
Developmental Stressors Induce Innate Immune Memory in Microglia and Contribute to Disease Risk

Abstract
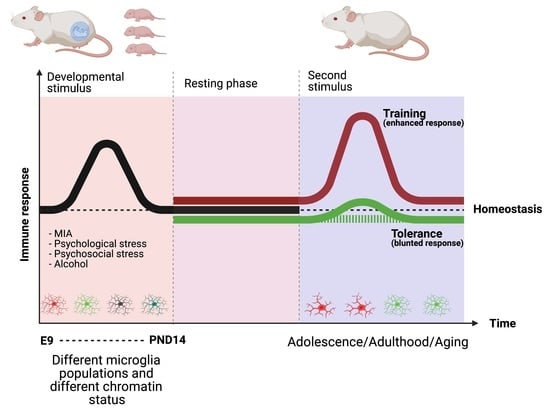
1. Developmental Stressors and Disease Risk
2. Developmental Stressors’ Impact on Brain Microglia
2.1. Impact of Prenatal Immune Activation on Microglia
2.2. Impact of Early Life Stress on Microglia
2.3. Impact of Developmental Ethanol on Microglia
2.4. Interventions for Developmental Stress Exposures
2.5. Transgenerational Transmission of Developmental Stressors
2.6. Summary
3. Microglia Innate Immune Memory
4. Molecular Mechanism Underlying Innate Immune Memory in Microglia
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MG | microglia |
| Stim | Stimulus |
| MIA | maternal immune activation |
| PIC | polyinosinic:polycytidylic acid |
| Hpc | Hippocampus |
| Cb | cerebellum |
| Ctx | cortex |
| LPS | lipopolysaccharide |
| FC | frontal cortex |
| Hypo | hypothalamus |
| P | postnatal day |
| E | embryonic day |
| FIRS | fetal inflammatory response syndrome |
| ELS | early life stress |
| H | hour |
| EtOH | ethanol |
| BMDM | bone marrow derived macrophages |
| PBMCs | peripheral blood mononuclear cells |
| APP23 | amyloid precursor protein |
| 5xFAD | 5 familiar Alzheimer’s disease mutation mouse model |
| Ercc1 | excision repair cross-complementation group 1 |
| A | amyloid beta |
References
- Ramsay, D.S.; Woods, S.C. Clarifying the roles of homeostasis and allostasis in physiological regulation. Psychol. Rev. 2014, 121, 225–247. [Google Scholar] [CrossRef] [PubMed]
- McEwen, B.S.; Akil, H. Revisiting the Stress Concept: Implications for Affective Disorders. J. Neurosci. 2020, 40, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Korte, S.M.; Koolhaas, J.M.; Wingfield, J.C.; McEwen, B.S. The Darwinian concept of stress: Benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neurosci. Biobehav. Rev. 2005, 29, 3–38. [Google Scholar] [CrossRef]
- Catale, C.; Gironda, S.; Lo Iacono, L.; Carola, V. Microglial Function in the Effects of Early-Life Stress on Brain and Behavioral Development. J. Clin. Med. 2020, 9, 468. [Google Scholar] [CrossRef]
- Guidi, J.; Lucente, M.; Sonino, N.; Fava, G.A. Allostatic Load and Its Impact on Health: A Systematic Review. Psychother. Psychosom. 2021, 90, 11–27. [Google Scholar] [CrossRef]
- Ullmann, E.; Perry, S.W.; Licinio, J.; Wong, M.L.; Dremencov, E.; Zavjalov, E.L.; Shevelev, O.B.; Khotskin, N.V.; Koncevaya, G.V.; Khotshkina, A.S.; et al. From Allostatic Load to Allostatic State—An Endogenous Sympathetic Strategy to Deal with Chronic Anxiety and Stress. Front. Behav. Neurosci. 2019, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Bekkering, S.; Domínguez-Andrés, J.; Joosten, L.A.B.; Riksen, N.P.; Netea, M.G. Trained Immunity: Reprogramming Innate Immunity in Health and Disease. Annu. Rev. Immunol. 2021, 39, 667–693. [Google Scholar] [CrossRef]
- Raineki, C.; Opendak, M.; Sarro, E.; Showler, A.; Bui, K.; McEwen, B.S.; Wilson, D.A.; Sullivan, R.M. During infant maltreatment, stress targets hippocampus, but stress with mother present targets amygdala and social behavior. Proc. Natl. Acad. Sci. USA 2019, 116, 22821–22832. [Google Scholar] [CrossRef]
- Marrocco, J.; Gray, J.D.; Kogan, J.F.; Einhorn, N.R.; O’Cinneide, E.M.; Rubin, T.G.; Carroll, T.S.; Schmidt, E.F.; McEwen, B.S. Early Life Stress Restricts Translational Reactivity in CA3 Neurons Associated with Altered Stress Responses in Adulthood. Front. Behav. Neurosci. 2019, 13, 157. [Google Scholar] [CrossRef]
- Hill, M.N.; Eiland, L.; Lee, T.T.Y.; Hillard, C.J.; McEwen, B.S. Early life stress alters the developmental trajectory of corticolimbic endocannabinoid signaling in male rats. Neuropharmacology 2019, 146, 154–162. [Google Scholar] [CrossRef]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64. [Google Scholar] [CrossRef]
- Eyles, D.W. How do established developmental risk-factors for schizophrenia change the way the brain develops. Transl. Psychiatry 2021, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Boyce, W.T.; Levitt, P.; Martinez, F.D.; McEwen, B.S.; Shonkoff, J.P. Genes, Environments, and Time: The Biology of Adversity and Resilience. Pediatrics 2021, 147, e20201651. [Google Scholar] [CrossRef] [PubMed]
- Monk, C.; Lugo-Candelas, C.; Trumpff, C. Prenatal Developmental Origins of Future Psychopathology: Mechanisms and Pathways. Annu. Rev. Clin. Psychol. 2019, 15, 317–344. [Google Scholar] [CrossRef]
- Dunn, G.A.; Nigg, J.T.; Sullivan, E.L. Neuroinflammation as a risk factor for attention deficit hyperactivity disorder. Pharmacol. Biochem. Behav. 2019, 182, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Meyer, U. Maternal Immune Activation and Neuropsychiatric Illness: A Translational Research Perspective. Am. J. Psychiatry 2018, 175, 1073–1083. [Google Scholar] [CrossRef]
- Green, M.J.; Watkeys, O.J.; Whitten, T.; Thomas, C.; Kariuki, M.; Dean, K.; Laurens, K.R.; Harris, F.; Carr, V.J. Increased incidence of childhood mental disorders following exposure to early life infection. Brain Behav. Immun. 2021, 97, 376–382. [Google Scholar] [CrossRef]
- Canetta, S.E.; Bao, Y.; Co, M.D.; Ennis, F.A.; Cruz, J.; Terajima, M.; Shen, L.; Kellendonk, C.; Schaefer, C.A.; Brown, A.S. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am. J. Psychiatry 2014, 171, 557–563. [Google Scholar] [CrossRef]
- Goines, P.; Haapanen, L.; Boyce, R.; Duncanson, P.; Braunschweig, D.; Delwiche, L.; Hansen, R.; Hertz-Picciotto, I.; Ashwood, P.; Van de Water, J. Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav. Immun. 2011, 25, 514–523. [Google Scholar] [CrossRef]
- Abdallah, M.W.; Hougaard, D.M.; Nørgaard-Pedersen, B.; Grove, J.; Bonefeld-Jørgensen, E.C.; Mortensen, E.L. Infections during pregnancy and after birth, and the risk of autism spectrum disorders: A register-based study utilizing a Danish historic birth cohort. Turk Psikiyatr. Derg. 2012, 23, 229–235. [Google Scholar] [CrossRef]
- Jones, K.L.; Van de Water, J. Maternal autoantibody related autism: Mechanisms and pathways. Mol. Psychiatry 2019, 24, 252–265. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Atladóttir, H.O.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Parner, E.T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, H.; Gressens, P.; Mallard, C. Inflammation during fetal and neonatal life: Implications for neurologic and neuropsychiatric disease in children and adults. Ann. Neurol. 2012, 71, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, M.D.; Frigon, C.; Graham, A.S.; Mueller, B.A. Maternal infection and risk of cerebral palsy in term and preterm infants. J. Perinatol. 2005, 25, 108–113. [Google Scholar] [CrossRef]
