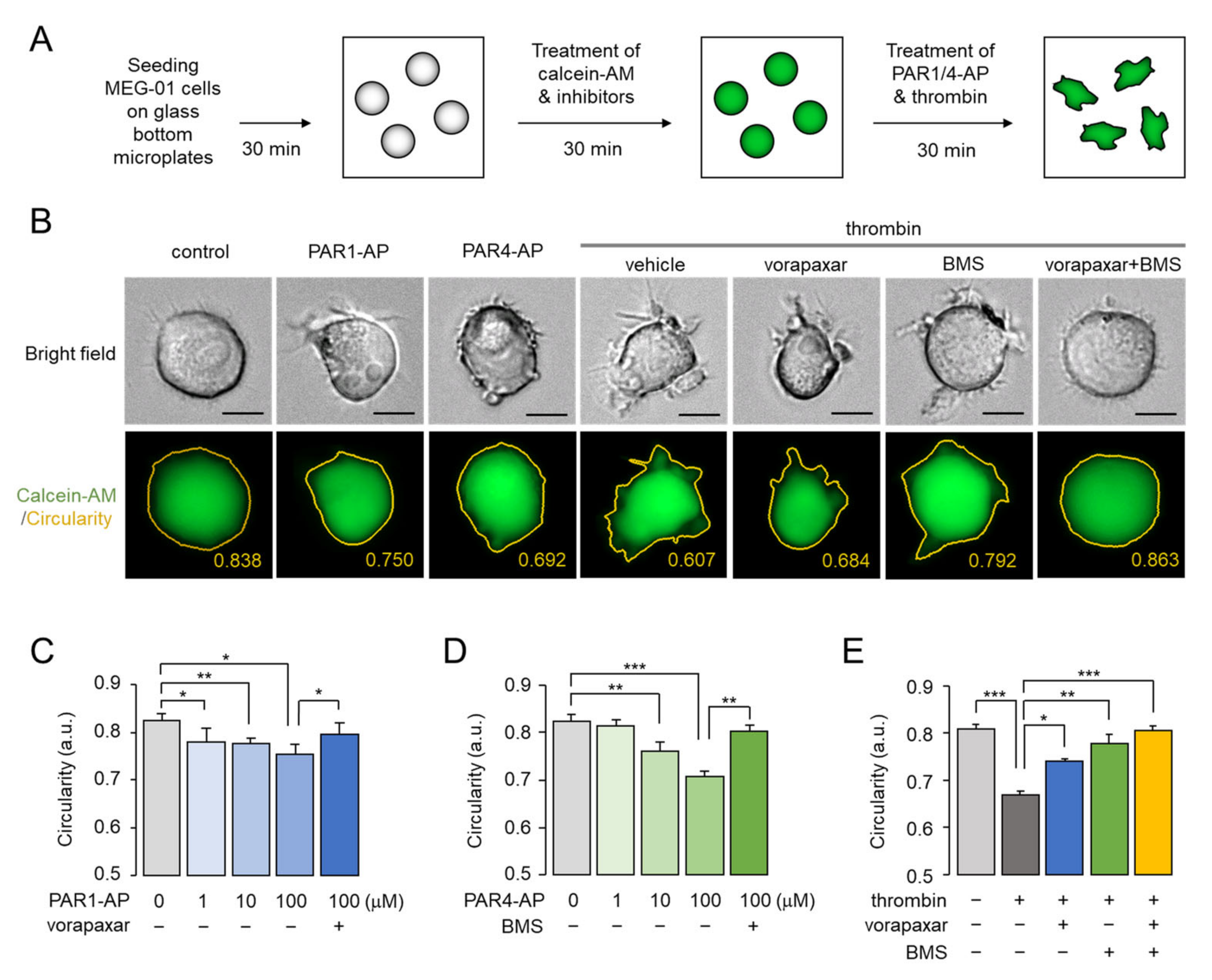
2.4. Assessment of PAR1- and PAR4-Mediated Morphological Changes in Living MEG-01 Cells
2.5. Prolonged Effect of PAR4-Mediated Morphological Changes in MEG-01 Cells
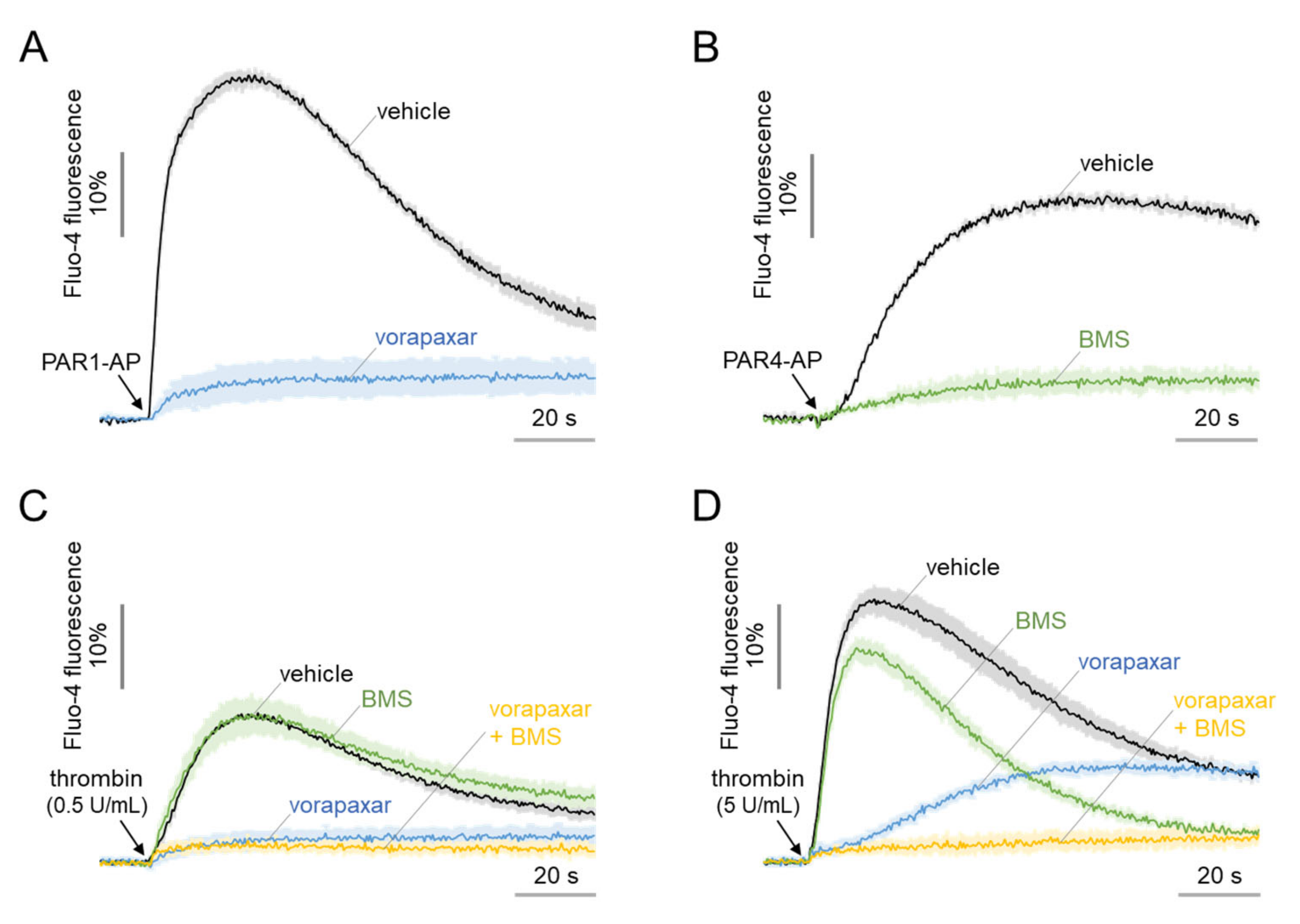
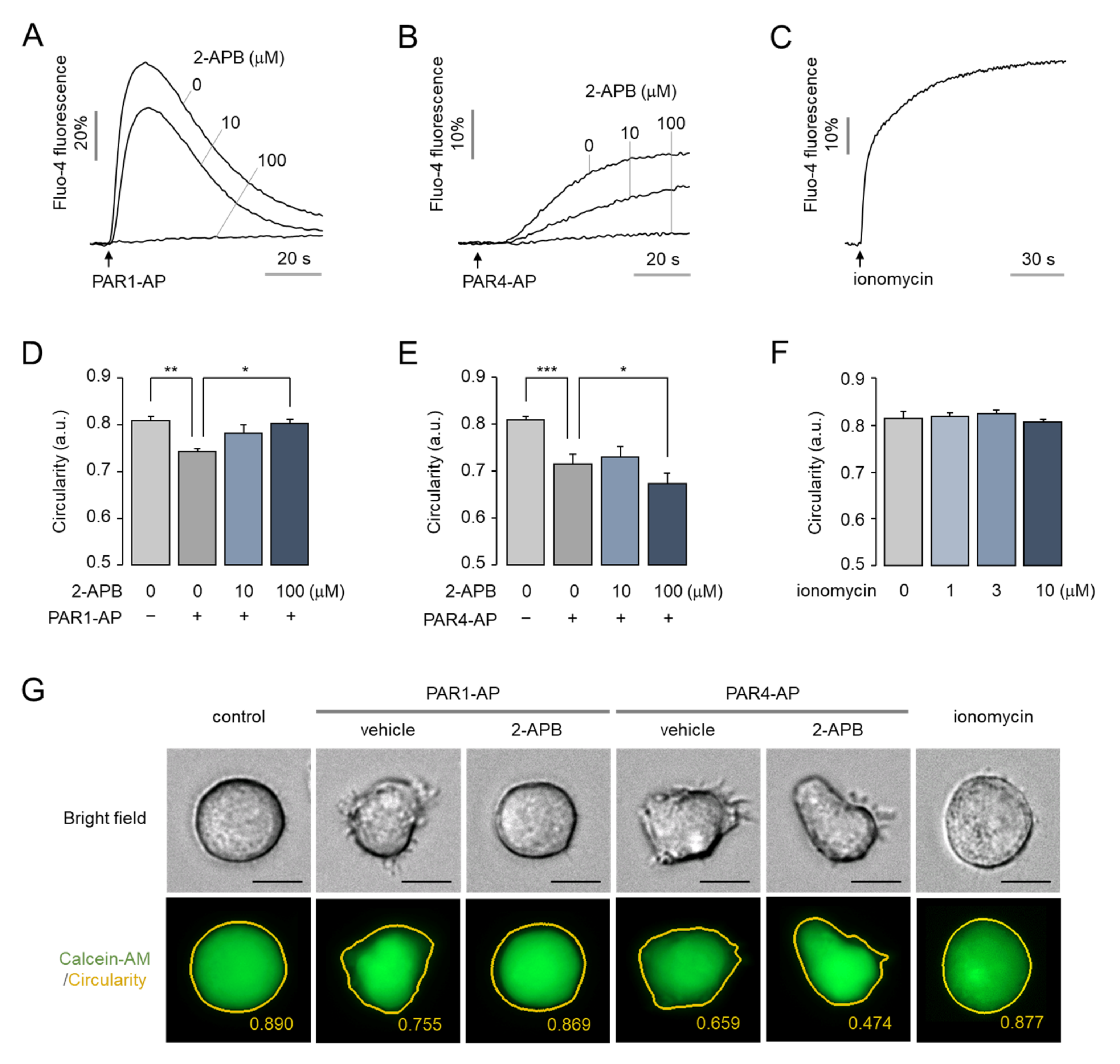
2.6. The Morphological Changes of MEG-01 Cells by PAR4 Activation Are Calcium-Independent and Strong, but PAR1 Activation Is Not
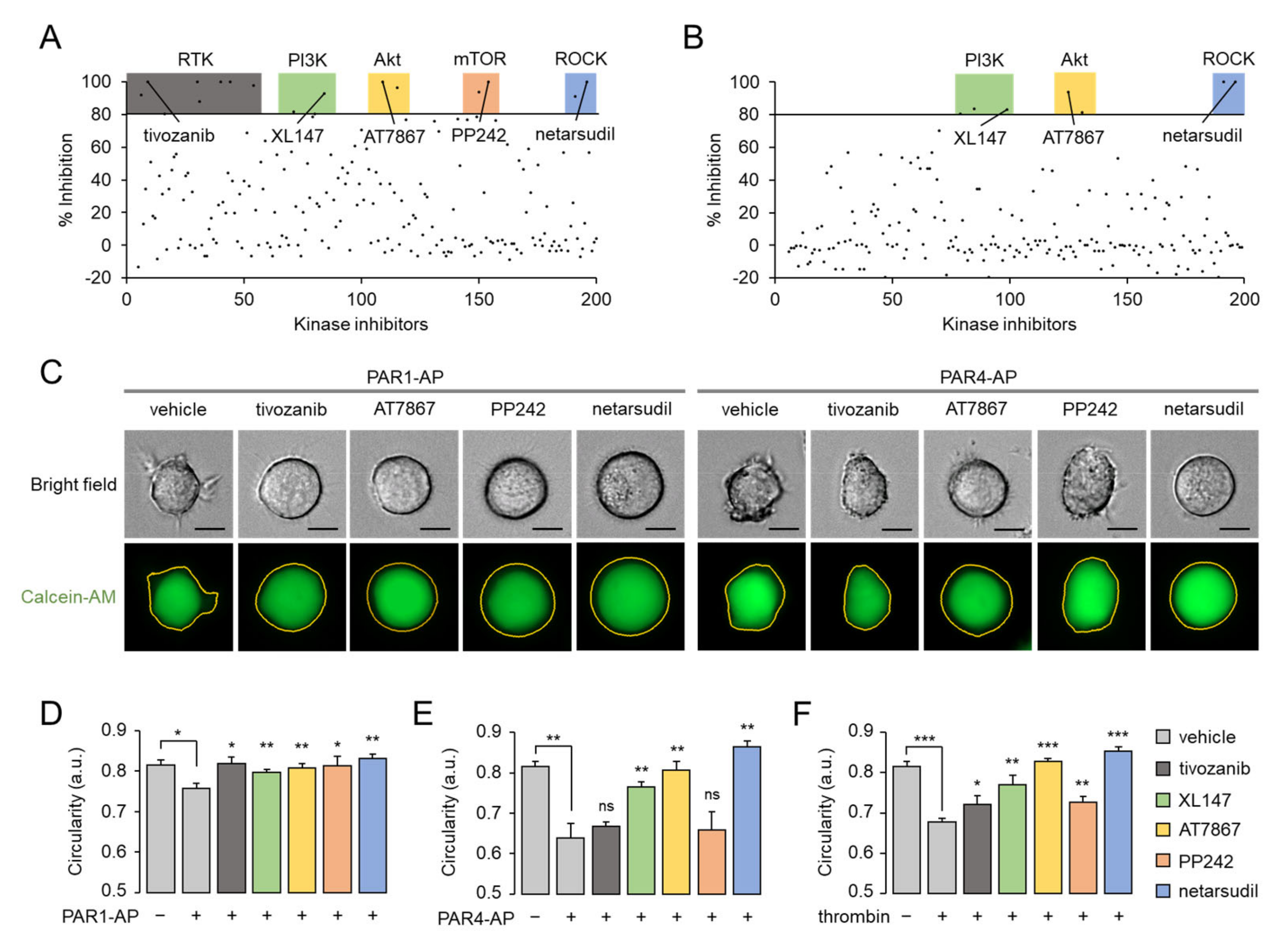
2.7. Thrombin-Induced Morphological Change Is Primarily Mediated through PI3K-Akt and Rho-ROCK Pathway via PAR4 Activation
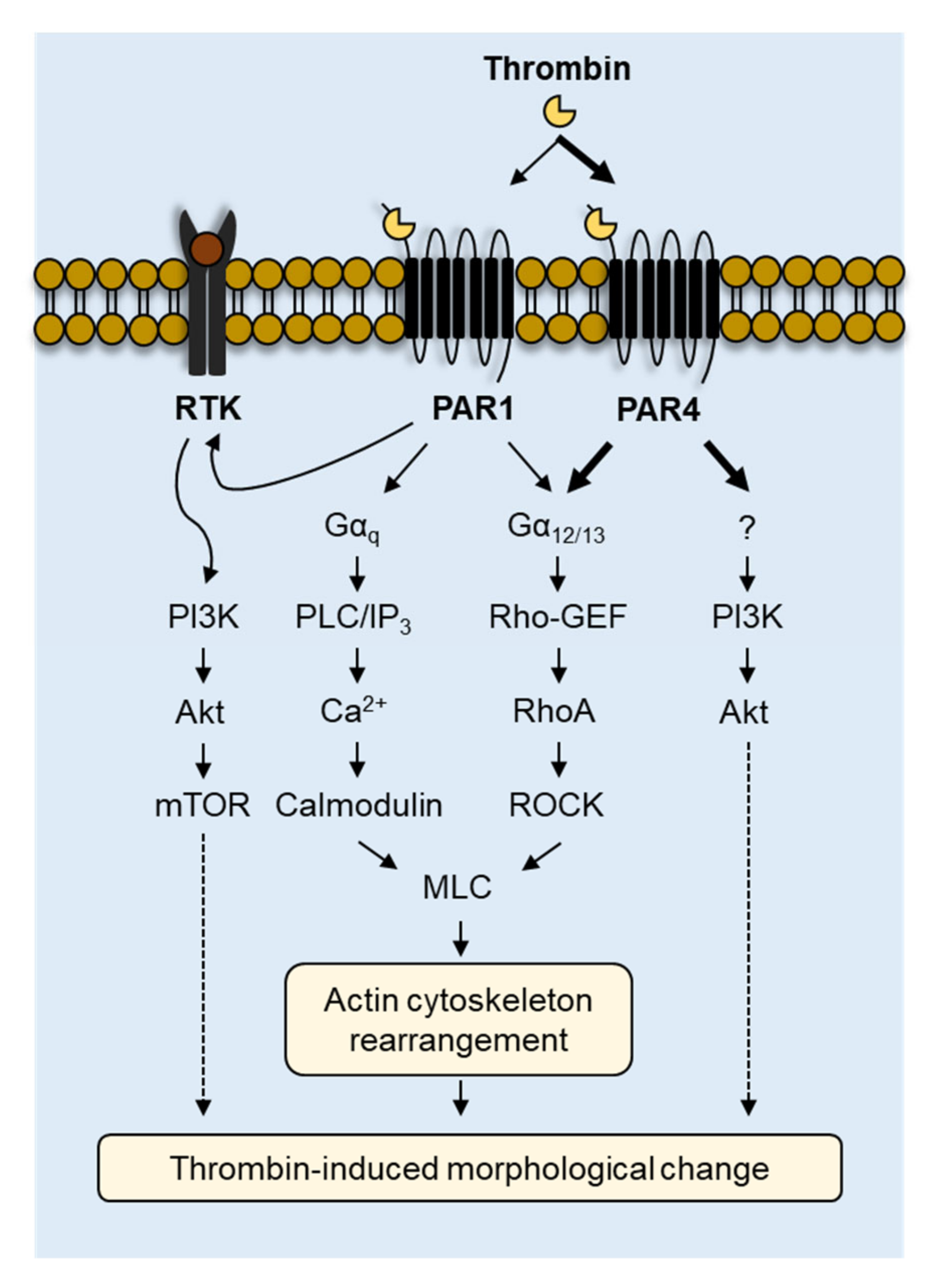
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cell Lines
4.2. Materials and Reagents
4.3. Immunoblotting
4.4. Reverse Transcription Polymerase Chain Reaction
4.5. Intracellular Calcium Measurement
4.6. Immunofluorescence Staining
4.7. Calcein-AM-Based Assessment of Morphological Change
4.8. Data and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | Activating peptide |
| GPCR | G protein-coupled receptor |
| MLC | Myosin light chain |
| PAR | Protease-activated receptor |
| PI3K | Phosphoinositide 3-kinase |
| ROCK | Rho-associated protein kinase |
| RTK | Receptor tyrosine kinase |
| HCS | High-content screening |
References
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet Signaling. In Antiplatelet Agents: Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 59–85. [Google Scholar] [CrossRef] [Green Version]
