
DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
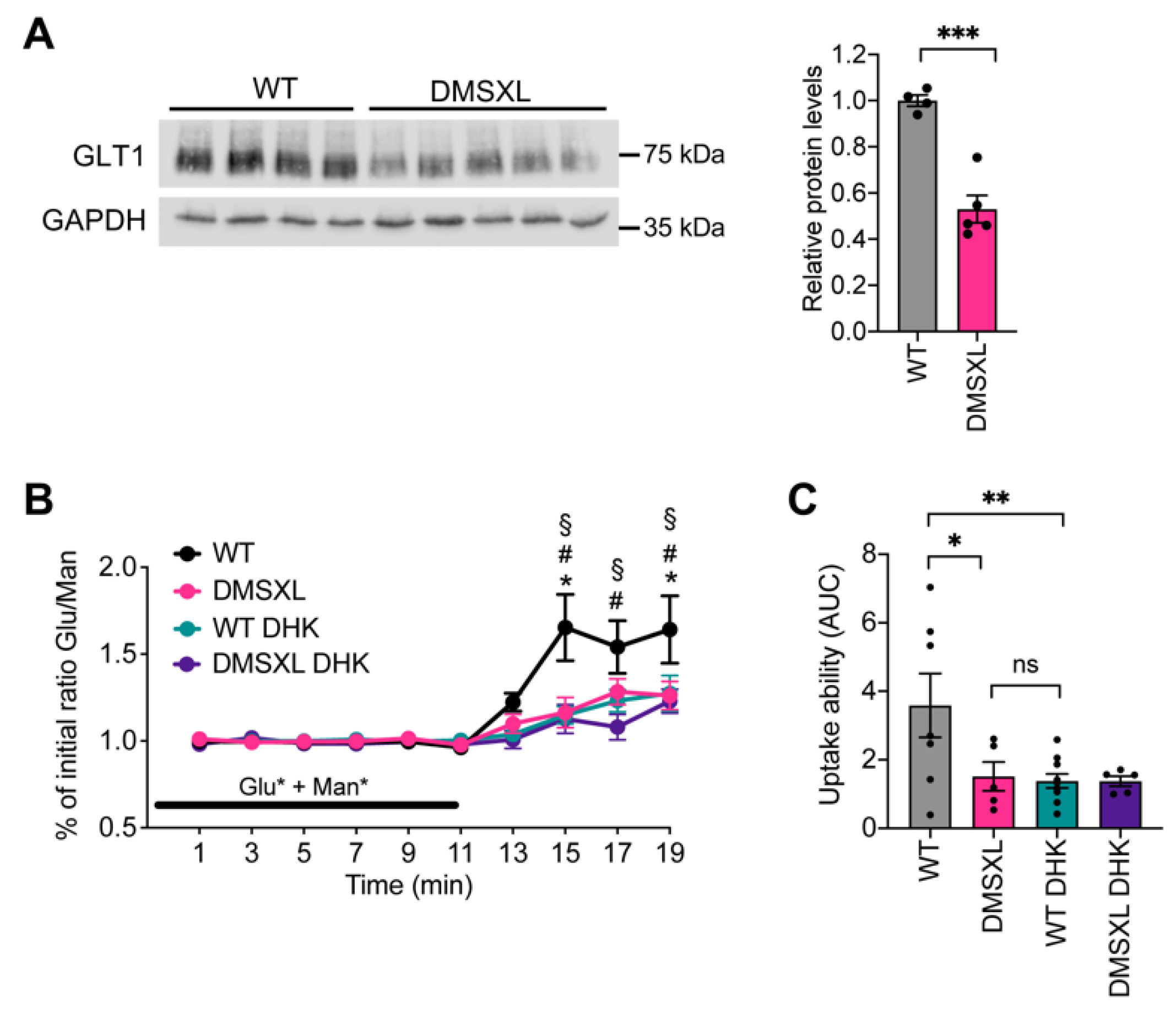
2.1. Glutamate Uptake Ability in the DMSXL Hippocampus
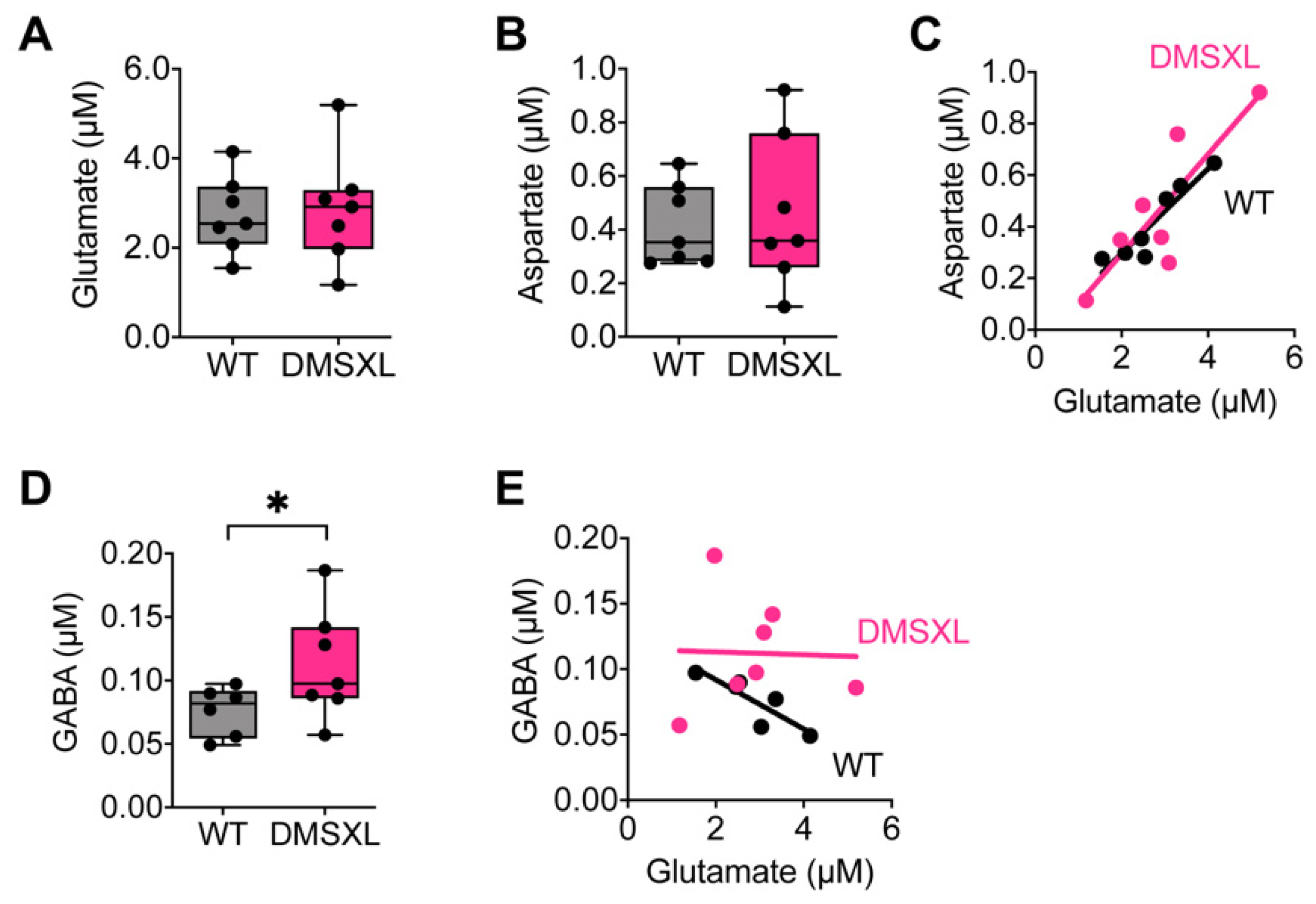
2.2. Neurotransmitter Extracellular Levels
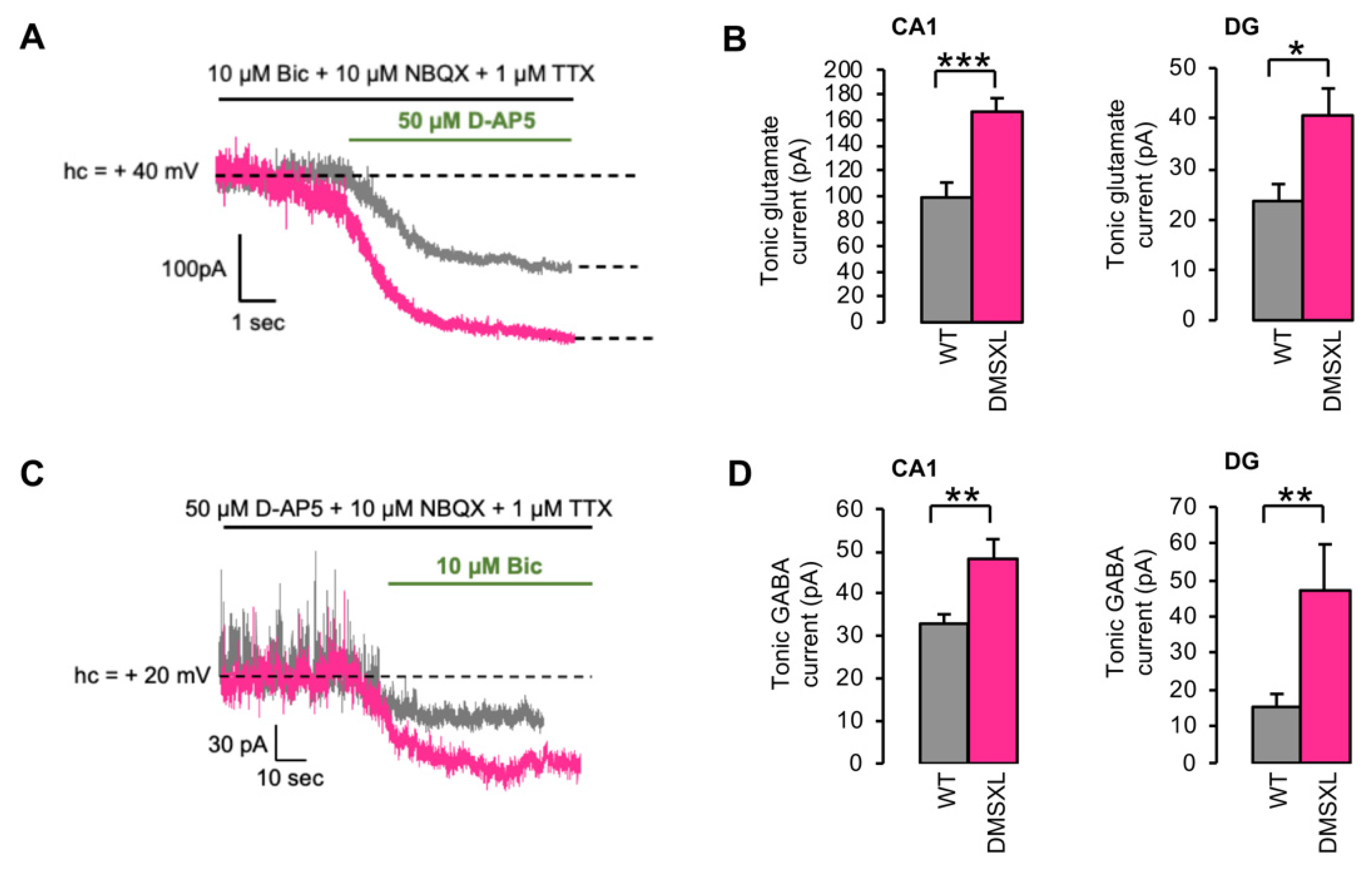
2.3. Tonically Activated Receptors by Ambient Neurotransmitters
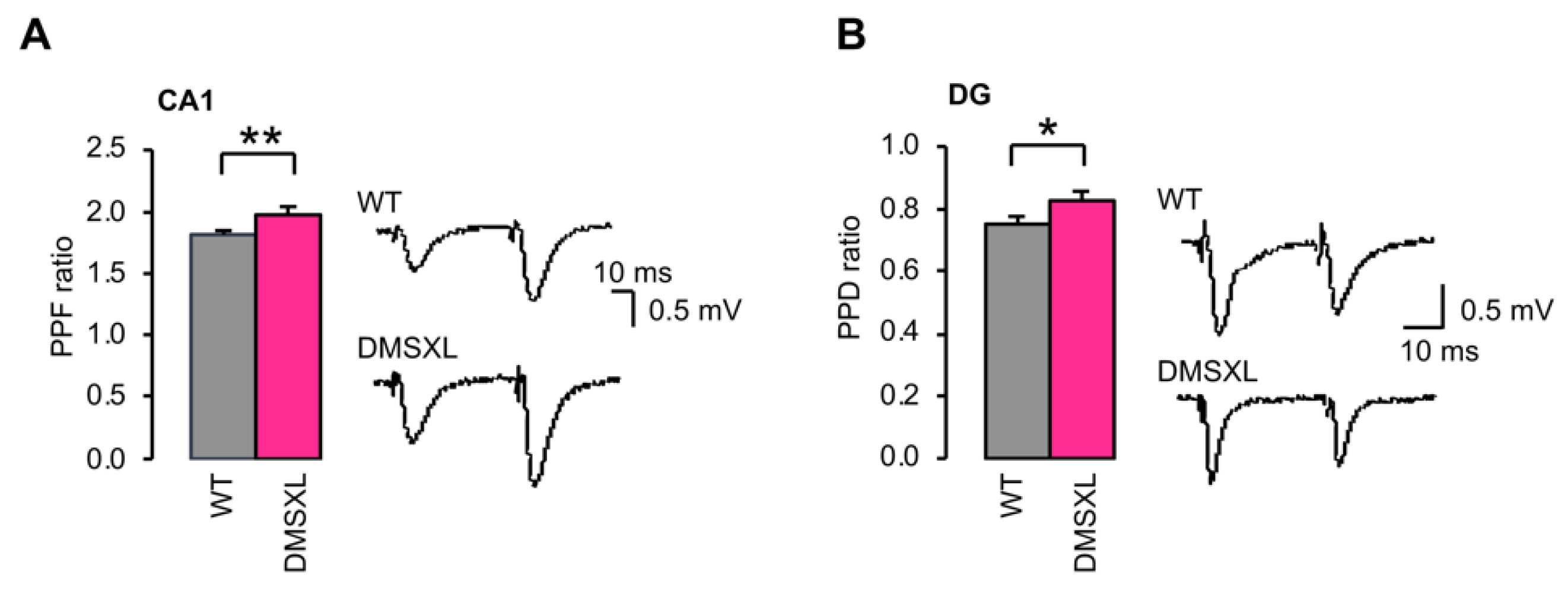
2.4. Basal Synaptic Transmission and Short-Term Synaptic Plasticity
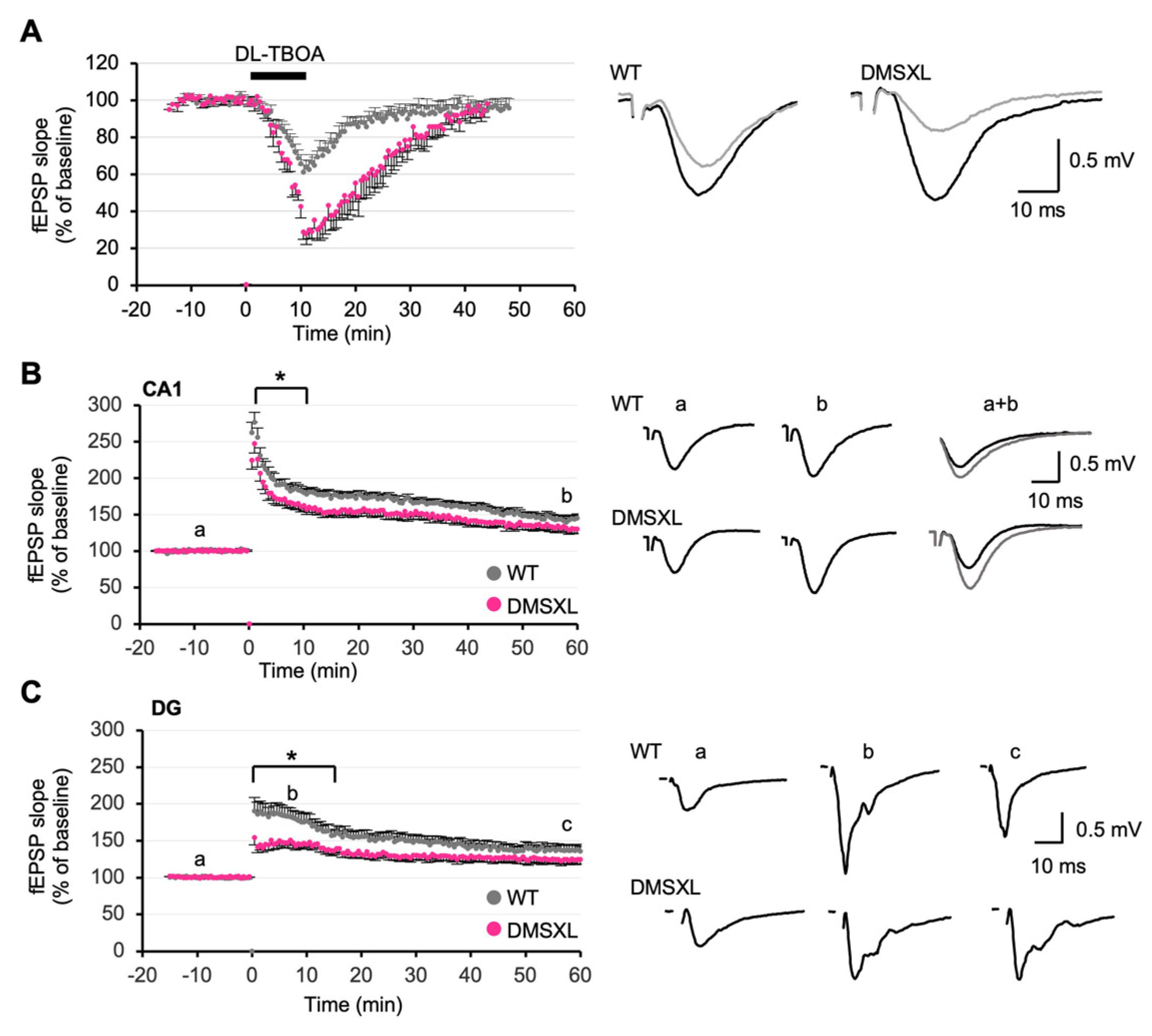
2.5. Effect of Glutamate Transporter Inhibition on Synaptic Plasticity
2.6. Long-Term Synaptic Potentiation
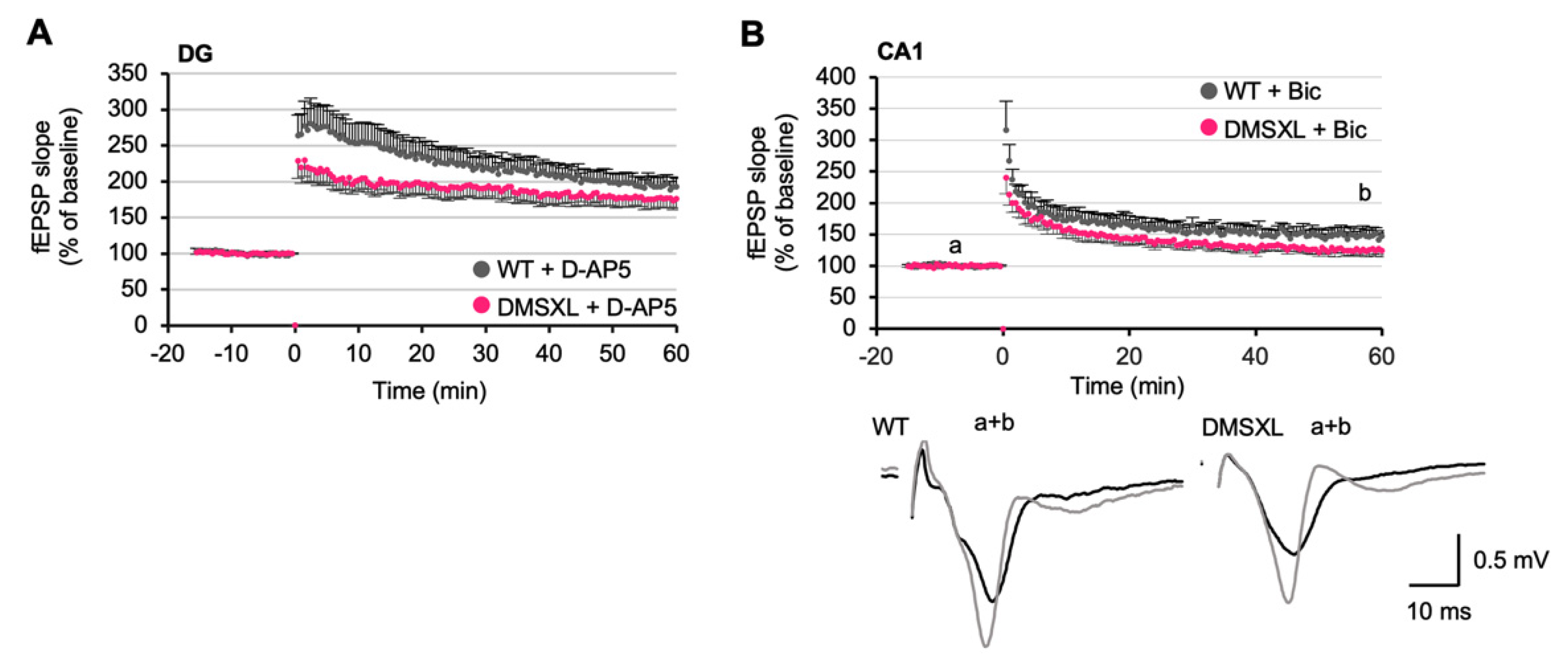
2.7. Effects of NMDA and GABA Antagonists on LTP
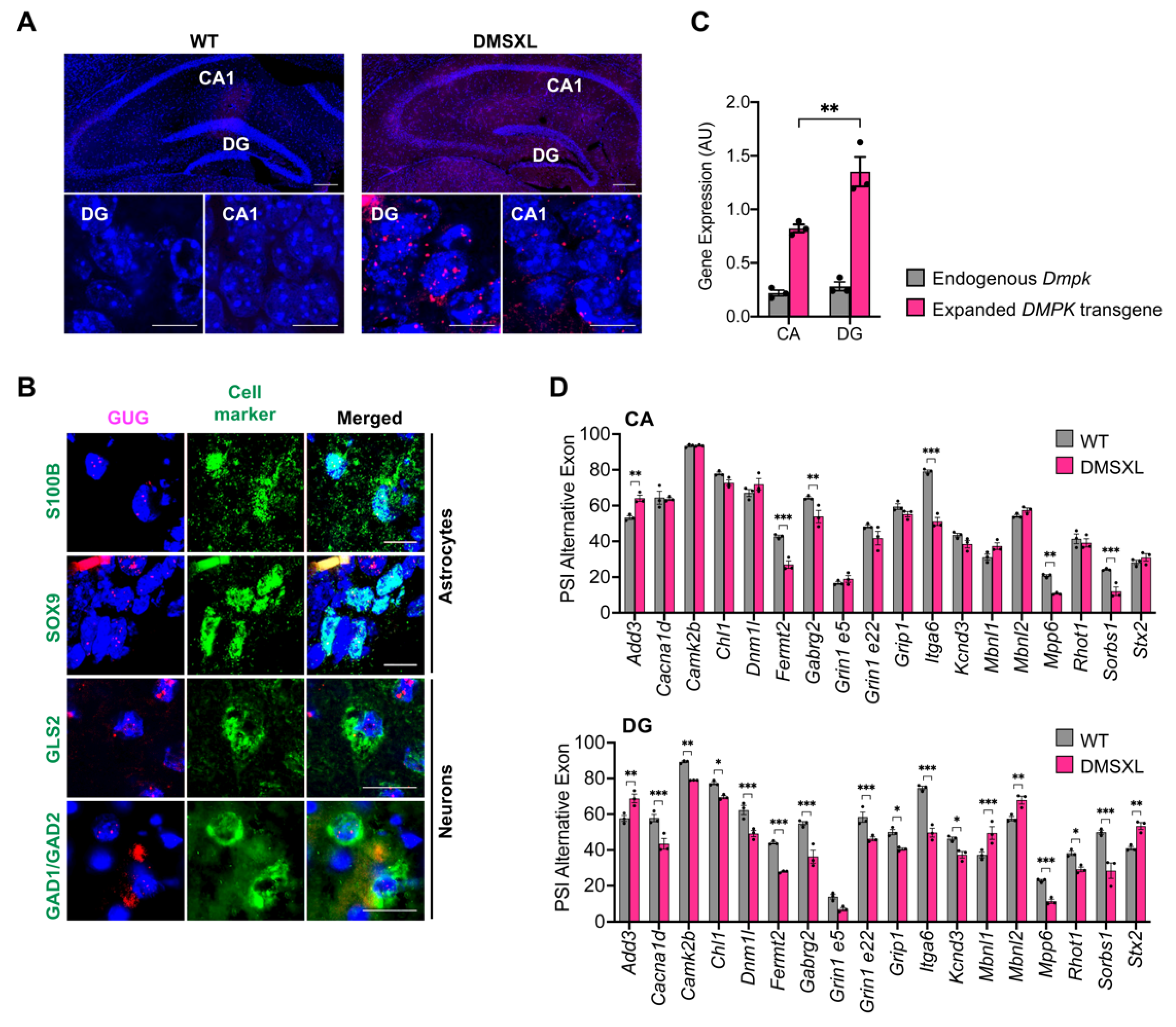
2.8. Regional Distribution of RNA Toxicity in the Hippocampus of DMSXL Mice
3. Discussion
3.1. Glutamate Uptake and Tonic Currents in DMSXL Hippocampus
3.2. Neurotransmission and Synaptic Plasticity in DMSXL Mice
3.3. RNA Spliceopathy in DMSXL Mouse Brains and Possible Effect on Altered Synaptic Dysfunction
3.4. Conclusion
4. Materials and Methods
4.1. Transgenic Mice
4.2. Reagents
4.3. Cornu Ammonis (CA) and Dentate Gyrus (DG) Dissection from Mouse Hippocampus
4.4. Reverse Transcription and PCR Analysis
4.5. Fluorescent In Situ Hybridization (FISH) and Immunofluorescence Analysis (IFA)
4.6. Western Blot Analysis
4.7. Ex Vivo Mouse Brain Slice Preparation
4.8. Extracellular Recordings
4.9. Synaptic Transmission
4.10. Synaptic Plasticity
4.11. Patch Clamp Recordings
4.12. Microdialysis Sampling
4.13. Dialysate Content Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Harper, P.S.P.S. Myotonic Dystrophy; W. B. Saunders: London, UK, 2001; ISBN 0702021520. [Google Scholar]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Meola, G.; Sansone, V. Cerebral involvement in myotonic dystrophies. Muscle Nerve 2007, 36, 294–306. [Google Scholar] [CrossRef]