- Humberg, A.; Fortmann, I.; Siller, B.; Kopp, M.V.; Herting, E.; Göpel, W.; Härtel, C. German Neonatal Network GCFLRAPIATBOLPRIMALC Preterm birth and sustained inflammation: Consequences for the neonate. Semin. Immunopathol. 2020, 42, 451–468. [Google Scholar] [CrossRef]
- Shi, Z.; Ma, L.; Luo, K.; Bajaj, M.; Chawla, S.; Natarajan, G.; Hagberg, H.; Tan, S. Chorioamnionitis in the Development of Cerebral Palsy: A Meta-analysis and Systematic Review. Pediatrics 2017, 139, e20163781. [Google Scholar] [CrossRef]
- Zhu, P.; Hao, J.H.; Tao, R.X.; Huang, K.; Jiang, X.M.; Zhu, Y.D.; Tao, F.B. Sex-specific and time-dependent effects of prenatal stress on the early behavioral symptoms of ADHD: A longitudinal study in China. Eur. Child Adolesc. Psychiatry 2015, 24, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.; Wainstock, T.; Braun, K.; Segal, M. Stress In Utero: Prenatal Programming of Brain Plasticity and Cognition. Biol. Psychiatry 2015, 78, 315–326. [Google Scholar] [CrossRef]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.M.; Green, J.C.; Desai, M.M.; Weitzman, C.C.; Rosenthal, M.S. Need and unmet need for care coordination among children with mental health conditions. Pediatrics 2014, 133, e530–e537. [Google Scholar] [CrossRef]
- Agorastos, A.; Pervanidou, P.; Chrousos, G.P.; Baker, D.G. Developmental Trajectories of Early Life Stress and Trauma: A Narrative Review on Neurobiological Aspects Beyond Stress System Dysregulation. Front. Psychiatry 2019, 10, 118. [Google Scholar] [CrossRef]
- Beversdorf, D.Q.; Manning, S.E.; Hillier, A.; Anderson, S.L.; Nordgren, R.E.; Walters, S.E.; Nagaraja, H.N.; Cooley, W.C.; Gaelic, S.E.; Bauman, M.L. Timing of prenatal stressors and autism. J. Autism Dev. Disord. 2005, 35, 471–478. [Google Scholar] [CrossRef]
- Khashan, A.S.; Abel, K.M.; McNamee, R.; Pedersen, M.G.; Webb, R.T.; Baker, P.N.; Kenny, L.C.; Mortensen, P.B. Higher risk of offspring schizophrenia following antenatal maternal exposure to severe adverse life events. Arch. Gen. Psychiatry 2008, 65, 146–152. [Google Scholar] [CrossRef]
- Kinney, D.K.; Munir, K.M.; Crowley, D.J.; Miller, A.M. Prenatal stress and risk for autism. Neurosci. Biobehav. Rev. 2008, 32, 1519–1532. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Olsen, J.; Vestergaard, M.; Obel, C. Attention-deficit/hyperactivity disorder in the offspring following prenatal maternal bereavement: A nationwide follow-up study in Denmark. Eur. Child Adolesc. Psychiatry 2010, 19, 747–753. [Google Scholar] [CrossRef]
- Ronald, A.; Pennell, C.E.; Whitehouse, A.J. Prenatal Maternal Stress Associated with ADHD and Autistic Traits in early Childhood. Front. Psychol. 2010, 1, 223. [Google Scholar] [CrossRef] [PubMed]
- Danese, A.; Moffitt, T.E.; Pariante, C.M.; Ambler, A.; Poulton, R.; Caspi, A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch. Gen. Psychiatry 2008, 65, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Wang, J.; Huang, K.; Wang, S.; Liu, H.; Wan, S.; Yan, S.; Hao, J.; Zhu, P.; Tao, F. Prenatal pregnancy-related anxiety predicts boys’ ADHD symptoms via placental C-reactive protein. Psychoneuroendocrinology 2020, 120, 104797. [Google Scholar] [CrossRef] [PubMed]
- Stoye, D.Q.; Blesa, M.; Sullivan, G.; Galdi, P.; Lamb, G.J.; Black, G.S.; Quigley, A.J.; Thrippleton, M.J.; Bastin, M.E.; Reynolds, R.M.; et al. Maternal cortisol is associated with neonatal amygdala microstructure and connectivity in a sexually dimorphic manner. eLife 2020, 9, e60729. [Google Scholar] [CrossRef]
- Chang, G. Maternal Substance Use: Consequences, Identification, and Interventions. Alcohol. Res. 2020, 40, 6. [Google Scholar] [CrossRef]
- Ross, E.J.; Graham, D.L.; Money, K.M.; Stanwood, G.D. Developmental consequences of fetal exposure to drugs: What we know and what we still must learn. Neuropsychopharmacology 2015, 40, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Forray, A. Substance use during pregnancy. F1000Research 2016, 5, 887. [Google Scholar] [CrossRef]
- Lees, B.; Mewton, L.; Jacobus, J.; Valadez, E.A.; Stapinski, L.A.; Teesson, M.; Tapert, S.F.; Squeglia, L.M. Association of Prenatal Alcohol Exposure With Psychological, Behavioral, and Neurodevelopmental Outcomes in Children From the Adolescent Brain Cognitive Development Study. Am. J. Psychiatry 2020, 177, 1060–1072. [Google Scholar] [CrossRef]
- O’Connor, M.J.; Paley, B. Psychiatric conditions associated with prenatal alcohol exposure. Dev. Disabil. Res. Rev. 2009, 15, 225–234. [Google Scholar] [CrossRef]
- Monnelly, V.J.; Hamilton, R.; Chappell, F.M.; Mactier, H.; Boardman, J.P. Childhood neurodevelopment after prescription of maintenance methadone for opioid dependency in pregnancy: A systematic review and meta-analysis. Dev. Med. Child Neurol. 2019, 61, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Bada, H.S.; Bann, C.M.; Whitaker, T.M.; Bauer, C.R.; Shankaran, S.; Lagasse, L.; Lester, B.M.; Hammond, J.; Higgins, R. Protective factors can mitigate behavior problems after prenatal cocaine and other drug exposures. Pediatrics 2012, 130, e1479–e1488. [Google Scholar] [CrossRef] [PubMed]
- Oei, J.L. Alcohol use in pregnancy and its impact on the mother and child. Addiction 2020, 115, 2148–2163. [Google Scholar] [CrossRef]
- Lester, B.M.; Lagasse, L.L. Children of addicted women. J. Addict. Dis 2010, 29, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Kirlic, N.; Newman, E.; Lagasse, L.L.; Derauf, C.; Shah, R.; Smith, L.M.; Arria, A.M.; Huestis, M.A.; Haning, W.; Strauss, A.; et al. Cortisol reactivity in two-year-old children prenatally exposed to methamphetamine. J. Stud. Alcohol. Drugs 2013, 74, 447–451. [Google Scholar] [CrossRef][Green Version]
- Lacagnina, M.J.; Rivera, P.D.; Bilbo, S.D. Glial and neuroimmune mechanisms as critical modulators of drug use and abuse. Neuropsychopharmacology 2017, 42, 156–177. [Google Scholar] [CrossRef]
- Danese, A.; Lewis, S.J. Psychoneuroimmunology of Early-Life Stress: The Hidden Wounds of Childhood Trauma. Neuropsychopharmacology 2017, 42, 99–114. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Q.; Wang, Y.; Hu, J.; Jiang, H.; Cheng, W.; Ma, Y.; Liu, M.; Sun, A.; Zhang, X.; et al. Duloxetine prevents the effects of prenatal stress on depressive-like and anxiety-like behavior and hippocampal expression of pro-inflammatory cytokines in adult male offspring rats. Int. J. Dev. Neurosci. 2016, 55, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Fonken, L.K.; Watkins, L.R.; Maier, S.F. Microglia: Neuroimmune-sensors of stress. Semin. Cell Dev. Biol. 2019, 94, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Cole, S.W. Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biol. Psychiatry 2012, 72, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Santana-Coelho, D.; Layne-Colon, D.; Valdespino, R.; Ross, C.C.; Tardif, S.D.; O’Connor, J.C. Advancing Autism Research From Mice to Marmosets: Behavioral Development of Offspring Following Prenatal Maternal Immune Activation. Front. Psychiatry 2021, 12, 705554. [Google Scholar] [CrossRef]
