
Computer Modeling Explains the Structural Reasons for the Difference in Reactivity of Amine Transaminases Regarding Prochiral Methylketones

 , ,
, ,  ,
, 
 and
and 
Abstract
:
1. Introduction
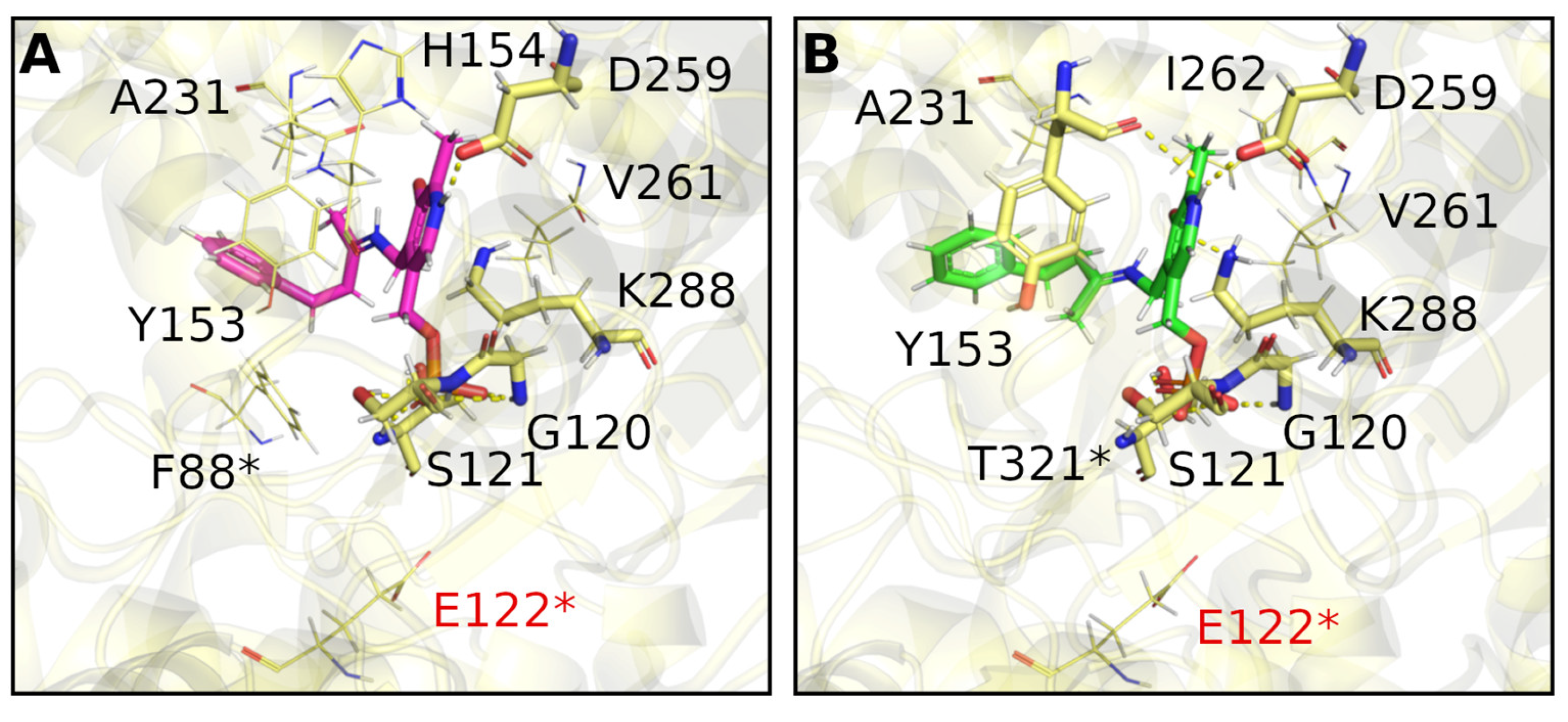
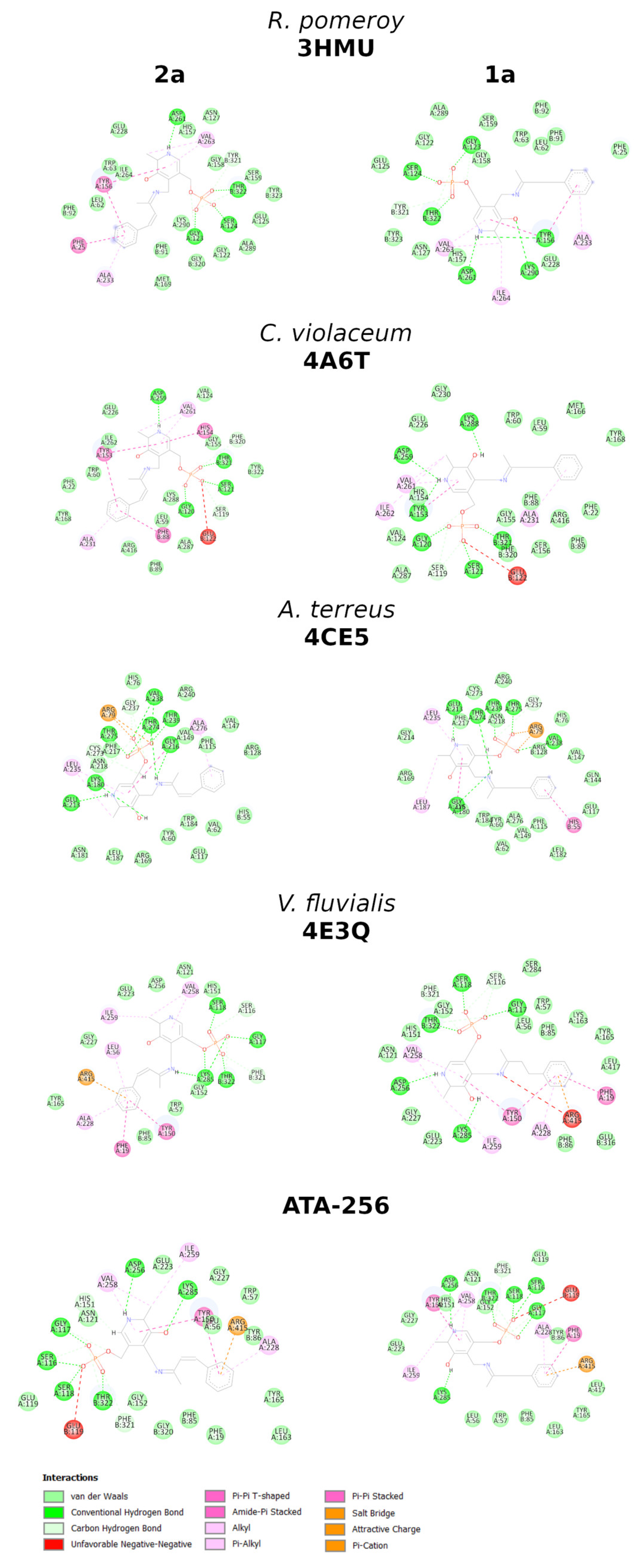
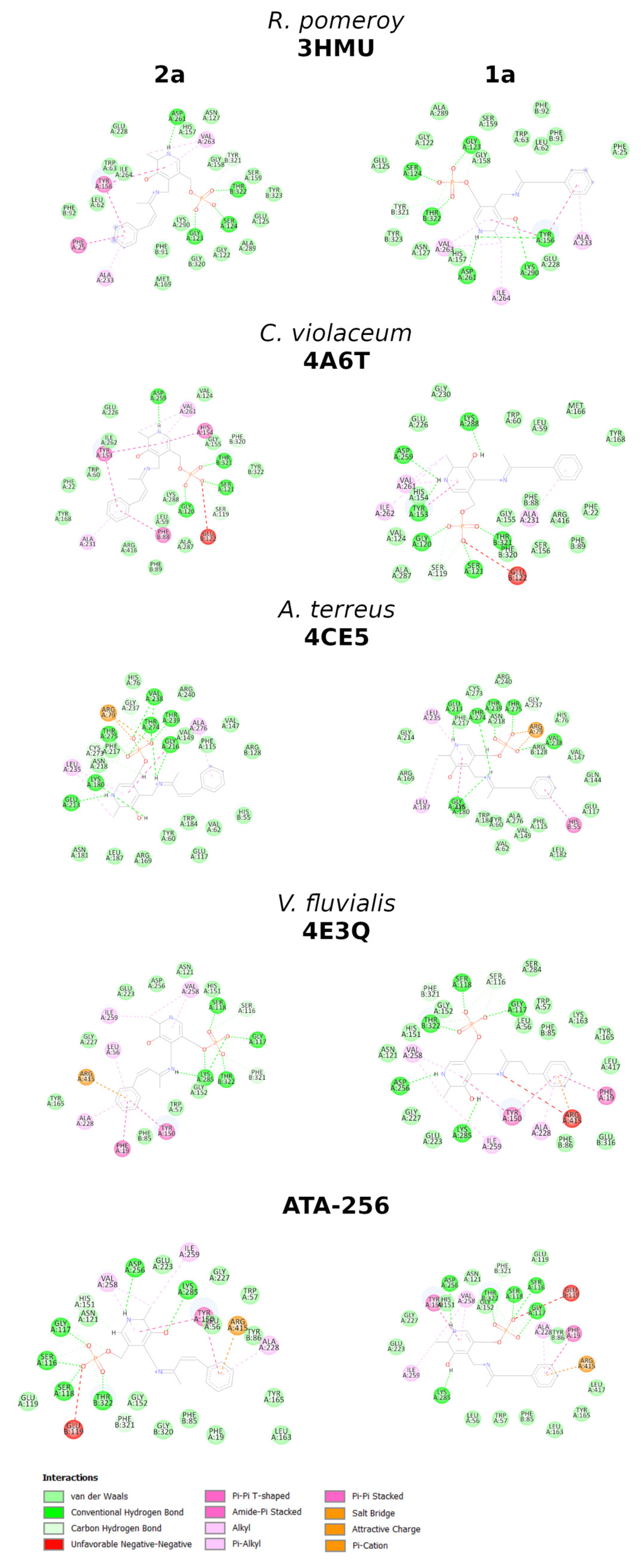
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Analytical Methods
3.3. Expression and Production of Cell-Free Extracts Containing ATAs
3.4. Bradford Assay
3.5. General Procedure for Reactions with the Crude Cell Lysate
3.6. General Procedure for Reactions with ATA-256
3.7. Reductive Amination for the Synthesis of the Racemic Amines
3.8. General Procedure for the Acetylation of Amines
3.9. General Procedure for in Silico Studies
3.10. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghislieri, D.; Turner, N.T. Biocatalytic approaches to the synthesis of enantiomerically pure chiral amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Liu, T.-L.; Wang, C.-J.; Zhang, X. Synthesis of chiral aliphatic amines through asymmetric hydrogenation. Angew. Chem. Int. Ed. 2013, 52, 8416–8419. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.S.; Park, E.S.; Shin, J.S. Features and technical applications of ω-transaminases. Appl. Microbiol. Biotechnol. 2012, 94, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.W.H.; de Souza, R.O.M.A.; Bornscheuer, U.T. Asymmetric synthesis of chiral halogenated amines using amine transaminases. ChemCatChem 2018, 10, 951–955. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Pereira, P.C. Biocatalysis engineering: The big picture. Chem. Soc. Rev. 2017, 46, 2678–2691. [Google Scholar] [CrossRef]
- Pellis, A.; Cantone, S.; Ebert, C.; Gardossi, L. Evolving biocatalysis to meet bioeconomy challenges and opportunities. New Biotechnol. 2018, 40, 154–169. [Google Scholar] [CrossRef]
- Turner, N.J.; O’Reilly, E. Biocatalytic retrosynthesis. Nat. Chem. Biol. 2013, 9, 285–288. [Google Scholar] [CrossRef]