- Okkersen, K.; Buskes, M.; Groenewoud, J.; Kessels, R.P.C.; Knoop, H.; van Engelen, B.; Raaphorst, J. The cognitive profile of myotonic dystrophy type 1: A systematic review and meta-analysis. Cortex 2017, 95, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Winblad, S.; Jensen, C.; Månsson, J.E.; Samuelsson, L.; Lindberg, C. Depression in Myotonic Dystrophy type 1: Clinical and neuronal correlates. Behav. Brain Funct. 2010, 6, 25. [Google Scholar] [CrossRef]
- Gourdon, G.; Meola, G. Myotonic dystrophies: State of the art of new therapeutic developments for the CNS. Front. Cell. Neurosci. 2017, 11, 101. [Google Scholar] [CrossRef]
- Minnerop, M.; Weber, B.; Schoene-Bake, J.C.; Roeske, S.; Mirbach, S.; Anspach, C.; Schneider-Gold, C.; Betz, R.C.; Helmstaedter, C.; Tittgemeyer, M.; et al. The brain in myotonic dystrophy 1 and 2: Evidence for a predominant white matter disease. Brain 2011, 134, 3527–3543. [Google Scholar] [CrossRef] [PubMed]
- Leddy, S.; Serra, L.; Esposito, D.; Vizzotto, C.; Giulietti, G.; Silvestri, G.; Petrucci, A.; Meola, G.; Lopiano, L.; Cercignani, M.; et al. Lesion distribution and substrate of white matter damage in myotonic dystrophy type 1: Comparison with multiple sclerosis. NeuroImage Clin. 2021, 29, 102562. [Google Scholar] [CrossRef]
- Okkersen, K.; Monckton, D.G.; Le, N.; Tuladhar, A.M.; Raaphorst, J.; Van Engelen, B.G.M. Brain imaging in myotonic dystrophy type 1. Neurology 2017, 89, 960–969. [Google Scholar] [CrossRef]