- Mueller, F.S.; Scarborough, J.; Schalbetter, S.M.; Richetto, J.; Kim, E.; Couch, A.; Yee, Y.; Lerch, J.P.; Vernon, A.C.; Weber-Stadlbauer, U. Behavioral, neuroanatomical, and molecular correlates of resilience and susceptibility to maternal immune activation. Mol. Psychiatry 2021, 26, 396–410. [Google Scholar] [CrossRef]
- Meyer, U.; Feldon, J. Neural basis of psychosis-related behaviour in the infection model of schizophrenia. Behav. Brain Res. 2009, 204, 322–334. [Google Scholar] [CrossRef]
- Mueller, F.S.; Richetto, J.; Hayes, L.N.; Zambon, A.; Pollak, D.D.; Sawa, A.; Meyer, U.; Weber-Stadlbauer, U. Influence of poly(I:C) variability on thermoregulation, immune responses and pregnancy outcomes in mouse models of maternal immune activation. Brain Behav. Immun. 2019, 80, 406–418. [Google Scholar] [CrossRef]
- Wu, W.L.; Hsiao, E.Y.; Yan, Z.; Mazmanian, S.K.; Patterson, P.H. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 2017, 62, 11–23. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; Patterson, P.H. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav. Immun. 2011, 25, 604–615. [Google Scholar] [CrossRef]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Yim, Y.S.; Park, A.; Berrios, J.; Lafourcade, M.; Pascual, L.M.; Soares, N.; Kim, J.Y.; Kim, S.; Kim, H.; Waisman, A. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 2017, 549, 482–487. [Google Scholar]
- Smolders, S.; Notter, T.; Smolders, S.M.T.; Rigo, J.M.; Brône, B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav. Immun. 2018, 73, 51–65. [Google Scholar] [CrossRef]
- Mattei, D.; Djodari-Irani, A.; Hadar, R.; Pelz, A.; de Cossío, L.F.; Goetz, T.; Matyash, M.; Kettenmann, H.; Winter, C.; Wolf, S.A. Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav. Immun. 2014, 38, 175–184. [Google Scholar] [CrossRef]
- Mattei, D.; Ivanov, A.; Ferrai, C.; Jordan, P.; Guneykaya, D.; Buonfiglioli, A.; Schaafsma, W.; Przanowski, P.; Deuther-Conrad, W.; Brust, P.; et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl. Psychiatry 2017, 7, e1120. [Google Scholar] [CrossRef]
- Hadar, R.; Dong, L.; del-Valle-Anton, L.; Guneykaya, D.; Voget, M.; Edemann-Callesen, H.; Schweibold, R.; Djodari-Irani, A.; Goetz, T.; Ewing, S.; et al. Deep brain stimulation during early adolescence prevents microglial alterations in a model of maternal immune activation. Brain Behav. Immun. 2017, 63, 71–80. [Google Scholar] [CrossRef]
- Ozaki, K.; Kato, D.; Ikegami, A.; Hashimoto, A.; Sugio, S.; Guo, Z.; Shibushita, M.; Tatematsu, T.; Haruwaka, K.; Moorhouse, A.J. Maternal immune activation induces sustained changes in fetal microglia motility. Sci. Rep. 2020, 10, 21378. [Google Scholar] [CrossRef]
- Schaafsma, W.; Basterra, L.B.; Jacobs, S.; Brouwer, N.; Meerlo, P.; Schaafsma, A.; Boddeke, E.W.G.M.; Eggen, B.J.L. Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol. Dis. 2017, 106, 291–300. [Google Scholar] [CrossRef]
- Clark, S.M.; Notarangelo, F.M.; Li, X.; Chen, S.; Schwarcz, R.; Tonelli, L.H. Maternal immune activation in rats blunts brain cytokine and kynurenine pathway responses to a second immune challenge in early adulthood. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 89, 286–294. [Google Scholar] [CrossRef]
- Chamera, K.; Szuster-Głuszczak, M.; Trojan, E.; Basta-Kaim, A. Maternal Immune Activation Sensitizes Male Offspring Rats to Lipopolysaccharide-Induced Microglial Deficits Involving the Dysfunction of CD200-CD200R and CX3CL1-CX3CR1 Systems. Cells 2020, 9, 1676. [Google Scholar] [CrossRef]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016, 233, 1637–1650. [Google Scholar] [CrossRef]
- Benmhammed, H.; El Hayek, S.; Nassiri, A.; Bousalham, R.; Mesfioui, A.; Ouichou, A.; El Hessni, A. Effects of lipopolysaccharide administration and maternal deprivation on anxiety and depressive symptoms in male and female Wistar rats: Neurobehavioral and biochemical assessments. Behav. Brain Res. 2019, 362, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, K.; Salazar, L.A. Epigenetics: A Missing Link Between Early Life Stress and Depression. Adv. Exp. Med. Biol. 2021, 1305, 117–128. [Google Scholar] [PubMed]
- Nishi, M. Effects of Early-Life Stress on the Brain and Behaviors: Implications of Early Maternal Separation in Rodents. Int. J. Mol. Sci. 2020, 21, 7212. [Google Scholar] [CrossRef]
- Huot, R.L.; Thrivikraman, K.V.; Meaney, M.J.; Plotsky, P.M. Development of adult ethanol preference and anxiety as a consequence of neonatal maternal separation in Long Evans rats and reversal with antidepressant treatment. Psychopharmacology 2001, 158, 366–373. [Google Scholar] [CrossRef]
- Huot, R.L.; Gonzalez, M.E.; Ladd, C.O.; Thrivikraman, K.V.; Plotsky, P.M. Foster litters prevent hypothalamic-pituitary-adrenal axis sensitization mediated by neonatal maternal separation. Psychoneuroendocrinology 2004, 29, 279–289. [Google Scholar] [CrossRef]
- Menard, J.L.; Champagne, D.L.; Meaney, M.J. Variations of maternal care differentially influence ‘fear’ reactivity and regional patterns of cFos immunoreactivity in response to the shock-probe burying test. Neuroscience 2004, 129, 297–308. [Google Scholar] [CrossRef]
- Gómez-González, B.; Escobar, A. Prenatal stress alters microglial development and distribution in postnatal rat brain. Acta Neuropathol. 2010, 119, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Pernía, O.; Carrero, P.; Garcia-Segura, L.M. Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J. Neuroinflamm. 2012, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Diz-Chaves, Y.; Astiz, M.; Bellini, M.J.; Garcia-Segura, L.M. Prenatal stress increases the expression of proinflammatory cytokines and exacerbates the inflammatory response to LPS in the hippocampal formation of adult male mice. Brain Behav. Immun. 2013, 28, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Ke, X.; Liu, Q.; Fu, Q.; Majnik, A.; Lane, R. Adverse early life environment increases hippocampal microglia abundance in conjunction with decreased neural stem cells in juvenile mice. Int. J. Dev. Neurosci. 2016, 55, 56–65. [Google Scholar] [CrossRef]
- Bittle, J.; Stevens, H.E. The role of glucocorticoid, interleukin-1β, and antioxidants in prenatal stress effects on embryonic microglia. J. Neuroinflamm. 2018, 15, 44. [Google Scholar] [CrossRef]