- Cutlan, R.; De Rose, S.; Isupov, M.N.; Littlechild, J.A.; Harmer, N.J. Using enzyme cascades in biocatalysis: Highlight on transaminases and carboxylic acid reductases. Biochim. Biophys. Acta—Proteins Proteom. 2020, 1868, 140322. [Google Scholar] [CrossRef]
- Steffen-Munsberg, F.; Vickers, C.; Kohls, H.; Land, H.; Mallin, H.; Nobili, A.; Skalden, L.; van den Bergh, T.; Joosten, H.J.; Berglund, P.; et al. Bioinformatic analysis of a PLP-dependent enzyme superfamily suitable for biocatalytic applications. Biotechnol. Adv. 2015, 33, 566–604. [Google Scholar] [CrossRef]
- Fuchs, M.; Farnberger, J.E.; Kroutil, W. The industrial age of biocatalytic transamination. Eur. J. Org. Chem. 2015, 2015, 6965–6982. [Google Scholar] [CrossRef] [Green Version]
- Rudat, J.; Brucher, B.R.; Syldatk, C. Transaminases for the synthesis of enantiopure beta-amino acids. AMB Expr. 2012, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Mathew, S.; Yun, H. ω-Transaminases for the production of optically pure amines and unnatural amino acids. ACS Catal. 2012, 2, 993–1001. [Google Scholar] [CrossRef]
- Simon, R.C.; Richter, N.; Busto, E.; Kroutil, W. Recent developments of cascade reactions involving ω-transaminases. ACS Catal. 2014, 4, 129–143. [Google Scholar] [CrossRef]
- Patil, M.D.; Grogan, G.; Bommarius, A.; Yun, H. Recent advances in ω-transaminase-mediated biocatalysis for the enantioselective synthesis of chiral amines. Catalysts 2018, 8, 254. [Google Scholar] [CrossRef] [Green Version]
- Höhne, M.; Schätzle, S.; Jochens, H.; Robins, K.; Bornscheuer, U.T. Rational assignment of key motifs for function guides in silico enzyme identification. Nat. Chem. Biol. 2010, 6, 807–813. [Google Scholar] [CrossRef]
- Dawood, A.W.H.; Bassut, J.; de Souza, R.O.M.A.; Bornscheuer, U.T. Combination of the Suzuki–Miyaura cross-coupling reaction with engineered transaminases. Chem. Eur. J. 2018, 24, 16009–16013. [Google Scholar] [CrossRef]
- Ferrandi, E.E.; Monti, D. Amine transaminases in chiral amines synthesis: Recent advances and challenges. World J. Microbiol. Biotechnol. 2018, 34, 1–10. [Google Scholar] [CrossRef]