- Weber, Y.G.; Roebling, R.; Kassubek, J.; Hoffmann, S.; Rosenbohm, A.; Wolf, M.; Steinbach, P.; Jurkat-Rott, K.; Walter, H.; Reske, S.N.; et al. Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology 2010, 74, 1108–1117. [Google Scholar] [CrossRef]
- Giorgio, A.; Dotti, M.T.; Battaglini, M.; Marino, S.; Mortilla, M.; Stromillo, M.L.; Bramanti, P.; Orrico, A.; Federico, A.; De Stefano, N. Cortical damage in brains of patients with adult-form of myotonic dystrophy type 1 and no or minimal MRI abnormalities. J. Neurol. 2006, 253, 1471–1477. [Google Scholar] [CrossRef]
- Weijs, R.; Okkersen, K.; van Engelen, B.; Küsters, B.; Lammens, M.; Aronica, E.; Raaphorst, J.; van Cappellen van Walsum, A.M. Human brain pathology in myotonic dystrophy type 1: A systematic review. Neuropathology 2021, 41, 3–20. [Google Scholar] [CrossRef]
- Serra, L.; Mancini, M.; Silvestri, G.; Petrucci, A.; Masciullo, M.; Spanò, B.; Torso, M.; Mastropasqua, C.; Giacanelli, M.; Caltagirone, C.; et al. Brain Connectomics’ Modification to Clarify Motor and Nonmotor Features of Myotonic Dystrophy Type 1. Neural Plast. 2016, 2016, 2696085. [Google Scholar] [CrossRef] [PubMed]
- Serra, L.; Silvestri, G.; Petrucci, A.; Basile, B.; Masciullo, M.; Makovac, E.; Torso, M.; Spanò, B.; Mastropasqua, C.; Harrison, N.A.; et al. Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurol. 2014, 71, 603–611. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- De Antonio, M.; Dogan, C.; Hamroun, D.; Mati, M.; Zerrouki, S.; Eymard, B.; Katsahian, S.; Bassez, G. Unravelling the myotonic dystrophy type 1 clinical spectrum: A systematic registry-based study with implications for disease classification. Rev. Neurol. 2016, 172, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Braz, S.O.; Acquaire, J.; Gourdon, G.; Gomes-Pereira, M. Of Mice and Men: Advances in the understanding of neuromuscular aspects of myotonic dystrophy. Front. Neurol. 