
Brain Tissue-Derived Extracellular Vesicle Mediated Therapy in the Neonatal Ischemic Brain

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results
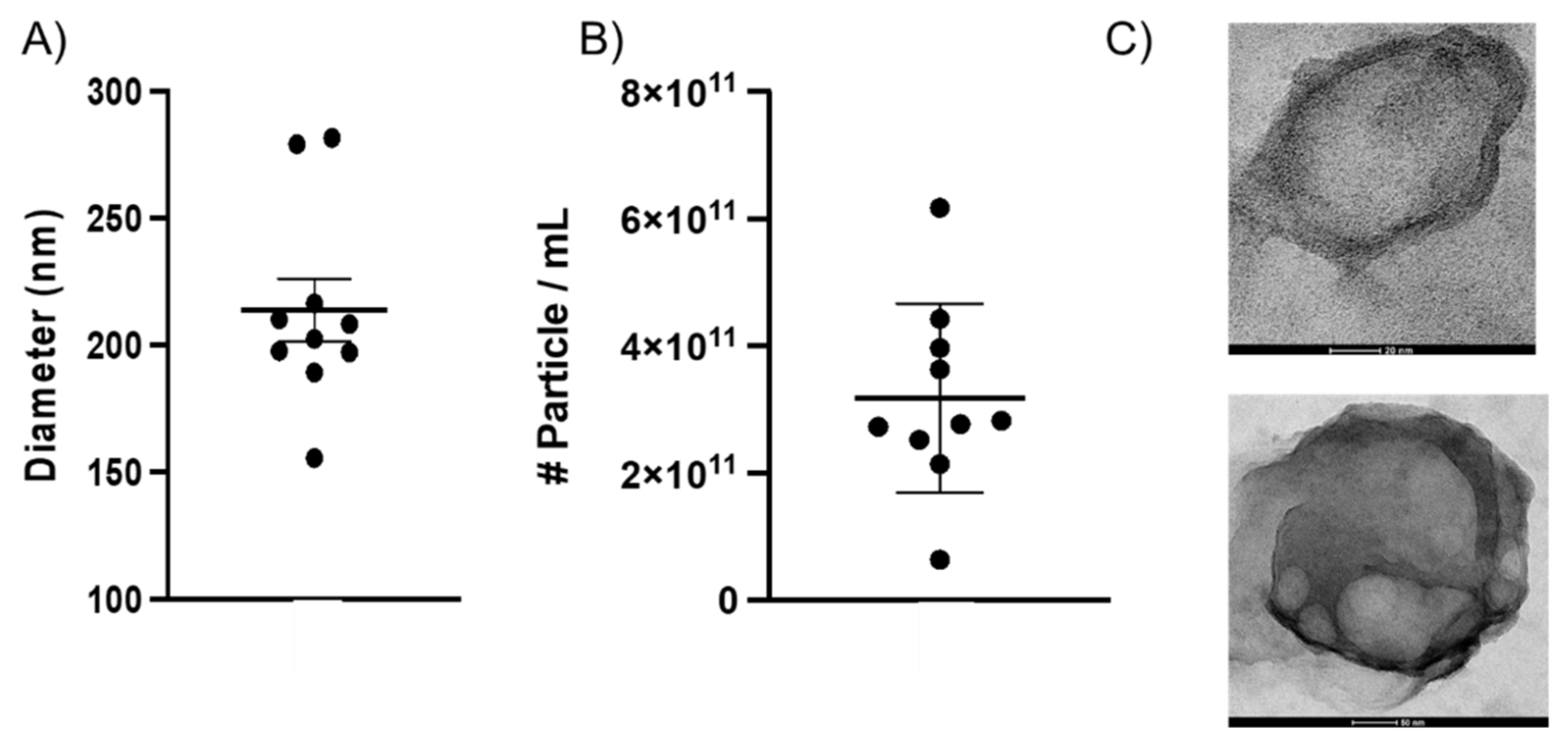
2.1. Characterizing BEVs Isolated from Whole Neonatal Brain Tissue
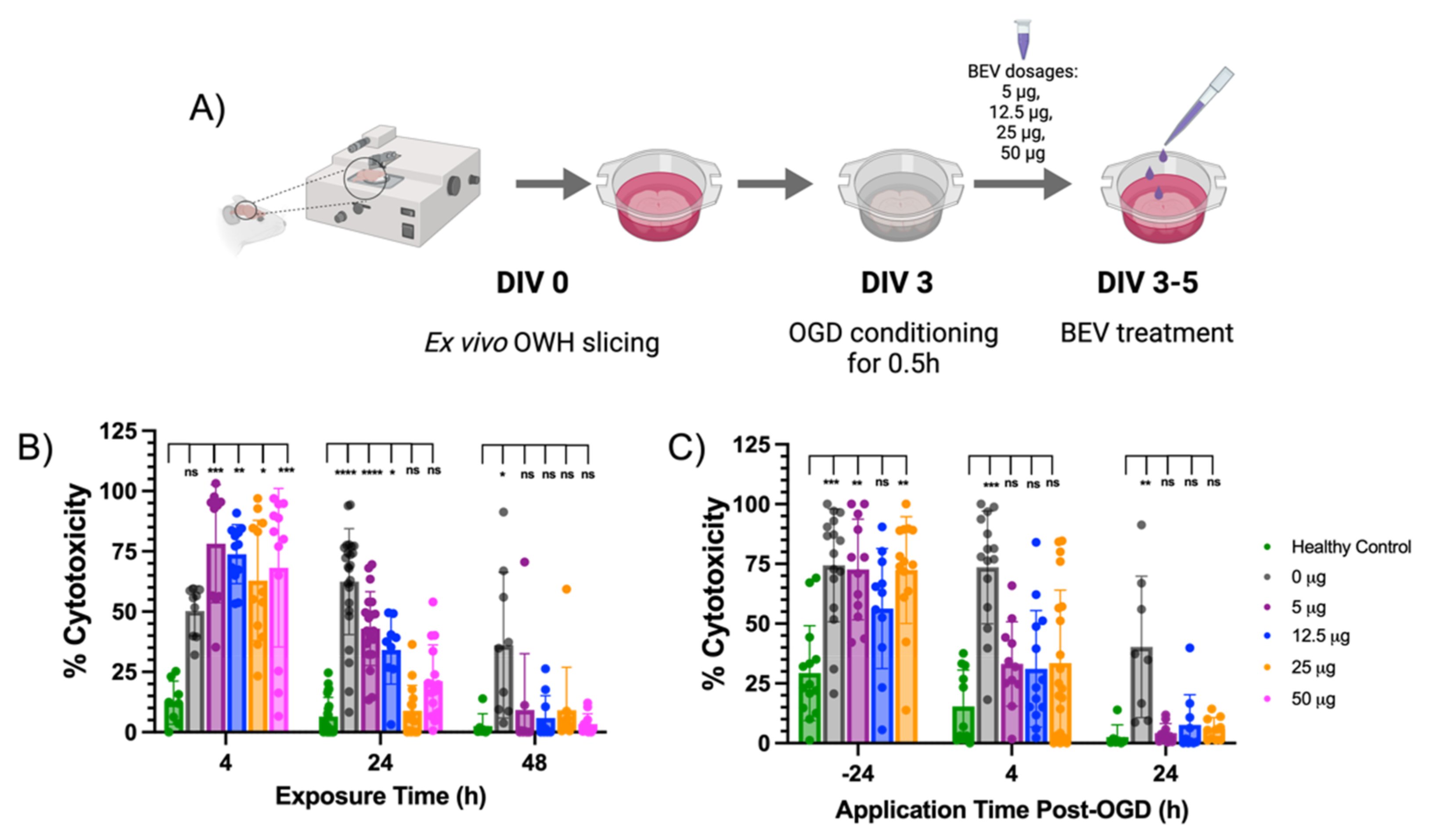
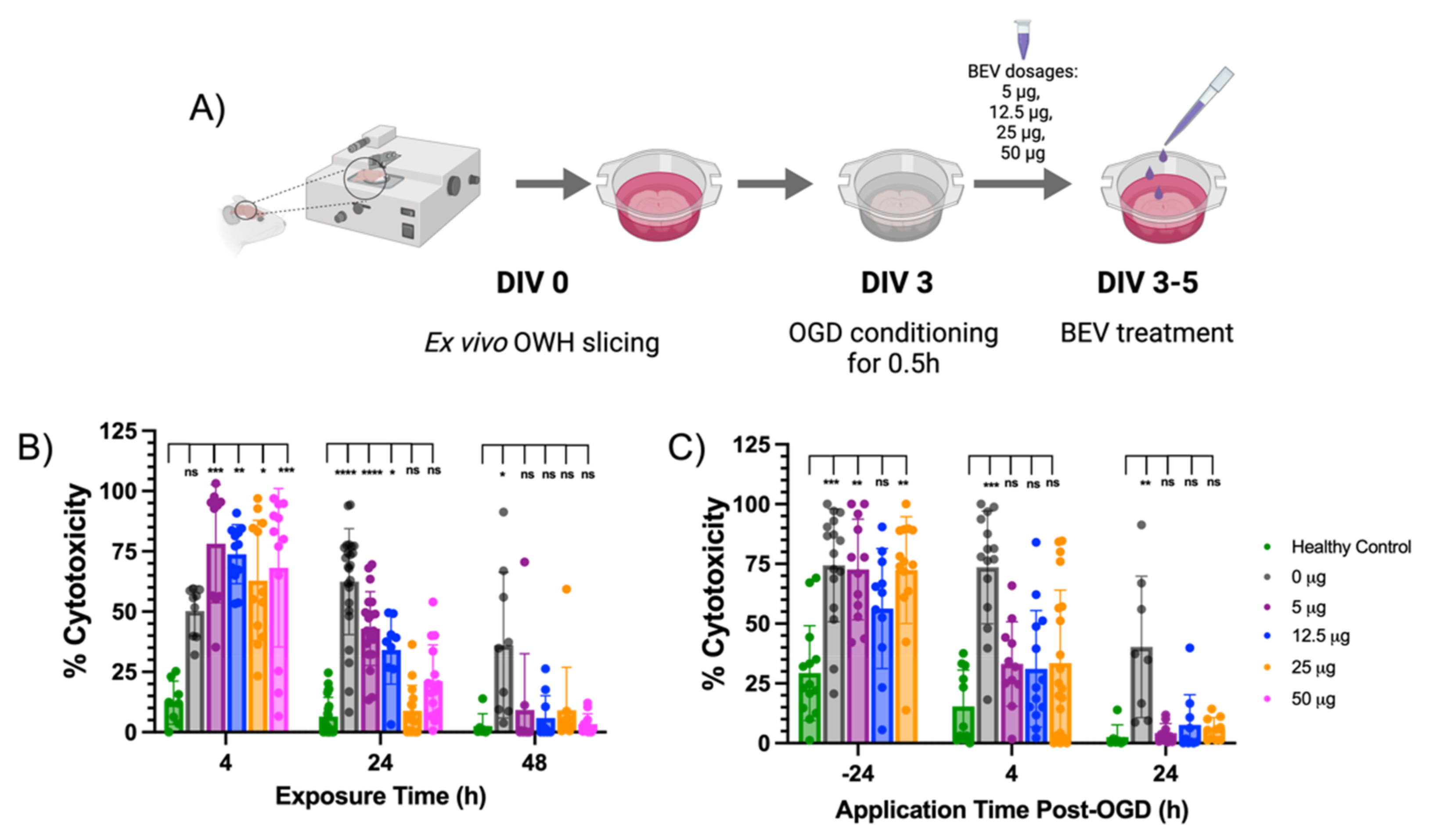
2.2. BEV-Mediated Therapeutic Effects on an Ex Vivo Ischemic Slice Model
2.3. RNA Expression Changes in Response to BEV Treatment
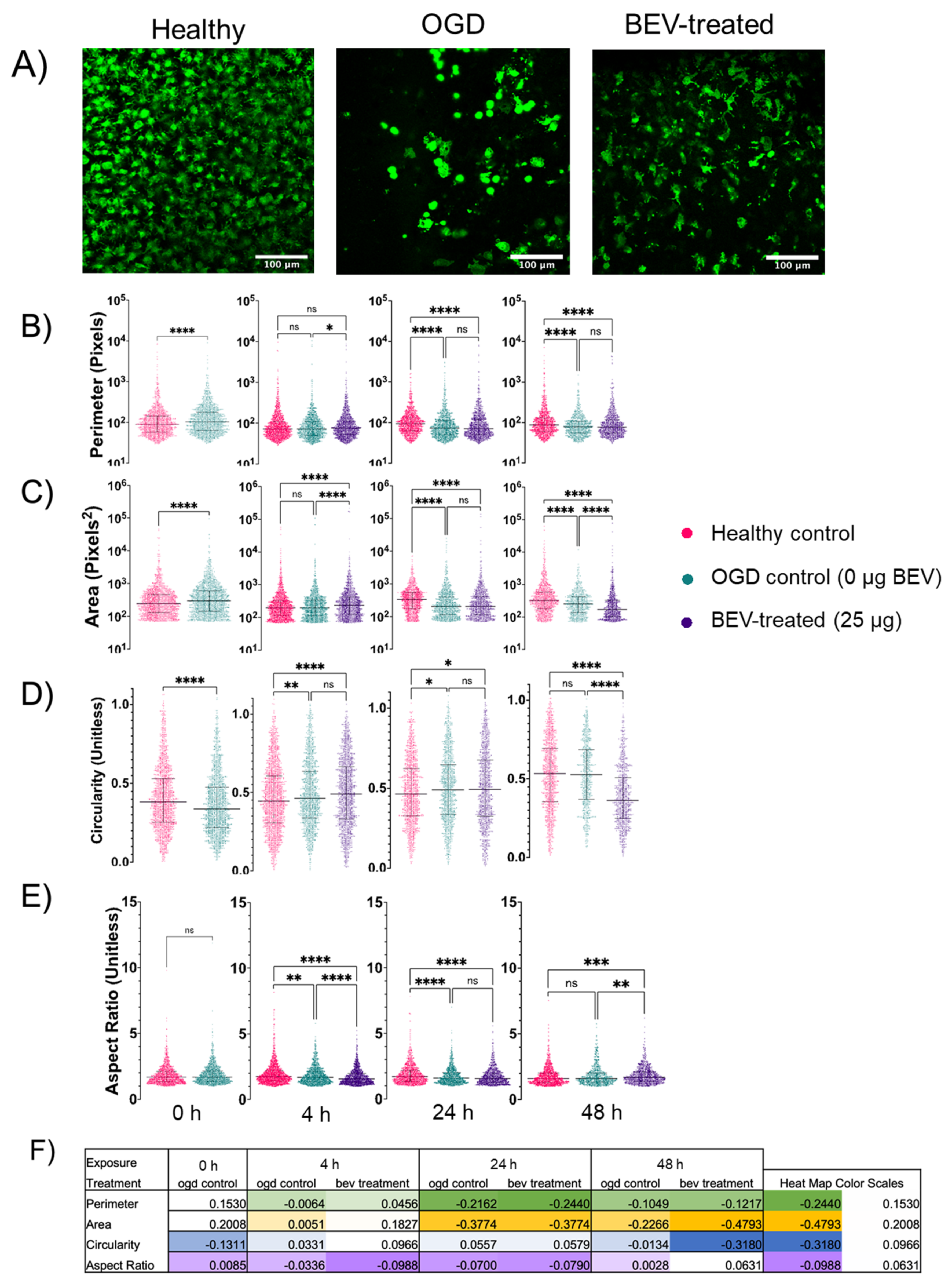
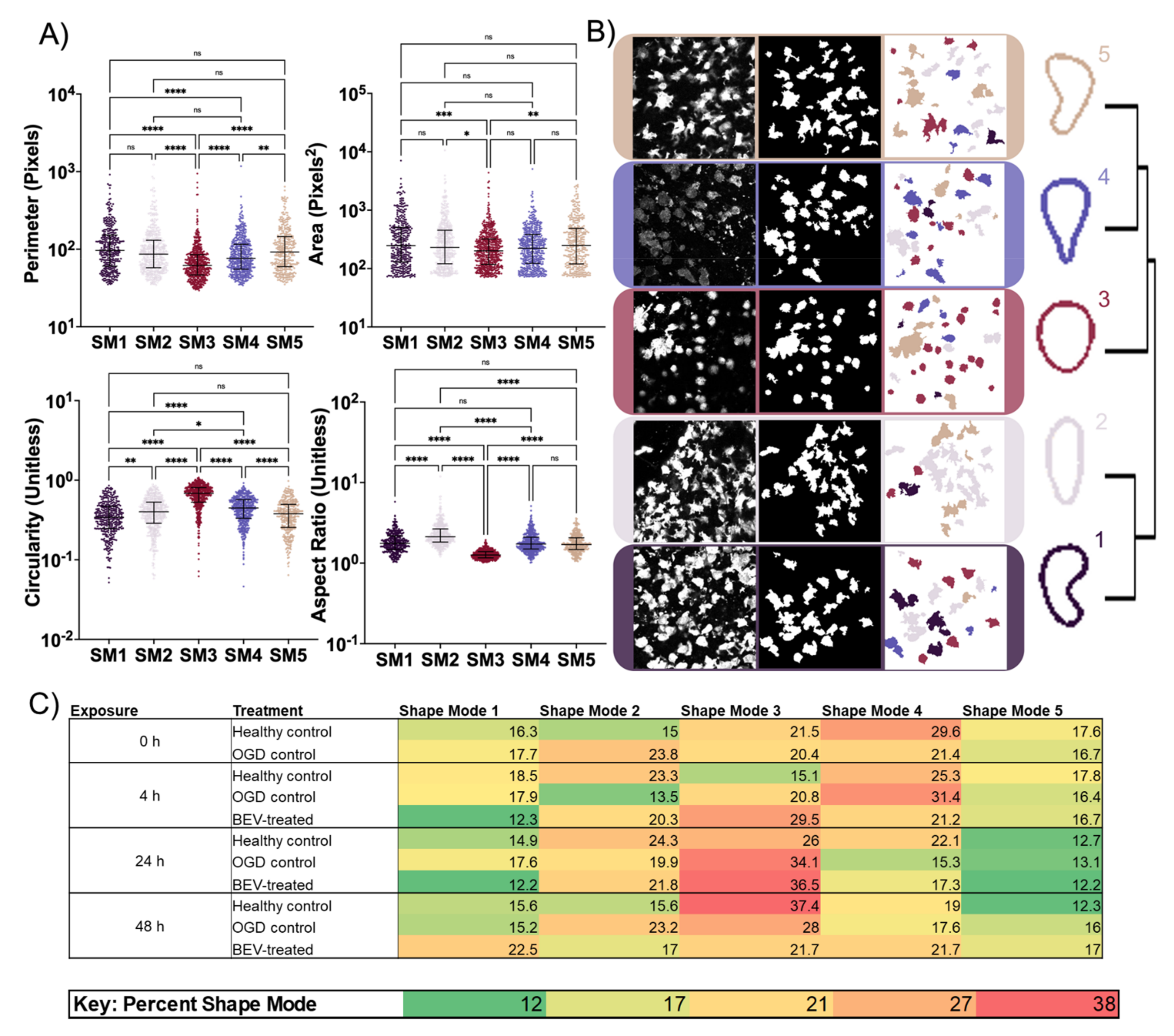
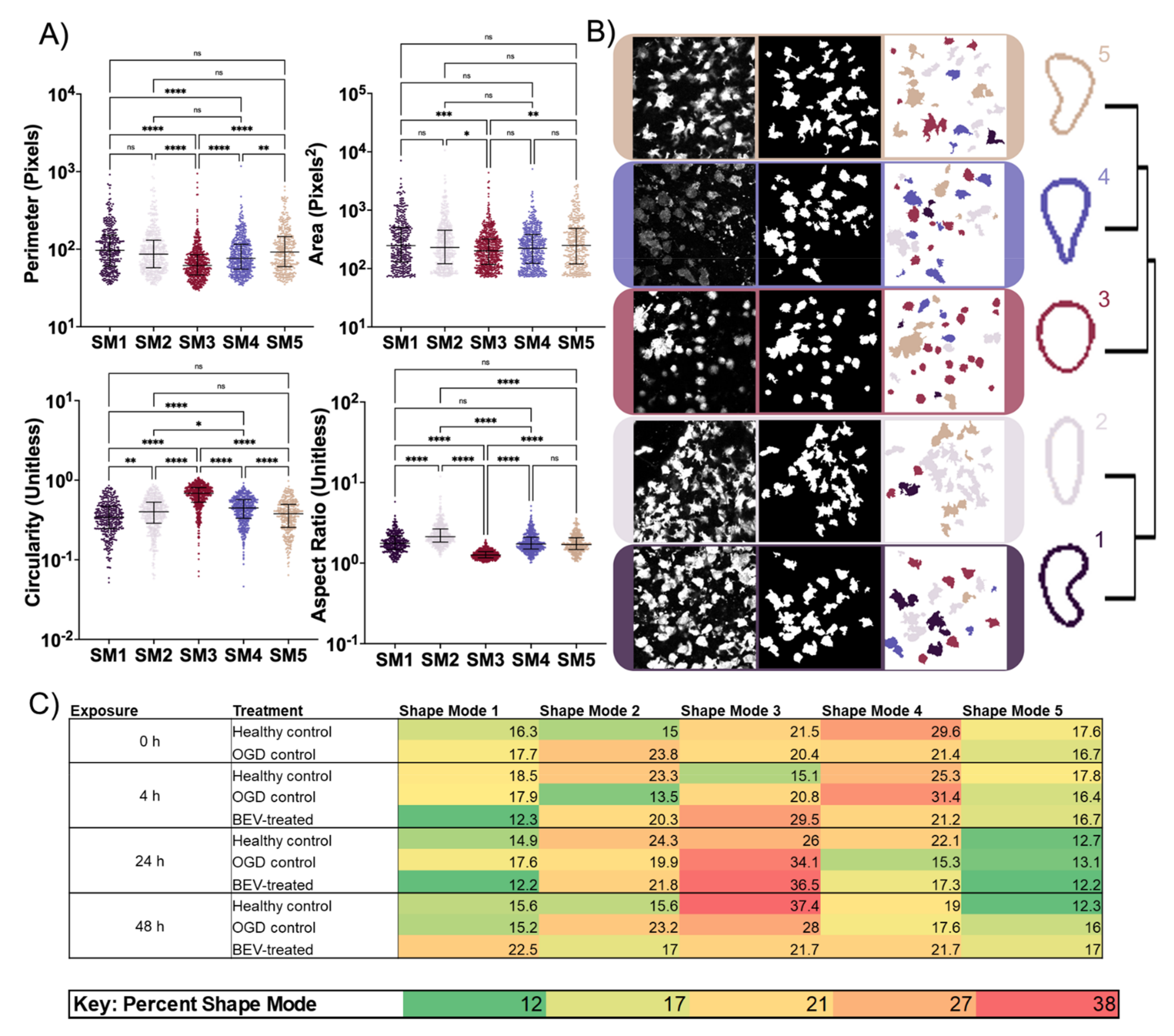
2.4. Morphological Response of Glial Cells to BEV Treatment
3. Discussion
4. Materials and Methods
4.1. Animal Care and Ethics
4.2. OWH Slicing Methodology
4.3. OGD Methodology
4.4. BEV Isolation Using a Combination of Methods
4.5. BEV Characterization
4.6. TEM Imaging
4.7. BEV Administration on Ex Vivo Slices
4.8. Confocal Imaging
4.9. Cell Morphology Analysis
4.10. Reverse Transcriptase Quantitative Polymerase Chain Reaction (RT-qPCR)
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mercuri, E.; Guzzetta, A.; Laroche, S.; Ricci, D.; van Haastert, I.; Simpson, A.; Luciano, R.; Bleakley, C.; Frisone, M.F.; Haataja, L.; et al. Neurologic examination of preterm infants at term age: Comparison with term infants. J. Pediatr. 2003, 142, 647–655. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Wood, M.J.A.; O’Loughlin, A.J.; Lakhal, S. Exosomes and the blood-brain barrier: Implications for neurological diseases. Ther. Deliv. 2011, 2, 1095–1099. [Google Scholar] [CrossRef]
- Douglas-Escobar, M.; Weiss, M.D. Hypoxic-Ischemic Encephalopathy. JAMA Pediatr. 2015, 169, 397. [Google Scholar] [CrossRef]