- Pavlidis, I.V.; Weiß, M.S.; Genz, M.; Spurr, P.; Hanlon, S.P.; Wirz, B.; Iding, H.; Bornscheuer, U.T. Identification of (S)-selective transaminases for the asymmetric synthesis of bulky chiral amines. Nat. Chem. 2016, 8, 1076–1082. [Google Scholar] [CrossRef]
- Biggelaar, L.; Soumillion, P.; Debecker, D. Enantioselective transamination in continuous flow mode with transaminase immobilized in a macrocellular silica monolith. Catalysts 2017, 7, 54. [Google Scholar] [CrossRef]
- Molnár, Z.; Farkas, E.; Lakó, Á.; Erdélyi, B.W.; Kroutil, W.; Vértessy, B.G.; Paizs, C.; Poppe, L. Immobilized whole-cell transaminase biocatalysts for continuous-flow kinetic resolution of amines. Catalysts 2019, 9, 438. [Google Scholar] [CrossRef] [Green Version]
- De María, P.D.; de Gonzalo, G.; Alcántara, A.R. Biocatalysis as useful tool in asymmetric synthesis: An assessment of recently granted patents (2014–2019). Catalysts 2019, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Taday, F.; Ryan, J.; Argent, S.P.; Caprio, V.; Maciá, B.; O’Reilly, E. Asymmetric construction of alkaloids by employing a key ω-transaminase cascade. Chem. Eur. J. 2020, 26, 3729–3732. [Google Scholar] [CrossRef] [PubMed]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvelage, S.; Dörr, M.; Höhne, M.; Bornscheuer, U.T. A systematic analysis of the substrate scope of (S)- and (R)-selective amine transaminases. Adv. Synth. Catal. 2017, 359, 4235–4243. [Google Scholar] [CrossRef]
- Koszelewski, D.; Göritzer, M.; Clay, D.; Seisser, B.; Kroutil, W. Synthesis of optically active amines employing recombinant ω-transaminases in E. coli cells. ChemCatChem 2010, 2, 73–77. [Google Scholar] [CrossRef]
- Mallin, H.; Höhne, M.; Bornscheuer, U.T. Immobilization of (R)- and (S)-amine transaminases on chitosan support and their application for amine synthesis using isopropylamine as donor. J. Biotechnol. 2014, 191, 32–37. [Google Scholar] [CrossRef]