2018, 9, 519. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Swanson, M.S. Short tandem repeat expansions and RNA-mediated pathogenesis in myotonic dystrophy. Int. J. Mol. Sci. 2019, 20, 3365. [Google Scholar] [CrossRef]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Hernández, O.; Guiraud-Dogan, C.; Sicot, G.; Huguet, A.; Luilier, S.; Steidl, E.; Saenger, S.; Marciniak, E.; Obriot, H.; Chevarin, C.; et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 2013, 136, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, Physiological, and Motor Performance Defects in DMSXL Mice Carrying >1000 CTG Repeats from the Human DM1 Locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Pereira, M.; Foiry, L.; Nicole, A.; Huguet, A.; Junien, C.; Munnich, A.; Gourdon, G. CTG trinucleotide repeat “big jumps”: Large expansions, small mice. PLoS Genet. 2007, 3, 0488–0491. [Google Scholar] [CrossRef]
- Sicot, G.; Servais, L.; Dinca, D.M.; Leroy, A.; Prigogine, C.; Medja, F.; Braz, S.O.; Huguet-Lachon, A.; Chhuon, C.; Nicole, A.; et al. Downregulation of the Glial GLT1 Glutamate Transporter and Purkinje Cell Dysfunction in a Mouse Model of Myotonic Dystrophy. Cell Rep. 2017, 19, 2718–2729. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.G.; Kegelman, T.P.; Su, Z.Z.; Das, S.K.; Dash, R.; Dasgupta, S.; Barral, P.M.; Hedvat, M.; Diaz, P.; et al. Role of Excitatory Amino Acid Transporter-2 (EAAT2) and glutamate in neurodegeneration: Opportunities for developing novel therapeutics. J. Cell. Physiol. 2011, 226, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-Mediated Alternative Splicing in the Developing Brain and Dysregulation in Myotonic Dystrophy. Neuron 2012, 75, 437–450. [Google Scholar] [CrossRef]