- Zhao, Q.; Peng, C.; Wu, X.; Chen, Y.; Wang, C.; You, Z. Maternal sleep deprivation inhibits hippocampal neurogenesis associated with inflammatory response in young offspring rats. Neurobiol. Dis. 2014, 68, 57–65. [Google Scholar] [CrossRef]
- Zhao, Q.; Xie, X.; Fan, Y.; Zhang, J.; Jiang, W.; Wu, X.; Yan, S.; Chen, Y.; Peng, C.; You, Z. Phenotypic dysregulation of microglial activation in young offspring rats with maternal sleep deprivation-induced cognitive impairment. Sci. Rep. 2015, 5, 9513. [Google Scholar] [CrossRef]
- Han, Y.; Wang, J.; Zhao, Q.; Xie, X.; Song, R.; Xiao, Y.; Kang, X.; Zhang, L.; Zhang, Y.; Peng, C.; et al. Pioglitazone alleviates maternal sleep deprivation-induced cognitive deficits in male rat offspring by enhancing microglia-mediated neurogenesis. Brain Behav. Immun. 2020, 87, 568–578. [Google Scholar] [CrossRef]
- Delpech, J.C.; Wei, L.; Hao, J.; Yu, X.; Madore, C.; Butovsky, O.; Kaffman, A. Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav. Immun. 2016, 57, 79–93. [Google Scholar] [CrossRef]
- Roque, A.; Ochoa-Zarzosa, A.; Torner, L. Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav. Immun. 2016, 55, 39–48. [Google Scholar] [CrossRef]
- Takatsuru, Y.; Nabekura, J.; Ishikawa, T.; Kohsaka, S.-I.; Koibuchi, N. Early-life stress increases the motility of microglia in adulthood. J. Physiol. Sci. 2015, 65, 187–194. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, L.; Sun, L.; Xie, Y.; Xiao, L.; Wang, H.; Wang, G. Brief postpartum separation from offspring promotes resilience to lipopolysaccharide challenge-induced anxiety and depressive-like behaviors and inhibits neuroinflammation in C57BL/6J dams. Brain Behav. Immun. 2021, 95, 190–202. [Google Scholar] [CrossRef]
- Thayer, Z.M.; Wilson, M.A.; Kim, A.W.; Jaeggi, A.V. Impact of prenatal stress on offspring glucocorticoid levels: A phylogenetic meta-analysis across 14 vertebrate species. Sci. Rep. 2018, 8, 4942. [Google Scholar] [CrossRef]
- Shi, D.D.; Zhang, Y.D.; Ren, Y.Y.; Peng, S.Y.; Yuan, T.F.; Wang, Z. Predictable maternal separation confers adult stress resilience via the medial prefrontal cortex oxytocin signaling pathway in rats. Mol. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Peña, C.J.; Kronman, H.G.; Walker, D.M.; Cates, H.M.; Bagot, R.C.; Purushothaman, I.; Issler, O.; Loh, Y.E.; Leong, T.; Kiraly, D.D.; et al. Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area OTX2. Science 2017, 356, 1185–1188. [Google Scholar] [CrossRef]
- Pascual, M.; Montesinos, J.; Montagud-Romero, S.; Forteza, J.; Rodríguez-Arias, M.; Miñarro, J.; Guerri, C. TLR4 response mediates ethanol-induced neurodevelopment alterations in a model of fetal alcohol spectrum disorders. J. Neuroinflamm. 2017, 14, 145. [Google Scholar] [CrossRef]
- Kane, C.J.; Phelan, K.D.; Han, L.; Smith, R.R.; Xie, J.; Douglas, J.C.; Drew, P.D. Protection of neurons and microglia against ethanol in a mouse model of fetal alcohol spectrum disorders by peroxisome proliferator-activated receptor-γ agonists. Brain Behav. Immun. 2011, 25 (Suppl. S1), S137–S145. [Google Scholar] [CrossRef] [PubMed]
- Drew, P.D.; Johnson, J.W.; Douglas, J.C.; Phelan, K.D.; Kane, C.J. Pioglitazone blocks ethanol induction of microglial activation and immune responses in the hippocampus, cerebellum, and cerebral cortex in a mouse model of fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2015, 39, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Topper, L.A.; Baculis, B.C.; Valenzuela, C.F. Exposure of neonatal rats to alcohol has differential effects on neuroinflammation and neuronal survival in the cerebellum and hippocampus. J. Neuroinflamm. 2015, 12, 160. [Google Scholar] [CrossRef] [PubMed]
- Ahlers, K.E.; Karaçay, B.; Fuller, L.; Bonthius, D.J.; Dailey, M.E. Transient activation of microglia following acute alcohol exposure in developing mouse neocortex is primarily driven by BAX-dependent neurodegeneration. Glia 2015, 63, 1694–1713. [Google Scholar] [CrossRef] [PubMed]
- Boschen, K.E.; Ruggiero, M.J.; Klintsova, A.Y. Neonatal binge alcohol exposure increases microglial activation in the developing rat hippocampus. Neuroscience 2016, 324, 355–366. [Google Scholar] [CrossRef]
- Komada, M.; Hara, N.; Kawachi, S.; Kawachi, K.; Kagawa, N.; Nagao, T.; Ikeda, Y. Mechanisms underlying neuro-inflammation and neurodevelopmental toxicity in the mouse neocortex following prenatal exposure to ethanol. Sci. Rep. 2017, 7, 4934. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Wang, X.; Xu, M.; Frank, J.A.; Luo, J. Minocycline attenuates ethanol-induced cell death and microglial activation in the developing spinal cord. Alcohol 2019, 79, 25–35. [Google Scholar] [CrossRef]
- Shrivastava, P.; Cabrera, M.A.; Chastain, L.G.; Boyadjieva, N.I.; Jabbar, S.; Franklin, T.; Sarkar, D.K. Mu-opioid receptor and delta-opioid receptor differentially regulate microglial inflammatory response to control proopiomelanocortin neuronal apoptosis in the hypothalamus: Effects of neonatal alcohol. J. Neuroinflamm. 2017, 14, 83. [Google Scholar] [CrossRef]
- Chastain, L.G.; Franklin, T.; Gangisetty, O.; Cabrera, M.A.; Mukherjee, S.; Shrivastava, P.; Jabbar, S.; Sarkar, D.K. Early life alcohol exposure primes hypothalamic microglia to later-life hypersensitivity to immune stress: Possible epigenetic mechanism. Neuropsychopharmacology 2019, 44, 1579–1588. [Google Scholar] [CrossRef]
- Linker, K.E.; Cross, S.J.; Leslie, F.M. Glial mechanisms underlying substance use disorders. Eur. J. Neurosci. 2019, 50, 2574–2589. [Google Scholar] [CrossRef]
- Stellwagen, D.; Kemp, G.M.; Valade, S.; Chambon, J. Glial regulation of synaptic function in models of addiction. Curr. Opin. Neurobiol. 2019, 57, 179–185. [Google Scholar] [CrossRef]
- Catale, C.; Bussone, S.; Lo Iacono, L.; Carola, V. Microglial alterations induced by psychoactive drugs: A possible mechanism in substance use disorder. Semin. Cell Dev. Biol. 2019, 94, 164–175. [Google Scholar] [CrossRef]
- Singh, G.; Segura, B.J.; Georgieff, M.K.; Gisslen, T. Fetal inflammation induces acute immune tolerance in the neonatal rat hippocampus. J. Neuroinflamm. 2021, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, Z.; Lin, W.; Yan, J.; Zhu, C.; Yin, D.; He, S.; Su, Y.; Xu, N.; Caldwell, R.W.; et al. Modulating microglia activation prevents maternal immune activation induced schizophrenia-relevant behavior phenotypes via arginase 1 in the dentate gyrus. Neuropsychopharmacology 2020, 45, 1896–1908. [Google Scholar] [CrossRef]
- Zhu, F.; Zheng, Y.; Liu, Y.; Zhang, X.; Zhao, J. Minocycline alleviates behavioral deficits and inhibits microglial activation in the offspring of pregnant mice after administration of polyriboinosinic-polyribocytidilic acid. Psychiatry Res. 2014, 219, 680–686. [Google Scholar] [CrossRef]