- Ferriero, D.M.; Fullerton, H.J.; Bernard, T.J.; Billinghurst, L.; Daniels, S.R.; DeBaun, M.R.; deVeber, G.; Ichord, R.N.; Jordan, L.C.; Massicotte, P.; et al. Management of Stroke in Neonates and Children: A Scientific Statement from the American Heart Association/American Stroke Association. Stroke 2019, 50, e51–e96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, L.J.; Shi, L.; Hoerder-Suabedissen, A.; Molnár, Z. Neonatal Hypoxia Ischaemia: Mechanisms, Models, and Therapeutic Challenges. Front. Cell. Neurosci. 2017, 11, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adami, R.R.; Grundy, M.E.; Poretti, A.; Felling, R.J.; Lemmon, M.; Graham, E.M. Distinguishing Arterial Ischemic Stroke from Hypoxic-Ischemic Encephalopathy in the Neonate at Birth. Obstet. Gynecol. 2016, 128, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurinczuk, J.J.; White-Koning, M.; Badawi, N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Human Dev. 2010, 86, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Hunt, R.; Tarnow-Mordi, W.; Inder, T.; Davis, P. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2007, CD003311. [Google Scholar] [CrossRef]
- Davidson, J.; Davies, A.; Wassink, G.; Bennet, L.; Gunn, A. Can we further optimize therapeutic hypothermia for hypoxic-ischemic encephalopathy? Neural Regen. Res. 2019, 14, 1678. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Gudbergsson, J.M.; Jønsson, K.; Simonsen, J.B.; Johnsen, K.B. Systematic review of targeted extracellular vesicles for drug delivery—Considerations on methodological and biological heterogeneity. J. Control. Release 2019, 306, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chopp, M. Exosome Therapy for Stroke. Stroke 2018, 49, 1083–1090. [Google Scholar] [CrossRef]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: Advantages and disadvantages. Am. J. Physiol.