- Parrot, S.; Touret, M.; Denoroy, L. In vivo determination of glutamate uptake by brain microdialysis. In Neuromethods; Humana Press: New York, NY, USA, 2018; Volume 130, pp. 431–467. [Google Scholar]
- Touret, M.; Parrot, S.; Denoroy, L.; Belin, M.F.; Didier-Bazes, M. Glutamatergic alterations in the cortex of genetic absence epilepsy rats. BMC Neurosci. 2007, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Wise, B.L.; Perkins, R.K.; Stevenson, E.; Scott, K.G. Penetration of C14-labelled mannitol from serum into cerebrospinal fluid and brain. Exp. Neurol. 1964, 10, 264–270. [Google Scholar] [CrossRef]
- Parrot, S.; Corscadden, A.; Lallemant, L.; Benyamine, H.; Comte, J.-C.; Huguet-lachon, A.; Gourdon, G.; Gomes-Pereira, M. Defects in Mouse Cortical Glutamate Uptake Can Be Unveiled In Vivo by a Two-in-One Quantitative Microdialysis. ACS Chem. Neurosci. 2022, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, K.; Galante, M.; Angulo, M.C.; Audinat, E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J. Physiol. 2007, 580, 373–383. [Google Scholar] [CrossRef]
- Dan Liu, D.; Yang, Q.; Li, S. tian Activation of extrasynaptic NMDA receptors induces LTD in rat hippocampal CA1 neurons. Brain Res. Bull. 2013, 93, 10–16. [Google Scholar] [CrossRef]
- Katagiri, H.; Tanaka, K.; Manabe, T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur. J. Neurosci. 2001, 14, 547–553. [Google Scholar] [CrossRef]
- Parrot, S.; Renaud, B.; Zimmer, L.; Denoroy, L. Monitoring Neurotransmitter Amino Acids by Microdialysis: Pharmacodynamic Applications. Appl. Microdialysis Pharm. Sci. 2011, 151–206. [Google Scholar] [CrossRef]