- Corripio, I.; Roldán, A.; McKenna, P.; Sarró, S.; Alonso-Solís, A.; Salgado, L.; Álvarez, E.; Molet, J.; Pomarol-Clotet, E.; Portella, M. Target selection for deep brain stimulation in treatment resistant schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 112, 110436. [Google Scholar] [CrossRef]
- Sendi, M.S.E.; Waters, A.C.; Tiruvadi, V.; Riva-Posse, P.; Crowell, A.; Isbaine, F.; Gale, J.T.; Choi, K.S.; Gross, R.E.; SMayberg, H.; et al. Intraoperative neural signals predict rapid antidepressant effects of deep brain stimulation. Transl. Psychiatry 2021, 11, 551. [Google Scholar] [CrossRef]
- Dougherty, D.D. Deep Brain Stimulation: Clinical Applications. Psychiatr. Clin. N. Am. 2018, 41, 385–394. [Google Scholar] [CrossRef]
- Lippmann, B.; Barmashenko, G.; Funke, K. Effects of repetitive transcranial magnetic and deep brain stimulation on long-range synchrony of oscillatory activity in a rat model of developmental schizophrenia. Eur. J. Neurosci. 2021, 53, 2848–2869. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, Q.; Wang, J.; Tang, M.; Huang, S.; Peng, K.; Han, Y.; Zhang, J.; Liu, G.; Fang, Q.; et al. Maternal immune activation-induced PPARγ-dependent dysfunction of microglia associated with neurogenic impairment and aberrant postnatal behaviors in offspring. Neurobiol. Dis. 2019, 125, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Miyaoka, T.; Wake, R.; Hashioka, S.; Hayashida, M.; Oh-Nishi, A.; Azis, I.A.; Izuhara, M.; Tsuchie, K.; Araki, T.; Arauchi, R.; et al. Remission of Psychosis in Treatment-Resistant Schizophrenia following Bone Marrow Transplantation: A Case Report. Front. Psychiatry 2017, 8, 174. [Google Scholar] [CrossRef]
- Andoh, M.; Shibata, K.; Okamoto, K.; Onodera, J.; Morishita, K.; Miura, Y.; Ikegaya, Y.; Koyama, R. Exercise Reverses Behavioral and Synaptic Abnormalities after Maternal Inflammation. Cell Rep. 2019, 27, 2817–2825. [Google Scholar] [CrossRef]
- Bale, T.L. Lifetime stress experience: Transgenerational epigenetics and germ cell programming. Dialogues Clin. Neurosci. 2014, 16, 297–305. [Google Scholar]
- Weber-Stadlbauer, U.; Richetto, J.; Labouesse, M.A.; Bohacek, J.; Mansuy, I.M.; Meyer, U. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol. Psychiatry 2017, 22, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Ronovsky, M.; Berger, S.; Zambon, A.; Reisinger, S.N.; Horvath, O.; Pollak, A.; Lindtner, C.; Berger, A.; Pollak, D.D. Maternal immune activation transgenerationally modulates maternal care and offspring depression-like behavior. Brain Behav. Immun. 2017, 63, 127–136. [Google Scholar] [CrossRef]
- Ambeskovic, M.; Babenko, O.; Ilnytskyy, Y.; Kovalchuk, I.; Kolb, B.; Metz, G.A.S. Ancestral Stress Alters Lifetime Mental Health Trajectories and Cortical Neuromorphology via Epigenetic Regulation. Sci. Rep. 2019, 9, 6389. [Google Scholar] [CrossRef]
- Weber-Stadlbauer, U.; Richetto, J.; Zwamborn, R.A.J.; Slieker, R.C.; Meyer, U. Transgenerational modification of dopaminergic dysfunctions induced by maternal immune activation. Neuropsychopharmacology 2021, 46, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Ambeskovic, M.; Roseboom, T.J.; Metz, G.A.S. Transgenerational effects of early environmental insults on aging and disease incidence. Neurosci. Biobehav. Rev. 2020, 117, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Taouk, L.; Schulkin, J. Transgenerational transmission of pregestational and prenatal experience: Maternal adversity, enrichment, and underlying epigenetic and environmental mechanisms. J. Dev. Orig. Health Dis. 2016, 7, 588–601. [Google Scholar] [CrossRef]
- Katzmarski, N.; Domínguez-Andrés, J.; Cirovic, B.; Renieris, G.; Ciarlo, E.; Le Roy, D.; Lepikhov, K.; Kattler, K.; Gasparoni, G.; Händler, K.; et al. Transmission of trained immunity and heterologous resistance to infections across generations. Nat. Immunol. 2021, 22, 1382–1390. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Kurtz, J. Specific memory within innate immune systems. Trends Immunol. 2005, 26, 186–192. [Google Scholar] [CrossRef]
- Rodrigues, J.; Brayner, F.A.; Alves, L.C.; Dixit, R.; Barillas-Mury, C. Hemocyte differentiation mediates innate immune memory in Anopheles gambiae mosquitoes. Science 2010, 329, 1353–1355. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W. Trained immunity: A memory for innate host defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Jalabi, W.; Shpargel, K.B.; Farabaugh, K.T.; Dutta, R.; Yin, X.; Kidd, G.J.; Bergmann, C.C.; Stohlman, S.A.; Trapp, B.D. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J. Neurosci. 2012, 32, 11706–11715. [Google Scholar] [CrossRef]
- Wendeln, A.C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef]
- Neher, J.J.; Cunningham, C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019, 40, 358–374. [Google Scholar] [CrossRef]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507. [Google Scholar] [CrossRef]
- Feng, Y.W.; Wu, C.; Liang, F.Y.; Lin, T.; Li, W.Q.; Jing, Y.H.; Dai, P.; Yu, H.X.; Lan, Y.; Pei, Z.; et al. hUCMSCs Mitigate LPS-Induced Trained Immunity in Ischemic Stroke. Front. Immunol. 2020, 11, 1746. [Google Scholar] [CrossRef]
- Palin, K.; Cunningham, C.; Forse, P.; Perry, V.H.; Platt, N. Systemic inflammation switches the inflammatory cytokine profile in CNS Wallerian degeneration. Neurobiol. Dis. 2008, 30, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Campion, S.; Lunnon, K.; Murray, C.L.; Woods, J.F.; Deacon, R.M.; Rawlins, J.N.; Perry, V.H. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 2009, 65, 304–312. [Google Scholar] [CrossRef]
- Pott Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic IL-1 exacerbates neurodegeneration and motor symptoms in a model of Parkinson’s disease. Brain 2008, 131, 1880–1894. [Google Scholar] [CrossRef] [PubMed]
- Ramaglia, V.; Hughes, T.R.; Donev, R.M.; Ruseva, M.M.; Wu, X.; Huitinga, I.; Baas, F.; Neal, J.W.; Morgan, B.P. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 965–970. [Google Scholar] [CrossRef]
- Raj, D.D.; Jaarsma, D.; Holtman, I.R.; Olah, M.; Ferreira, F.M.; Schaafsma, W.; Brouwer, N.; Meijer, M.M.; de Waard, M.C.; van der Pluijm, I.; et al. Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol. Aging 2014, 35, 2147–2160. [Google Scholar] [CrossRef]