- Offermanns, S. Activation of Platelet Function Through G Protein–Coupled Receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Investig. 1999, 103, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeley, S.; Covic, L.; Jacques, S.L.; Sudmeier, J.; Baleja, J.D.; Kuliopulos, A. Structural Basis for Thrombin Activation of a Protease-Activated Receptor: Inhibition of Intramolecular Liganding. Chem. Biol. 2003, 10, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling During Platelet Adhesion and Activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Mao, Y.; Thomas, D.; Kim, S.; Daniel, J.L.; Kunapuli, S.P. RhoA downstream of Gq and G12/13 pathways regulates protease-activated receptor-mediated dense granule release in platelets. Biochem. Pharmacol. 2009, 77, 835–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, M.T.; Schmaier, A.H. Interaction of Thrombin with PAR1 and PAR4 at the Thrombin Cleavage Site. Biochemistry 2007, 46, 8603–8610. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.-K.H.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Duvernay, M.T.; Temple, K.J.; Maeng, J.G.; Blobaum, A.L.; Stauffer, S.R.; Lindsley, C.W.; Hamm, H.E. Contributions of Protease-Activated Receptors PAR1 and PAR4 to Thrombin-Induced GPIIbIIIa Activation in Human Platelets. Mol. Pharmacol. 2017, 91, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Boknäs, N.; Faxälv, L.; Centellas, D.S.; Wallstedt, M.; Ramström, S.; Grenegård, M.; Lindahl, T.L. Thrombin-induced platelet activation via PAR4: Pivotal role for exosite II. Thromb. Haemost. 2014, 112, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, M.J.; Weiss, E.J.; Faruqi, T.R.; Coughlin, S.R. Protease-activated Receptors 1 and 4 Are Shut Off with Distinct Kinetics after Activation by Thrombin. J. Biol. Chem. 2000, 275, 25216–25221. [Google Scholar] [CrossRef] [Green Version]