—Cell Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesenberry, P.J.; Aliotta, J.; Deregibus, M.C.; Camussi, G. Role of extracellular RNA-carrying vesicles in cell differentiation and reprogramming. Stem Cell Res. Ther. 2015, 6, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, M.M.; Kaiser, J.; Schwab, M.E. Extracellular Vesicles: Multimodal Envoys in Neural Maintenance and Repair. Trends Neurosci. 2018, 41, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Fröhlich, D.; Kuo, W.P.; Amphornrat, J.; Thilemann, S.; Saab, A.S.; Kirchhoff, F.; Möbius, W.; Goebbels, S.; Nave, K.-A.; et al. Neurotransmitter-Triggered Transfer of Exosomes Mediates Oligodendrocyte-Neuron Communication. PLoS Biol. 2013, 11, e1001604. [Google Scholar] [CrossRef] [Green Version]
- Dickens, A.M.; Tovar-y-Romo, L.B.; Yoo, S.-W.; Trout, A.L.; Bae, M.; Kanmogne, M.; Megra, B.; Williams, D.W.; Witwer, K.W.; Gacias, M.; et al. Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal. 2017, 10, eaai7696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otero-Ortega, L.; Laso-García, F.; Gómez-de Frutos, M.C.; Diekhorst, L.; Martínez-Arroyo, A.; Alonso-López, E.; García-Bermejo, M.L.; Rodríguez-Serrano, M.; Arrúe-Gonzalo, M.; Díez-Tejedor, E.; et al. Low dose of extracellular vesicles identified that promote recovery after ischemic stroke. Stem Cell Res. Ther. 2020, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Xin, D.; Li, T.; Chu, X.; Ke, H.; Yu, Z.; Cao, L.; Bai, X.; Liu, D.; Wang, Z. Mesenchymal stromal cell-derived extracellular vesicles modulate microglia/macrophage polarization and protect the brain against hypoxia-ischemic injury in neonatal mice by targeting delivery of miR-21a-5p. Acta Biomater. 2020, 113, 597–613. [Google Scholar] [CrossRef]
- Zhang, Y.; Chopp, M.; Zhang, Z.G.; Katakowski, M.; Xin, H.; Qu, C.; Ali, M.; Mahmood, A.; Xiong, Y. Systemic administration of cell-free exosomes generated by human bone marrow derived mesenchymal stem cells cultured under 2D and 3D conditions improves functional recovery in rats after traumatic brain injury. Neurochem. Int. 2017, 111, 69–81. [Google Scholar] [CrossRef]