- Fesko, K.; Steiner, K.; Breinbauer, R.; Schwab, H.; Schürmann, M.; Strohmeier, G.A. Investigation of one-enzyme systems in the ω-transaminase-catalyzed synthesis of chiral amines. J. Mol. Catal. B Enzym. 2013, 96, 103–110. [Google Scholar] [CrossRef]
- Schätzle, S.; Steffen-Munsberg, F.; Thontowi, A.; Höhne, M.; Robins, K.; Bornscheuer, U.T. Enzymatic asymmetric synthesis of enantiomerically pure aliphatic, aromatic and arylaliphatic amines with (R)-selective amine transaminases. Adv. Synth. Catal. 2011, 353, 2439–2445. [Google Scholar] [CrossRef]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, C.M.; Gourlay, L.J.; Dominguez, B.; Paradisi, F. An (R)-selective transaminases from Thermomyces stellatus: Stabilizing the tetrameric form. Front. Bioeng. Biotechnol. 2020, 8, 707. [Google Scholar] [CrossRef]
- Mutti, F.G.; Fuchs, C.S.; Pressnitz, D.; Turrini, N.G.; Sattler, J.H.; Lerchner, A.; Skerra, A.; Kroutil, W. Amination of ketones by employing two new (S)-selective ω-transaminases and the His-tagged ω-TA from Vibrio fluvialis. Eur. J. Org. Chem. 2012, 2012, 1003–1007. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Rozzell, D.; Kroutil, W. Asymmetric synthesis of optically pure pharmacologically relevant amines employing ω-transaminases. Adv. Synth. Catal. 2008, 17, 2761–2766. [Google Scholar] [CrossRef]
- Höhne, M.; Kühl, S.; Robins, K.; Bornscheuer, U.T. Efficient asymmetric synthesis of chiral amines by combining transaminase and pyruvate decarboxylase. ChemBioChem 2008, 3, 363–365. [Google Scholar] [CrossRef]
- Crowe, M.; Foulkes, M.; Francese, G.; Grimler, D.; Kuesters, R.; Laumen, K.; Li, Y.; Lin, C.; Nazor, J.; Ruch, T.; et al. Chemical Process for Preparing Spiroindolones and Intermediates Thereof. U.S. Patent 9,598,712 B2, 21 March 2017. [Google Scholar]
- Schmidt, N.G.; Simon, R.C.; Kroutil, W. Biocatalytic asymmetric synthesis of optically pure aromatic propargilic amines employing ω-transaminases. Adv. Synth. Catal. 2015, 357, 1815–1821. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; Lavandera, I.; Gotor-Fernandez, V. Sequential two-step steroselective amination of allylic alcohols through the combination of laccases and amine transaminases. ChemBioChem 2020, 21, 200–211. [Google Scholar] [CrossRef]