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem. Int. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Jackman, S.L.; Regehr, W.G. The Mechanisms and Functions of Synaptic Facilitation. Neuron 2017, 94, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Potier, B.; Poindessous-Jazat, F.; Dutar, P.; Billard, J.M. NMDA receptor activation in the aged rat hippocampus. Exp. Gerontol. 2000, 35, 1185–1199. [Google Scholar] [CrossRef]
- Barnes, C.A.; Rao, G.; Shen, J. Age-related decrease in the n-methyl-D-aspartate(R)-mediated excitatory postsynaptic potential in hippocampal region CA1. Neurobiol. Aging 1997, 18, 445–452. [Google Scholar] [CrossRef]
- Nägerl, U.V.; Eberhorn, N.; Cambridge, S.B.; Bonhoeffer, T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 2004, 44, 759–767. [Google Scholar] [CrossRef]
- Michel, L.; Huguet-Lachon, A.; Gourdon, G. Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS ONE 2015, 10, e0137620. [Google Scholar] [CrossRef]
- Gonçalves-Ribeiro, J.; Pina, C.C.; Sebastião, A.M.; Vaz, S.H. Glutamate Transporters in Hippocampal LTD/LTP: Not Just Prevention of Excitotoxicity. Front. Cell. Neurosci. 2019, 13, 357. [Google Scholar] [CrossRef]
- Androuin, A.; Potier, B.; Nägerl, U.V.; Cattaert, D.; Danglot, L.; Thierry, M.; Youssef, I.; Triller, A.; Duyckaerts, C.; El Hachimi, K.H.; et al. Evidence for altered dendritic spine compartmentalization in Alzheimer’s disease and functional effects in a mouse model. Acta Neuropathol. 2018, 135, 839–854. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takagi, T.; Ishii, S.; Suzuki, H.; Miyakawa, T. Attenuated bidirectional short-term synaptic plasticity in the dentate gyrus of Schnurri-2 knockout mice, a model of schizophrenia 11 Medical and Health Sciences 1109 Neurosciences 11 Medical and Health Sciences 1103 Clinical Sciences. Mol. Brain 2018, 11, 56–59. [Google Scholar] [CrossRef]