- Deng, M.; Xiao, H.; Peng, H.; Yuan, H.; Xu, Y.; Zhang, G.; Tang, J.; Hu, Z. Preservation of neuronal functions by exosomes derived from different human neural cell types under ischemic conditions. Eur. J. Neurosci. 2018, 47, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Budnik, V.; Ruiz-Cañada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic Administration of Exosomes Released from Mesenchymal Stromal Cells Promote Functional Recovery and Neurovascular Plasticity After Stroke in Rats. J. Cereb. Blood Flow Metab. 2013, 33, 1711–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benakis, C.; Garcia-Bonilla, L.; Iadecola, C.; Anrather, J. The role of microglia and myeloid immune cells in acute cerebral ischemia. Front. Cell. Neurosci. 2015, 8, 461. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, L.; Turchinovich, A.; Mahairaki, V.; Troncoso, J.C.; Pletniková, O.; Haughey, N.J.; Vella, L.J.; Hill, A.F.; Zheng, L.; et al. Influence of species and processing parameters on recovery and content of brain tissue-derived extracellular vesicles. J. Extracell. Vesicles 2020, 9, 1785746. [Google Scholar] [CrossRef]
- Royo, F.; Théry, C.; Falcón-Pérez, J.M.; Nieuwland, R.; Witwer, K.W. Methods for Separation and Characterization of Extracellular Vesicles: Results of a Worldwide Survey Performed by the ISEV Rigor and Standardization Subcommittee. Cells 2020, 9, 1955. [Google Scholar] [CrossRef] [PubMed]
- Tzaridis, T.; Bachurski, D.; Liu, S.; Surmann, K.; Babatz, F.; Gesell Salazar, M.; Völker, U.; Hallek, M.; Herrlinger, U.; Vorberg, I.; et al. Extracellular Vesicle Separation Techniques Impact Results from Human Blood Samples: Considerations for Diagnostic Applications. Int. J. Mol. Sci. 2021, 22, 9211. [Google Scholar] [CrossRef] [PubMed]