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease tolerance as a defense strategy. Science 2012, 335, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Vachharajani, V.; McCall, C.E. Epigenetic and metabolic programming of innate immunity in sepsis. Innate Immun. 2019, 25, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qin, Y.; Dai, A.; Zhang, Y.; Xue, H.; Ni, H.; Han, L.; Zhu, L.; Yuan, D.; Tao, T.; et al. SMAD4 is Involved in the Development of Endotoxin Tolerance in Microglia. Cell. Mol. Neurobiol. 2016, 36, 777–788. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, X.; Xia, L.P.; Lv, H.; Lou, F.; Ren, Y.; He, Z.Y.; Luo, X.G. Peripheral immune tolerance alleviates the intracranial lipopolysaccharide injection-induced neuroinflammation and protects the dopaminergic neurons from neuroinflammation-related neurotoxicity. J. Neuroinflamm. 2017, 14, 223. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Xie, X.; Liu, Y.; Luo, X. Peripheral Blood Monocyte Tolerance Alleviates Intraperitoneal Lipopolysaccharides-Induced Neuroinflammation in Rats Via Upregulating the CD200R Expression. Neurochem. Res. 2017, 42, 3019–3032. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Norgard, M.A.; Levasseur, P.R.; Olson, B.; Burfeind, K.G.; Buenafe, A.C.; Zhu, X.; Jeng, S.; McWeeney, S.K.; Marks, D.L. Persistent Toll-like receptor 7 stimulation induces behavioral and molecular innate immune tolerance. Brain Behav. Immun. 2019, 82, 338–353. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Lang, G.P.; Haas, F.; Williams, D.L.; Hudalla, H.; Bauer, M.; Groth, M.; Wetzker, R.; Bauer, R. Memory-Like Inflammatory Responses of Microglia to Rising Doses of LPS: Key Role of PI3Kγ. Front. Immunol. 2019, 10, 2492. [Google Scholar] [CrossRef] [PubMed]
- Lajqi, T.; Stojiljkovic, M.; Williams, D.L.; Hudalla, H.; Bauer, M.; Witte, O.W.; Wetzker, R.; Bauer, R.; Schmeer, C. Memory-Like Responses of Brain Microglia Are Controlled by Developmental State and Pathogen Dose. Front. Immunol. 2020, 11, 546415. [Google Scholar] [CrossRef]
- Ciernia, A.V.; Link, V.M.; Careaga, M.; LaSalle, J.M.; Ashwood, P. Genetic variants drive altered epigenetic regulation of endotoxin response in BTBR macrophages. Brain Behav. Immun. 2020, 89, 20–31. [Google Scholar] [CrossRef]
- Barrett, T.J.; Corr, E.M.; van Solingen, C.; Schlamp, F.; Brown, E.J.; Koelwyn, G.J.; Lee, A.H.; Shanley, L.C.; Spruill, T.M.; Bozal, F.; et al. Chronic stress primes innate immune responses in mice and humans. Cell Rep. 2021, 36, 109595. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Low, D.; Silvin, A.; Chen, J.; Grisel, P.; Schulte-Schrepping, J.; Blecher, R.; Ulas, T.; Squarzoni, P.; Hoeffel, G.; et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 2018, 172, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Moorlag, S.J.C.F.M.; Khan, N.; Novakovic, B.; Kaufmann, E.; Jansen, T.; van Crevel, R.; Divangahi, M.; Netea, M.G. β-Glucan Induces Protective Trained Immunity against Mycobacterium tuberculosis Infection: A Key Role for IL-1. Cell Rep. 2020, 31, 107634. [Google Scholar] [CrossRef]
- Marakalala, M.J.; Williams, D.L.; Hoving, J.C.; Engstad, R.; Netea, M.G.; Brown, G.D. Dectin-1 plays a redundant role in the immunomodulatory activities of β-glucan-rich ligands in vivo. Microbes Infect. 2013, 15, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Benn, C.S.; Joosten, L.A.; Jacobs, C.; van Loenhout, J.; Xavier, R.J.; Aaby, P.; van der Meer, J.W.; et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J. Innate Immun. 2014, 6, 152–158. [Google Scholar] [CrossRef]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.-C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef]
- Domínguez-Andrés, J.; Novakovic, B.; Li, Y.; Scicluna, B.P.; Gresnigt, M.S.; Arts, R.J.W.; Oosting, M.; Moorlag, S.J.C.F.M.; Groh, L.A.; Zwaag, J.; et al. The Itaconate Pathway Is a Central Regulatory Node Linking Innate Immune Tolerance and Trained Immunity. Cell Metab. 2019, 29, 211–220. [Google Scholar] [CrossRef]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.; Rao, N.A.; Aghajanirefah, A.; et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef] [PubMed]
- Arts, R.J.; Novakovic, B.; Ter Horst, R.; Carvalho, A.; Bekkering, S.; Lachmandas, E.; Rodrigues, F.; Silvestre, R.; Cheng, S.C.; Wang, S.Y.; et al. Glutaminolysis and Fumarate Accumulation Integrate Immunometabolic and Epigenetic Programs in Trained Immunity. Cell Metab. 2016, 24, 807–819. [Google Scholar] [CrossRef]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; Ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.C.; Chou, C.H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Andrés, J.; Joosten, L.A.; Netea, M.G. Induction of innate immune memory: The role of cellular metabolism. Curr. Opin. Immunol. 2019, 56, 10–16. [Google Scholar] [CrossRef]
- Schaafsma, W.; Zhang, X.; van Zomeren, K.C.; Jacobs, S.; Georgieva, P.B.; Wolf, S.A.; Kettenmann, H.; Janova, H.; Saiepour, N.; Hanisch, U.K.; et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav. Immun. 2015, 48, 205–221. [Google Scholar] [CrossRef]
- Bernier, L.P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat. Commun. 2020, 11, 1559. [Google Scholar] [CrossRef]
- Zhang, X.; Kracht, L.; Lerario, A.M.; Dubbelaar, M.L.; Brouwer, N.; Wesseling, E.M.; Boddeke, E.W.G.M.; Eggen, B.J.L.; Kooistra, S.M. Epigenetic Regulation of Innate Immune Memory in Microglia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Catale, C.; Bussone, S.; Lo Iacono, L.; Viscomi, M.T.; Palacios, D.; Troisi, A.; Carola, V. Exposure to different early-life stress experiences results in differentially altered DNA methylation in the brain and immune system. Neurobiol. Stress 2020, 13, 100249. [Google Scholar]
- Baghel, M.S.; Singh, B.; Patro, N.; Khanna, V.K.; Patro, I.K.; Thakur, M.K. Poly (I:C) Exposure in Early Life Alters Methylation of DNA and Acetylation of Histone at Synaptic Plasticity Gene Promoter in Developing Rat Brain Leading to Memory Impairment. Ann. Neurosci. 2019, 26, 35–41. [Google Scholar] [CrossRef]
- Chamera, K.; Kotarska, K.; Szuster-Głuszczak, M.; Trojan, E.; Skórkowska, A.; Pomierny, B.; Krzyżanowska, W.; Bryniarska, N.; Basta-Kaim, A. The prenatal challenge with lipopolysaccharide and polyinosinic:polycytidylic acid disrupts CX3CL1-CX3CR1 and CD200-CD200R signalling in the brains of male rat offspring: A link to schizophrenia-like behaviours. J. Neuroinflamm. 2020, 17, 247. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, K.; Missault, S.; Fransen, E.; Raeymaekers, L.; Willems, R.; Drinkenburg, W.; Timmermans, J.P.; Kumar-Singh, S.; Dedeurwaerdere, S. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav. Brain Res. 2014, 258, 179–186. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Riksen, N.P.; Netea, M.G. Immunometabolic control of trained immunity. Mol. Asp. Med. 2021, 77, 100897. [Google Scholar] [CrossRef]
- Ifrim, D.C.; Quintin, J.; Meerstein-Kessel, L.; Plantinga, T.S.; Joosten, L.A.; van der Meer, J.W.; van de Veerdonk, F.L.; Netea, M.G. Defective trained immunity in patients with STAT-1-dependent chronic mucocutaneaous candidiasis. Clin. Exp. Immunol. 2015, 181, 434–440. [Google Scholar] [CrossRef]