- Holinstat, M.; Voss, B.; Bilodeau, M.L.; McLaughlin, J.N.; Cleator, J.; Hamm, H.E. PAR4, but Not PAR1, Signals Human Platelet Aggregation via Ca2+ Mobilization and Synergistic P2Y12 Receptor Activation. J. Biol. Chem. 2006, 281, 26665–26674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-F.; Lee, J.-J.; Chang, C.-C.; Lin, K.-H.; Wang, S.-H.; Sheu, J.-R. Platelet protease-activated receptor (PAR)4, but not PAR1, associated with neutral sphingomyelinase responsible for thrombin-stimulated ceramide-NF-κB signaling in human platelets. Haematology 2013, 98, 793–801. [Google Scholar] [CrossRef]
- Duvernay, M.; Young, S.; Gailani, D.; Schoenecker, J.; Hamm, H.E. Protease-Activated Receptor (PAR) 1 and PAR4 Differentially Regulate Factor V Expression from Human Platelets. Mol. Pharmacol. 2013, 83, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, S.L.; Hamilton, J.R. Protease-activated receptor 4: From structure to function and back again. J. Cereb. Blood Flow Metab. 2016, 173, 2952–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudinga, G.R.; Khan, G.J.; Kong, Y. Protease-Activated Receptor 4 (PAR4): A Promising Target for Antiplatelet Therapy. Int. J. Mol. Sci. 2018, 19, 573. [Google Scholar] [CrossRef] [Green Version]
- Moskalensky, A.E.; Litvinenko, A.L. The platelet shape change: Biophysical basis and physiological consequences. Platelets 2019, 30, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.Z.S.; Daniel, J.L.; Kunapuli, S.P. Platelet Shape Change Is Mediated by both Calcium-dependent and -independent Signaling Pathways. J. Biol. Chem. 1999, 274, 28293–28300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwig, J.H. Mechanisms of actin rearrangements mediating platelet activation. J. Cell Biol. 1992, 118, 1421–1442. [Google Scholar] [CrossRef] [Green Version]