- Gandham, S.; Su, X.; Wood, J.; Nocera, A.L.; Alli, S.C.; Milane, L.; Zimmerman, A.; Amiji, M.; Ivanov, A.R. Technologies and Standardization in Research on Extracellular Vesicles. Trends Biotechnol. 2020, 38, 1066–1098. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, R.; Wood, T.R.; Nance, E. Superoxide dismutase reduces monosodium glutamate-induced injury in an organotypic whole hemisphere brain slice model of excitotoxicity. J. Biol. Eng. 2020, 14, 3. [Google Scholar] [CrossRef]
- Joseph, A.; Liao, R.; Zhang, M.; Helmbrecht, H.; McKenna, M.; Filteau, J.R.; Nance, E. Nanoparticle-microglial interaction in the ischemic brain is modulated by injury duration and treatment. Bioeng. Transl. Med. 2020, 5, e10175. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.R.; Hildahl, K.; Helmbrecht, H.; Corry, K.A.; Moralejo, D.H.; Kolnik, S.E.; Prater, K.E.; Juul, S.E.; Nance, E. A ferret brain slice model of oxygen-glucose deprivation captures regional responses to perinatal injury and treatment associated with specific microglial phenotypes. Bioeng. Transl. Med. 2021, e10265. [Google Scholar] [CrossRef]
- Humpel, C. Organotypic Brain Slice Cultures: A Review. Neuroscience 2015, 305, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Sisa, C.; Kholia, S.; Naylor, J.; Herrera Sanchez, M.B.; Bruno, S.; Deregibus, M.C.; Camussi, G.; Inal, J.M.; Lange, S.; Hristova, M. Mesenchymal Stromal Cell Derived Extracellular Vesicles Reduce Hypoxia-Ischaemia Induced Perinatal Brain Injury. Front. Physiol. 2019, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Doeppner, T.R.; Herz, J.; Görgens, A.; Schlechter, J.; Ludwig, A.-K.; Radtke, S.; Miroschedji, K.d.; Horn, P.A.; Giebel, B.; Hermann, D.M. Extracellular Vesicles Improve Post-Stroke Neuroregeneration and Prevent Postischemic Immunosuppression. Stem Cells Transl. Med. 2015, 4, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Extracellular membrane vesicles as vehicles for