- Wang, P.Y.; Chang, K.T.; Lin, Y.M.; Kuo, T.Y.; Wang, G.S. Ubiquitination of MBNL1 Is Required for Its Cytoplasmic Localization and Function in Promoting Neurite Outgrowth. Cell Rep. 2018, 22, 2294–2306. [Google Scholar] [CrossRef]
- Marteyn, A.; Maury, Y.; Gauthier, M.M.; Lecuyer, C.; Vernet, R.; Denis, J.A.; Pietu, G.; Peschanski, M.; Martinat, C. Mutant human embryonic stem cells reveal neurite and synapse formation defects in type 1 myotonic dystrophy. Cell Stem Cell 2011, 8, 434–444. [Google Scholar] [CrossRef]
- Quintero-Mora, M.L.; Depardon, F.; Waring, J.; Korneluk, R.G.; Cisneros, B. Expanded CTG repeats inhibit neuronal differentiation of the PC12 cell line. Biochem. Biophys. Res. Commun. 2002, 295, 289–294. [Google Scholar] [CrossRef]
- Otero, B.A.; Poukalov, K.; Hildebrandt, R.P.; Thornton, C.A.; Jinnai, K.; Fujimura, H.; Kimura, T.; Hagerman, K.A.; Sampson, J.B.; Day, J.W.; et al. Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep. 2021, 34, 108634. [Google Scholar] [CrossRef] [PubMed]
- González-Barriga, A.; Lallemant, L.; Dincã, D.M.; Braz, S.O.; Polvèche, H.; Magneron, P.; Pionneau, C.; Huguet-Lachon, A.; Claude, J.-B.; Chhuon, C.; et al. Integrative Cell Type-Specific Multi-Omics Approaches Reveal Impaired Programs of Glial Cell Differentiation in Mouse Culture Models of DM1. Front. Cell. Neurosci. 2021, 15, 126. [Google Scholar] [CrossRef] [PubMed]