- Moers, A.; Nieswandt, B.; Massberg, S.; Wettschureck, N.; Grüner, S.; Konrad, I.; Schulte, V.; Aktas, B.; Gratacap, M.-P.; Simon, M., I; et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 2003, 9, 1418–1422. [Google Scholar] [CrossRef] [Green Version]
- Aslan, J.E.; Mccarty, O.J.T. Rho GTPases in platelet function. J. Thromb. Haemost. 2013, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Covic, L.; Gresser, A.L.; Kuliopulos, A. Biphasic Kinetics of Activation and Signaling for PAR1 and PAR4 Thrombin Receptors in Platelets. Biochemistry 2000, 39, 5458–5467. [Google Scholar] [CrossRef]
- Mathur, G.; Mott, S.L.; Collins, L.; Nelson, G.A.; Knudson, C.M.; Schlueter, A.J. Factors influencing platelet clumping during peripheral blood hematopoietic stem cell collection. Transfusion 2017, 57, 1142–1151. [Google Scholar] [CrossRef] [Green Version]
- Sim, X.; Poncz, M.; Gadue, P.; French, D.L. Understanding platelet generation from megakaryocytes: Implications for in vitro–derived platelets. Blood 2016, 127, 1227–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paniccia, R.; Priora, R.; Liotta, A.A.; Abbate, R. Platelet function tests: A comparative review. Vasc. Health Risk Manag. 2015, 11, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, M.; Morishima, Y.; Ohno, R.; Kato, Y.; Hirabayashi, N.; Nagura, H.; Saito, H. Establishment of a novel human megakaryoblastic leukemia cell line, MEG-01, with positive Philadelphia chromosome. Blood 1985, 66, 1384–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogura, M.; Morishima, Y.; Okumura, M.; Hotta, T.; Takamoto, S.; Ohno, R.; Hirabayashi, N.; Nagura, H.; Saito, H. Functional