brain cell-to-cell interactions in physiological as well as pathological conditions. BioMed Res. Int. 2015, 2015, 152926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ye, Y.; Su, X.; He, J.; Bai, W.; He, X. MSCs-Derived Exosomes and Neuroinflammation, Neurogenesis and Therapy of Traumatic Brain Injury. Front. Cell. Neurosci. 2017, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Lu, J.; Shao, A.; Zhang, J.H.; Zhang, J. Glial Cells: Role of the Immune Response in Ischemic Stroke. Front. Immunol. 2020, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.B. Perinatal Ischemic Stroke. Stroke 2007, 38, 742–745. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Jiang, Y.; Sun, Y. Astrocyte-derived exosomes carry microRNA-17-5p to protect neonatal rats from hypoxic-ischemic brain damage via inhibiting BNIP-2 expression. Neurotoxicology 2021, 83, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Xin, D.; Li, T.; Chu, X.; Ke, H.; Liu, D.; Wang, Z. MSCs-extracellular vesicles attenuated neuroinflammation, synapse damage and microglial phagocytosis after hypoxia-ischemia injury by preventing osteopontin expression. Pharmacol. Res. 2021, 164, 105322. [Google Scholar] [CrossRef] [PubMed]
- Brenna, S.; Altmeppen, H.C.; Mohammadi, B.; Rissiek, B.; Schlink, F.; Ludewig, P.; Krisp, C.; Schluter, H.; Failla, A.V.; Schneider, C.; et al. Characterization of brain-derived extracellular vesicles reveals changes in cellular origin after stroke and enrichment of the prion protein with a potential role in cellular uptake. J. Extracell. Vesicles 2020, 9, 1809065. [Google Scholar] [CrossRef] [PubMed]
- Dubbelaar, M.L.; Kracht, L.; Eggen, B.J.L.; Boddeke, E.W.G.M. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753. [Google Scholar] [CrossRef] [PubMed]
- Savage, J.C.; Carrier, M.; Tremblay, M.-È. Morphology of Microglia Across Contexts of Health and Disease. In Microglia; Springer: New York, NY, USA, 2019; pp. 13–26. [Google Scholar]