- Kang, K.; Park, S.H.; Chen, J.; Qiao, Y.; Giannopoulou, E.; Berg, K.; Hanidu, A.; Li, J.; Nabozny, G.; Kang, K.; et al. Interferon-γ Represses M2 Gene Expression in Human Macrophages by Disassembling Enhancers Bound by the Transcription Factor MAF. Immunity 2017, 47, 235–250. [Google Scholar] [CrossRef]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Bachu, M.; Park, S.H.; Kang, K.; Bae, S.; Park-Min, K.H.; Ivashkiv, L.B. IFN-γ selectively suppresses a subset of TLR4-activated genes and enhancers to potentiate macrophage activation. Nat. Commun. 2019, 10, 3320. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Scicluna, B.P.; Arts, R.J.; Gresnigt, M.S.; Lachmandas, E.; Giamarellos-Bourboulis, E.J.; Kox, M.; Manjeri, G.R.; Wagenaars, J.A.; Cremer, O.L.; et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 2016, 17, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada González, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Model | Tissue Phenotype | Microglia Phenotype | Reactivity | Citation | |||
|---|---|---|---|---|---|---|---|
| 2nd Stim | Response | ||||||
| MIA | Rat, E15, PIC | ↑IL-1β and TNFα (RNA) in whole Hpc | ↑MG density in NAc, ↑Iba1 (protein) in the Cb and Hpc | none | [67] | Mattei 2014 | |
| MIA | Mouse, E15, PIC | ↑Tspo binding and IL-6 (protein) in whole Hpc | ↑Iba1 and CD18 (protein) in Cb and Hpc, ↓MG phagocytosis in Hpc | none | [68] | Mattei 2017 | |
| MG RNA-seq: ↓genes inflammatory response, phagocytosis, and cell migration. ↑genes for synaptic plasticity, VEGF signaling, and glial cell migration | |||||||
| MIA | Rat, E15, PIC | ↑MG density and soma size in NAc and Hpc, ↑MHCII (protein) in Ctx | none | [69] | Hadar 2017 | ||
| MIA | Mouse, E12 or E15, PIC | ↑MG motility (velocity) at E18, ↓MG motility (velocity) at P10 | LPS | ↑MG directional motility at P42 (after E12 MIA) | [70] | Ozaki 2020 | |
| MIA | Mouse, E15–16–17, LPS | LPS | ↓IL-1β, TNFα, and IL-6 (RNA) in MG from the whole brain | [71] | Schaafsma 2017 | ||
| ↑IL-1β (RNA) in MG from Hpc | |||||||
| ↑IL-1β, TNFα, and IL-6 (RNA) in whole Hpc | |||||||
| MIA | Rat, E15, PIC | At P35: ↑TNFα ↓IL-4 and IL-10 (RNA) in whole brain | LPS at P35 | ↑IL-1β, ↓IL-6, IL-4 and IL-10 (RNA) in the whole brain | [72] | Clark 2019 | |
| LPS at P60 | ↓IL-6, TNFα, IFNγ, and IL-10 (RNA) in the whole brain | ||||||
| MIA | Rat, E7, E10, E13, E16, E19, LPS | ↑CD200R (RNA), ↓CD200R (protein) in FC | LPS | mild changes in gene expression of inflammatory molecules in FC and Hpc in MIA-responsive and MIA-non-responsive mice | [73] | Chamera 2020 | |
| MIA | FIRS: Rat, E20, intra-amniotic LPS | LPS at P5 | ↓activated MG density, ↑IL-1β, ↓IL-6, TNFα, Cxcl10, Ccl2 (RNA) in the Hpc | [109] | Singh 2021 | ||
| ELS | Rat, mild random stress, E4–20 | ↑IL-6 (RNA) in Hpc | [54] | Zhang 2016 | |||
| ELS | Rat, maternal forced swim, E10–20 | ↑ramified MG density and ↓ameboid MG density | none | [81] | Gomez-Gonzalez 2010 | ||
| ELS | Mouse, maternal restraint stress, E12 –E20 | ↑IL-1β (RNA) in Hpc | ↑MG density, ↑MG activated morphology | LPS | ↑IL-6, TNFα, IP10 (RNA) in the Hpc, ↑MG with activated morphology | [82] | Diz-Chavez 2012 |
| ELS | Mouse, maternal restraint stress, E12 –E20 | ↑IL-1β and TNFα (RNA) in Hpc | ↑MG activated morphology in Hpc | LPS | ↑TNFα in the Hpc, ↑MG density in Hpc | [83] | Diz-Chavez 2013 |
| ELS | Mouse, stress environment, E13–17 | ↑Aif1 and Tlr9 (RNA & protein) in Hpc | ↑MG density in Hpc | none | [84] | Cohen 2016 | |
| ELS | Mouse, maternal restraint stress, E12 –E20 | ↑MG total density, ↑density of ameboid MG | none | [85] | Bittle 2018 | ||
| ELS | Mouse, IL-1β, E12–13 | ↑MG total density, ↑density of ameboid MG | |||||
| ELS | Mouse, corticosterone, E12–13 | ↑MG total density, ↑density of ameboid MG | |||||
| ELS | Rat, 72H maternal sleep deprivation, E4, E9, or E18 | ↑IL-1β, TNFα, IL-6 (RNA), ↓IL-10 (RNA) in the Hpc | ↑MG activated morphology | none | [86] | Zhao 2014 | |
| ELS | Rat, 72H maternal sleep deprivation, E18 | ↑IL-1β, TNFα, IL-6, CD68, iNOS, ↓IL-10, IL-4, Ym1, Arg1, Cd206 in the Hpc | ↑MG density, ↑Iba1 (protein) in Hpc | none | [87] | Zhao 2015 | |
| ELS | Rat, 72H maternal sleep deprivation, E18 | ↑IL-1β, TNFα, IL-6, ↓IL-10, IL-4, Ym1, Arg1 in the Hpc | ↑MG density with activated morphology | none | [88] | Han 2020 | |
| ELS | Mouse, maternal seperation, P1–21 | ↑MG density and MG density with activated morphology at P14, ↑MG phagocytosis at P28 in Hpc, MG RNA-seq: altered immune modulators | none | [89] | Delpech 2016 | ||
| ELS | Rat, maternal separation, P1–14 | ↑IL-1β, TNFα in Hpc | ↑MG density with activated morphology | 3H maternal separation | ↑IL-1β, ↓TNFα in Hpc, ↑TNFα and IL-6 in Hypo, ↑corticosterone | [90] | Roque 2016 |
| ELS | Mouse, maternal seperation, P2–14 | ↑MG motility | [91] | Takasturu 2015 | |||
| ELS | Mouse, 180 min maternal separation, P1–21 | LPS | ↑Iba1 in Hpc | [92] | Wu 2021 | ||
| ELS | Mouse, 15 min maternal separation, P1–21 | ↓Iba1, Nlrp3, IL-18, NFκB in the Hpc | |||||
| EtOH | Mouse, 10% drink, preconception to P21 | ↑IL-1β, Cxcl1, MCP1, MIP1α, IL-17, CD11b, and MHCII (protein) in whole Ctx | ↑Iba1 (protein) in the Ctx | none | [96] | Pascual 2017 | |
| EtOH | Mouse, 3.5g/kg EtOH, P2–9 | ↑MG density in the Cb | none | [97] | Kane 2011 | ||
| EtOH | Mouse, 4 g/kg EtOH, P4–9 | ↑IL-1β and TNFα (RNA) in Ctx, Hpc, and Cb | ↑MG activated morphology in the Hpc, Ctx, Cb | none | [98] | Drew 2015 | |
| EtOH | Rat, 4h EtOH vapor, P3–5 | ↑IL-1β and TNFα (RNA) in Cb | ↑MG activated morphology in the Cb | none | [99] | Topper 2015 | |
| EtOH | Mouse, 3–5 g/kg EtOH, P7–8 | ↑IL-1β and TNFα (RNA) in Ctx | ↑density of ameboid MG, ↑MG activated morphology, ↑Itgb2, P2ry12 (RNA), ↑CD68 (protein) in the Ctx | none | [100] | Ahlers 2015 | |
| EtOH | Mouse, 2 g/kg EtOH, E6–E18 | ↑TNFα, IL-12a, IL-10 (RNA), ↓IL-6 and TGFβ (RNA) in the Ctx | ↑MG density, ↑MG activated morphology in the Ctx | none | [102] | Komada 2017 | |
| EtOH | Mouse, 2.5 g/kg EtOH, P5 | ↑MCP1 and IL-6 (protein) in the spinal cord | ↑Iba1, CD68, and P2×7 (protein) in MG in the spinal cord | none | [103] | Ren 2019 | |
| EtOH | Rat, 2.5 g/kg EtOH, P2–6 | ↑TNFα, MCP1, Csf1r, and TLR4 (RNA) in Hypo | ↑MG density, ↑MG activated morphology, ↑Iba1 protein intensity in the Hypo | none | [104] | Shrivastava 2017 | |
| EtOH | Rat, 2.5 mg/kg EtOH, P2–6 | ↑TNFα, IL-6, Csf1r, and TLR4 (RNA) in Hypo | ↑activated MG density in the Hypo | LPS | ↑IL-6 and TNFα (RNA) in MG, ↑activated MG density in the Hypo | [105] | Chastain 2019 |