- Kaulmann, U.; Smithies, K.; Smith, M.E.B.; Hailes, H.C.; Ward, J.M. Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzym. Microbiol. Technol. 2007, 41, 628–637. [Google Scholar] [CrossRef]
- Gomm, A.; Grigoriou, S.; Peel, C.; Ryan, J.; Mujtaba, N.; Clarke, T.; Kulcinskaja, E.; O’Reilly, E. Application of “smart” amine donors for rapid screening and scale-up of transaminase-mediated biotransformations. Eur. J. Org. Chem. 2018, 2018, 5282–5284. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Miriyala, B.; Bhattacharyya, S.; Williamson, J.S. Chemoselective reductive alkylation of ammonia with carbonyl compounds: Synthesis of primary and symmetrical secondary amines. Tetrahedron 2004, 60, 1463–1471. [Google Scholar] [CrossRef]
- Lyskowski, A.; Gruber, C.; Steinkellner, G.; Schürmann, M.; Schwab, H.; Gruber, K.; Steiner, K. Crystal Structure of an (R)-selective ω-transaminase from Aspergillus terreus. PLoS ONE 2014, 9, e87350. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.S.; Damas, J.M.; Chen, Z.; Brissos, V.; Martins, L.O.; Soares, C.M.; Lindley, P.F.; Bento, I. The role of Asp116 in the reductive cleavage of dioxygen to water inCotA laccase: Assistance during the proton-transfer mechanism. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Midelfort, K.S.; Kumar, R.; Han, S.; Karmilowicz, M.J.; McConnell, K.; Gehlhaar, D.K.; Mistry, A.; Chang, J.S.; Anderson, M.; Villalobos, A.; et al. Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng. Des. Sel. 2013, 26, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atomstructure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Schrodinger, L. The PyMOL Molecular Graphics System, version 2.6.0a0; Schrodinger, LLC: New York, NY, USA, 2015. Available online: https://pymol.org/2/(accessed on 20 August 2021).