- Hanisch, U.-K. Functional diversity of microglia—How heterogeneous are they to begin with? Front. Cell. Neurosci. 2013, 7, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhya, R.; Zingg, W.; Shetty, S.; Shetty, A.K. Astrocyte-derived extracellular vesicles: Neuroreparative properties and role in the pathogenesis of neurodegenerative disorders. J. Control. Release 2020, 323, 225–239. [Google Scholar] [CrossRef]
- Vella, L.J.; Scicluna, B.J.; Cheng, L.; Bawden, E.G.; Masters, C.L.; Ang, S.; Willamson, N.; McLean, C.; Barnham, K.J.; Hill, A.F. A rigorous method to enrich for exosomes from brain tissue. J. Extracell. Vesicles 2017, 6, 1348885. [Google Scholar] [CrossRef]
- Zhang, M.; Vojtech, L.; Ye, Z.; Hladik, F.; Nance, E. Quantum Dot Labeling and Visualization of Extracellular Vesicles. ACS Appl. Nano Mater. 2020, 3, 7211–7222. [Google Scholar] [CrossRef]
- Webber, J.; Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2013, 2, 19861. [Google Scholar] [CrossRef]
- Van Der Walt, S.; Schönberger, J.L.; Nunez-Iglesias, J.; Boulogne, F.; Warner, J.D.; Yager, N.; Gouillart, E.; Yu, T. Scikit-image: Image processing in Python. PeerJ 2014, 2, e453. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kongsui, R.; Beynon, S.B.; Johnson, S.J.; Walker, F.R. Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. J. Neuroinflamm. 2014, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Phillip, J.M.; Han, K.-S.; Chen, W.-C.; Wirtz, D.; Wu, P.-H. A robust unsupervised machine-learning method to quantify the morphological heterogeneity of cells and nuclei. Nat. Protoc. 2021, 16, 754–774. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Helmbrecht, H.; Nance, E. Nance Lab Cell Morphology Flows GitHub Page. Available online: https://github.com/Nance-Lab/cellmorphflows (accessed on 20 November 2021).