and morphological differentiation induction of a human megakaryoblastic leukemia cell line (MEG-01s) by phorbol diesters. Blood 1988, 72, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Satoh, M.; Kuno, H.; Yoshida, T.; Kondo, H.; Takeuchi, M. Platelet-like particle formation in the human megakaryoblastic leukaemia cell lines, MEG-01 and MEG-01s. Br. J. Haematol. 1998, 100, 436–444. [Google Scholar] [CrossRef]
- Freixer, G.; Zekri-Nechar, K.; Zamorano-León, J.J.; Hugo-Martínez, C.; Butta, N.V.; Monzón, E.; Recio, M.-J.; Giner, M.; López-Farré, A. Pro-apoptotic properties and mitochondrial functionality in platelet-like-particles generated from low Aspirin-incubated Meg-01 cells. Platelets 2021, 32, 1063–1072. [Google Scholar] [CrossRef]
- Isakari, Y.; Sogo, S.; Ishida, T.; Kawakami, T.; Ono, T.; Taki, T.; Kiwada, H. Gene Expression Analysis during Platelet-Like Particle Production in Phorbol Myristate Acetate-Treated MEG-01 Cells. Biol. Pharm. Bull. 2009, 32, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Bian, W.; Chen, W.; Jiang, X.; Qu, H.; Jiang, J.; Yang, J.; Liang, X.; Zhao, B.; Sun, Y.; Zhang, C. Downregulation of Long Non-coding RNA Nuclear Paraspeckle Assembly Transcript 1 Inhibits MEG-01 Differentiation and Platelet-Like Particles Activity. Front. Genet. 2020, 11, 571467. [Google Scholar] [CrossRef]
- Banerjee, A.; Tripathi, A.; Duggal, S.; Banerjee, A.; Vrati, S. Dengue virus infection impedes megakaryopoiesis in MEG-01 cells where the virus envelope protein interacts with the transcription factor TAL-1. Sci. Rep. 2020, 10, 19587. [Google Scholar] [CrossRef] [PubMed]