| Cell-Type | Model | Immune Paradigm | 1st Stim | Reactivity | Metabolic Intervention | Citation | ||
|---|---|---|---|---|---|---|---|---|
| 2nd Stim | Response | |||||||
| Monocytes | Human primary monocytes | Training | b-glucan | N/A | ↑ TNFα, IL-6, HIFα and mTOR pathway, ↑ glycolysis and H3K27Ac hallmark in relevant promoter regions | Training is blocked with metformin and wortmanin | [160] | Cheng 2014 |
| Monocytes | Human primary monocytes | Training | b-glucan or fumarate | LPS | ↑ TNFα and IL-6, ↑ glycolysis, glutaminolysis and cholesterol synthesis | Training is blocked by metformin | [161] | Arts 2016 |
| Monocytes | Tolerance | LPS | LPS | ↓ glycolysis, glutamynolysis, and cholesterol synthesis | ||||
| Monocytes | Human primary monocytes | Training | b-glucan | LPS | ↑ TNFα, ↑ glycolysis, cholesterol synthesis pathway, and TCA cycle, ↑ H3K27Ac hallamrk in relevant promoter regions | Training is blocked by fluvastatin (through cholesterol synthesis pathway) | [162] | Bekkering 2018 |
| Monocytes | Training | Mevalonate | LPS | ↑ TNFα, ↑ glycolysis and TCA cycle, ↑ H3K27Ac hallamark in relevant promoter regions | ||||
| Monocytes | Human primary monocytes from patients with IgD syndrome | Training | Accumulation of mevalonate caused by mutations in mevalonate kinase | LPS | ↑ TNFα, IL1β, and IL6, ↑ glycolysis and mTOR pathway | |||
| Monocytes | Human BMDMs | Tolerance | LPS | LPS | ↓ TNFα, IL1β, IL12, and IL6 | Tolerance is blocked in the absence of Glutamine and a-ketoglutarate | [163] | Liu 2017 |
| Monocytes | Human monocytes from sepsis patients | Tolerance | Sepsis | LPS | Trancriptomic data determined ↓ of ample array of inflammatory interleukines and chemokines. Although their expression is ↑ at basal levels (w/o LPS stimulation) | [164] | Shalova 2015 | |
| Monocytes | PBMCs from chronic mucocutaneous candidiais patients | Impaired Training | STAT1 mutation | b-glucan, LPS | Equal TNFα and IL-6 as stimulated control | [176] | Ifrim 2015 | |
| Monocytes | PBMCs from hyper- immunoglobulinaemia E syndrome | Training | STAT3 mutation | b-glucan, LPS | ↑ TNFα, IL6 | |||
| Monocytes | Human primary monocytes from LPS exposed human | Tolerance | LPS | LPS | Epigenetic and transcriptomic data determined ↓ signatures of permissive hallmarks | [179] | Novakovick 2016 | |
| Monocytes | Human primary monocytes from LPS exposed human | Impaired Tolerance | LPS | b-glucan+LPS | Epigenetic and transcriptomic data determined ↑ signatures of permissive marks | |||
| Monocytes | Human primary monocytes | Training | b-glucan | LPS | Epigenetic and transcriptomic data determined ↑ signatures of permissive marks | |||
| Monocytes | Human primary monocytes | Tolerance | LPS | LPS | Epigenetic and transcriptomic data determined ↓ signatures of permissive hallmarks | |||
| Monocytes | PBMCs from patients with sepsis | Tolerance | Sepsis | LPS | ↓ TNFα, IL1β, and IL-6, ↓ glycolysis | IFNγ (rescued tolerance) | [182] | Cheng 2016 |
| Microglia | APP23 mice | Training | 1×LPS | AD model/ Ab amyloid accumulation | ↓ IL-10 in brain, ↓ accumulation of Aβ, Epigenetic and transcriptomic data determined an involvement of HIFα, and ↑ signature of permissive marks | [134] | Wendeln 2018 | |
| Microglia | Tolerance | 4×LPS | AD model/ Ab amyloid accumulation | ↓ IL-1β in brain, ↑ accumulation of Ab, Epigenetic and transcriptomic data determined ↓ signature of permissive marks | ||||
| Microglia | Stroke mouse model | Training | 1×LPS | Ischemia | ↑ IL-1β, ↓ IL-10 in brain, Epigenetic and transcriptomic data determined an involvement of HIFa and ↑ signature of permissive marks | |||
| Microglia | Tolerance | 4×LPS | Ischemia | ↓ IL-1β in brain | ||||
| Microglia | Mouse primary microglia culture | Training | 1×Ab | ↑ IL-1β, ↑ glycolysis and ↓ TCA | [136] | Baik 2019 | ||
| Microglia | Mouse Primary microglia culture | Tolerance | 3×Ab | ↓ IL-1β, ↓ glycolysis | ||||
| Microglia | 5×FAD mice | Tolerance | Endogenous Ab aggregates | Aβ iv injection | ↓ IL-1β, TNFα, and CCL2 among others, ↓ motility | |||
| Microglia | 5×FAD mice | Impaired Tolerance | Intervention with IFNγ | ↑ TNFα, ↑ glycolysis, ↑ phagocytic capacity | IFNγ (rescued tolerance) | |||
| Microglia | Mouse primary microglia | Training | Low dose LPS | LPS | ↑ TNFα and IL-6 | [149] | Lajqi 2019 | |
| Microglia | Training | b-dectin | LPS | ↑ TNFα and IL-6 | ||||
| Microglia | Tolerance | High dose LPS | LPS | ↓ TNFα and IL-6 | ||||
| Microglia | P6–P12 mouse pups | Training | Low dose LPS | LPS | ↑ TNFα, IL-6, IL-1β, iNOS | [150] | Lajqi 2020 | |
| Microglia | Adult mice | Training | Low dose LPS | LPS | ↑ TNFα, IL-6, IL-1β, iNOS | |||
| Microglia | Aging mice | Ablated Training | Low dose LPS | LPS | Equal TNFα, IL-6, IL-1β, iNOs as in stimulated control | |||
| Microglia | P6–P12 mouse pups | Tolerance | High dose LPS | LPS | ↓ TNFα, IL-6, IL-1β, iNOS | |||
| Microglia | Adult mice | Tolerance | High dose LPS | LPS | ↓ TNFα, IL-6, IL-1β, iNOS | |||
| Microglia | Aging mice | Impaired Tolerance | High dose LPS | LPS | Equal TNFα, IL-6, IL-1β, iNOS as in stimulated control | |||
| Microglia | Mouse primary microglia culture | Tolerance | LPS | LPS | ↓ IL-1β, TNFα, IL-6 | Tolerance is Relb mediated | [167] | Schaafsma 2015 |
| Microglia | Wild type mice | Tolerance | LPS | LPS | ↓ IL-1β, TNFα, IL-6 | Tolerance is Relb mediated | ||
| Microglia | Wild type mice | Tolerance | LPS | LPS | Epigenetic and transcriptomic data determined ↓ signatures of permissive marks | [169] | Zhang 2021 | |
| Microglia | Ercc1 KO mice | Training | Aging | LPS | Epigenetic and transcriptomic data determined ↑ signatures of permissive marks | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carloni, E.; Ramos, A.; Hayes, L.N. Developmental Stressors Induce Innate Immune Memory in Microglia and Contribute to Disease Risk. Int. J. Mol. Sci. 2021, 22, 13035. https://doi.org/10.3390/ijms222313035
Carloni E, Ramos A, Hayes LN. Developmental Stressors Induce Innate Immune Memory in Microglia and Contribute to Disease Risk. International Journal of Molecular Sciences. 2021; 22(23):13035. https://doi.org/10.3390/ijms222313035
Chicago/Turabian StyleCarloni, Elisa, Adriana Ramos, and Lindsay N. Hayes. 2021. "Developmental Stressors Induce Innate Immune Memory in Microglia and Contribute to Disease Risk" International Journal of Molecular Sciences 22, no. 23: 13035. https://doi.org/10.3390/ijms222313035
APA StyleCarloni, E., Ramos, A., & Hayes, L. N. (2021). Developmental Stressors Induce Innate Immune Memory in Microglia and Contribute to Disease Risk. International Journal of Molecular Sciences, 22(23), 13035. https://doi.org/10.3390/ijms222313035