- Ziemka-Nalecz, M.; Jaworska, J.; Zalewska, T. Insights into the Neuroinflammatory Responses after Neonatal Hypoxia-Ischemia. J. Neuropathol. Exp. Neurol. 2017, 76, 644–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NCBI Reference Sequence | Forward Primer | Reverse Primer |
|---|---|---|---|
| GAPDH | NM_017008.4 | ACTCCCATTCTTCCACCTTTG | ACTCCCATTCTTCCACCTTTG |
| IL-4 | NM_201270.1 | GTCACTGACTGTAGAGAGCTATTG | CTGTCGTTACATCCGTGGATAC |
| IL-6 | NM_012589.2 | GAAGTTAGAGTCACAGAAGGAGTG | GTTTGCCGAGTAGACCTCATAG |
| IL-9 | NM_001105747.1 | GAAGGACGACCCATCATCAAA | ACGGTGTGGTACAATCATCAG |
| IL-10 | NM_012854.2 | AGTGGAGCAGGTGAAGAATG | GAGTGTCACGTAGGCTTCTATG |
| IL-11 | NM_133519.5 | CTAGCACTTCAAAGGTCCTCAA | ACACCTTGAACCTTGCTATCTC |
| Ki67 | NM_001271366.1 | CACACACAAAGAGCCCATAGA | GATTCCTCCTGCCGGTTAAA |
| Nκβ | NM_001276711.1 | GGTTACGGGAGATGTGAAGATG | GTGGATGATGGCTAAGTGTAGG |
| CD68 | NM_001031638.1 | CTTGGCTCTCTCATTCCCTTAC | TGTATTCCACTGCCATGTAGTT |
| Vimentin | NM_031140.1 | CTTCCCTGAACCTGAGAGAAAC | GTCTCTGGTTTCAACCGTCTTA |
| GFAP | NM_017009.2 | AAAGACACTGAAACAGGAGAGAG | GGACTGAGCAACCAGGAATAG |
| Synapsin | NM_001110782.2 | GGACGGAAGGGATCACATTATT | ACCACAAGTTCCACGATGAG |
| CD11b | NM_012711.1 | GAGCACCATCTGGGACATAAA | GGCATCAGAGTCCACATCAA |
| iNOS | NM_012611.3 | TGGAGCGAGTTGTGGATTG | CCTCTTGTCTTTGACCCAGTAG |
| Casp-3 | NM_012922.2 | GAGCTTGGAACGCTAAGA | CTGACTTGCTCCCATGTAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.P.; Helmbrecht, H.; Ye, Z.; Adebayo, T.; Hashi, N.; Doan, M.-A.; Nance, E. Brain Tissue-Derived Extracellular Vesicle Mediated Therapy in the Neonatal Ischemic Brain. Int. J. Mol. Sci. 2022, 23, 620. https://doi.org/10.3390/ijms23020620
Nguyen NP, Helmbrecht H, Ye Z, Adebayo T, Hashi N, Doan M-A, Nance E. Brain Tissue-Derived Extracellular Vesicle Mediated Therapy in the Neonatal Ischemic Brain. International Journal of Molecular Sciences. 2022; 23(2):620. https://doi.org/10.3390/ijms23020620
Chicago/Turabian StyleNguyen, Nam Phuong, Hawley Helmbrecht, Ziming Ye, Tolulope Adebayo, Najma Hashi, My-Anh Doan, and Elizabeth Nance. 2022. "Brain Tissue-Derived Extracellular Vesicle Mediated Therapy in the Neonatal Ischemic Brain" International Journal of Molecular Sciences 23, no. 2: 620. https://doi.org/10.3390/ijms23020620
APA StyleNguyen, N. P., Helmbrecht, H., Ye, Z., Adebayo, T., Hashi, N., Doan, M.-A., & Nance, E. (2022). Brain Tissue-Derived Extracellular Vesicle Mediated Therapy in the Neonatal Ischemic Brain. International Journal of Molecular Sciences, 23(2), 620. https://doi.org/10.3390/ijms23020620