- Dhenge, A.; Kuhikar, R.; Kale, V.; Limaye, L. Regulation of differentiation of MEG01 to megakaryocytes and platelet-like particles by Valproic acid through Notch3 mediated actin polymerization. Platelets 2019, 30, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Hayakawa, M.; Kamoshita, N.; Yasumoto, A.; Suzuki-Inoue, K.; Yatomi, Y.; Ohmori, T. Establishment of a megakaryoblastic cell line for conventional assessment of platelet calcium signaling. Int. J. Hematol. 2020, 111, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.C.; Seiffert, D.; Bird, J.E.; Watson, C.A.; Bostwick, J.S.; Giancarli, M.; Allegretto, N.; Hua, J.; Harden, D.; Guay, J.; et al. Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci. Transl. Med. 2017, 9, eaaf5294. [Google Scholar] [CrossRef]
- Bender, M.; Palankar, R. Platelet Shape Changes during Thrombus Formation: Role of Actin-Based Protrusions. Hämostaseologie 2021, 41, 014–021. [Google Scholar] [CrossRef]
- Bauer, M.; Retzer, M.; Wilde, J.I.; Maschberger, P.; Essler, M.; Aepfelbacher, M.; Watson, S.P.; Siess, W. Dichotomous regulation of myosin phosphorylation and shape change by Rho-kinase and calcium in intact human platelets. Blood 1999, 94, 1665–1672. [Google Scholar] [CrossRef]
- Xu, W.-F.; Andersen, H.; Whitmore, T.E.; Presnell, S.R.; Yee, D.P.; Ching, A.; Gilbert, T.; Davie, E.W.; Foster, D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. USA 1998, 95, 6642–6646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodie, S.L.; Ford, I.; Greaves, M.; Nixon, G. Thrombin-Induced Activation of RhoA in Platelet Shape Change. Biochem. Biophys. Res. Commun. 2001, 287, 71–76. [Google Scholar] [CrossRef]