- BIOVIA, Dassault Systèmes. Discovery Studio Modelling Environment, version 21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2019. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/(accessed on 1 January 2016).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
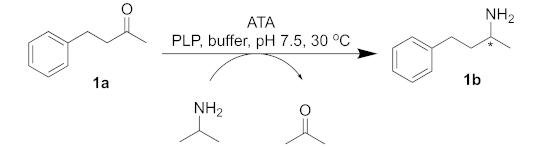 | |||
|---|---|---|---|
| Entry | ATA | Conv. (%) | (% ee) |
| 1 | C. violaceum | 80 | 97 (S) |
| 2 | R. pomeroyi | >99 | >99 (S) |
| 3 | A. terreus | 93 | >99 (R) |
| 4 | M. vanbaalenii | >99 | >99 (R) |
| 5 | V. fluvialis | 97 | 78 (S) |
| 6 | ATA-256 | >99 | >99 (S) |
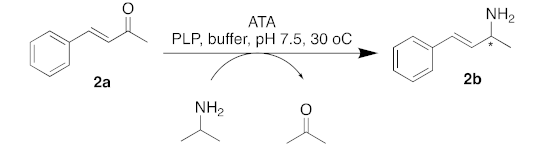 | |||
|---|---|---|---|
| Entry. | ATA | Conv. (%) | (% ee) |
| 1 | C. violaceum | 0 | - |
| 2 | R. pomeroyi | 9 | >99 (S) |
| 3 | A. terreus | 3 | >99 (R) |
| 4 | M. vanbaalenii | 4 | >99 (R) |
| 5 | V. fluvialis | 5 | 90 (S) |
| 6 | ATA-256 | 87 | >99 (S) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, I.S.; Farias, A.B.; Horta, B.A.C.; Milagre, H.M.S.; de Souza, R.O.M.A.; Bornscheuer, U.T.; Milagre, C.D.F. Computer Modeling Explains the Structural Reasons for the Difference in Reactivity of Amine Transaminases Regarding Prochiral Methylketones. Int. J. Mol. Sci. 2022, 23, 777. https://doi.org/10.3390/ijms23020777
Teixeira IS, Farias AB, Horta BAC, Milagre HMS, de Souza ROMA, Bornscheuer UT, Milagre CDF. Computer Modeling Explains the Structural Reasons for the Difference in Reactivity of Amine Transaminases Regarding Prochiral Methylketones. International Journal of Molecular Sciences. 2022; 23(2):777. https://doi.org/10.3390/ijms23020777
Chicago/Turabian StyleTeixeira, Iris S., André B. Farias, Bruno A. C. Horta, Humberto M. S. Milagre, Rodrigo O. M. A. de Souza, Uwe T. Bornscheuer, and Cintia D. F. Milagre. 2022. "Computer Modeling Explains the Structural Reasons for the Difference in Reactivity of Amine Transaminases Regarding Prochiral Methylketones" International Journal of Molecular Sciences 23, no. 2: 777. https://doi.org/10.3390/ijms23020777
APA StyleTeixeira, I. S., Farias, A. B., Horta, B. A. C., Milagre, H. M. S., de Souza, R. O. M. A., Bornscheuer, U. T., & Milagre, C. D. F. (2022). Computer Modeling Explains the Structural Reasons for the Difference in Reactivity of Amine Transaminases Regarding Prochiral Methylketones. International Journal of Molecular Sciences, 23(2), 777. https://doi.org/10.3390/ijms23020777