- Jensen, B.O.; Selheim, F.; Døskeland, S.O.; Gear, A.R.L.; Holmsen, H. Protein kinase A mediates inhibition of the thrombin-induced platelet shape change by nitric oxide. Blood 2004, 104, 2775–2782. [Google Scholar] [CrossRef]
- Parrales, A.; López, E.; López-Colomé, A. Thrombin activation of PI3K/PDK1/Akt signaling promotes cyclin D1 upregulation and RPE cell proliferation. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.; Li, Q.; Shen, J.; Ren, L.; Liu, X.; Wang, Q.; He, S.; Wu, Q.; Hu, H.; Mao, X.; et al. Modulation of Platelet Activation and Thrombus Formation Using a Pan-PI3K Inhibitor S14161. PLoS ONE 2014, 9, e102394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, 8. [Google Scholar] [CrossRef]
- Cramer, E.M.; Masse, J.M.; Caen, J.P.; Garcia, I.; Breton-Gorius, J.; Debili, N.; Vainchenker, W. Effect of thrombin on maturing human megakaryocytes. Am. J. Pathol. 1993, 143, 1498. [Google Scholar]
- Pleines, I.; Hagedorn, I.; Gupta, S.; May, F.; Chakarova, L.; van Hengel, J.; Offermanns, S.; Krohne, G.; Kleinschnitz, C.; Brakebusch, C.; et al. Megakaryocyte-specific RhoA deficiency causes macrothrombocytopenia and defective platelet activation in hemostasis and thrombosis. Blood 2012, 119, 1054–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.J.; Ismat, F.A.; Wang, Z.; Cerra, M.; Narayan, H.; Raftis, J.; Gray, T.J.; Connell, S.; Garonzik, S.; Ma, X.; et al. PAR4 (Protease-Activated Receptor 4) Antagonism With BMS-986120 Inhibits Human Ex Vivo Thrombus Formation. Arter. Thromb. Vasc. Biol. 2018, 38, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.-K.; Park, H.; Noh, J.-Y.; Lim, K.-M.; Chung, J.-H. Platelet Shape Changes and Cytoskeleton Dynamics as Novel Therapeutic Targets for Anti-Thrombotic Drugs. Biomol. Ther. 2017, 25, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Botía, P.; Acebes-Huerta, A.; Seghatchian, J.; Gutiérrez, L. On the Quest for In Vitro Platelet Production by Re-Tailoring the Concepts of Megakaryocyte Differentiation. Medicina 2020, 56, 671. [Google Scholar] [CrossRef] [PubMed]
- Mountford, J.K.; Petitjean, C.; Putra, H.K.; McCafferty, J.A.; Setiabakti, N.M.; Lee, H.; Tønnesen, L.L.; McFadyen, J.D.; Schoenwaelder, S.; Eckly, A.; et al. The class II PI 3-kinase, PI3KC2α, links platelet internal membrane structure to shear-dependent adhesive function. Nat. Commun. 2015, 6, 6535. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, Y.; Jeon, H.; Namkung, W. PAR4-Mediated PI3K/Akt and RhoA/ROCK Signaling Pathways Are Essential for Thrombin-Induced Morphological Changes in MEG-01 Cells. Int. J. Mol. Sci. 2022, 23, 776. https://doi.org/10.3390/ijms23020776
Heo Y, Jeon H, Namkung W. PAR4-Mediated PI3K/Akt and RhoA/ROCK Signaling Pathways Are Essential for Thrombin-Induced Morphological Changes in MEG-01 Cells. International Journal of Molecular Sciences. 2022; 23(2):776. https://doi.org/10.3390/ijms23020776
Chicago/Turabian StyleHeo, Yunkyung, Hyejin Jeon, and Wan Namkung. 2022. "PAR4-Mediated PI3K/Akt and RhoA/ROCK Signaling Pathways Are Essential for Thrombin-Induced Morphological Changes in MEG-01 Cells" International Journal of Molecular Sciences 23, no. 2: 776. https://doi.org/10.3390/ijms23020776
APA StyleHeo, Y., Jeon, H., & Namkung, W. (2022). PAR4-Mediated PI3K/Akt and RhoA/ROCK Signaling Pathways Are Essential for Thrombin-Induced Morphological Changes in MEG-01 Cells. International Journal of Molecular Sciences, 23(2), 776. https://doi.org/10.3390/ijms23020776