- Velázquez-Bernardino, P.; García-Sierra, F.; Hernández-Hernández, O.; Bermúdez De León, M.; Gourdon, G.; Gomes-Pereira, M.; Cisneros, B. Myotonic dystrophy type 1-associated CTG repeats disturb the expression and subcellular distribution of microtubule-associated proteins MAP1A, MAP2, and MAP6/STOP in PC12 cells. Mol. Biol. Rep. 2012, 39, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Lin, Y.M.; Wang, L.H.; Kuo, T.Y.; Cheng, S.J.; Wang, G.S. Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum. Mol. Genet. 2017, 26, 2247–2257. [Google Scholar] [CrossRef][Green Version]
- Black, D.L.; Grabowski, P.J. Alternative pre-mRNA splicing and neuronal function. Prog. Mol. Subcell. Biol. 2003, 31, 187–216. [Google Scholar] [CrossRef]
- Grossmann, D.; Berenguer-Escuder, C.; Chemla, A.; Arena, G.; Krüger, R. The Emerging Role of RHOT1/Miro1 in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2020, 11, 587. [Google Scholar] [CrossRef]
- Hofer, N.T.; Pinggera, A.; Nikonishyna, Y.V.; Tuluc, P.; Fritz, E.M.; Obermair, G.J.; Striessnig, J. Stabilization of negative activation voltages of Cav1.3 L-Type Ca2 + -channels by alternative splicing. Channels 2021, 15, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Truchet, B.; Manrique, C.; Sreng, L.; Chaillan, F.A.; Roman, F.S.; Mourre, C. Kv4 potassium channels modulate hippocampal EPSP-spike potentiation and spatial memory in rats. Learn. Mem. 2012, 19, 282–293. [Google Scholar] [CrossRef]
- Lou, H.; Park, J.J.; Cawley, N.X.; Sarcon, A.; Sun, L.; Adams, T.; Loh, Y.P. Carboxypeptidase E Cytoplasmic Tail Mediates Localization of Synaptic Vesicles to the Pre-active Zone in Hypothalamic Presynaptic Terminals. J. Neurochem. 2010, 114, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Band, A.M.; Kuismanen, E. Localization of plasma membrane t-SNAREs syntaxin 2 and 3 in intracellular compartments. BMC Cell Biol. 2005, 6, 26. [Google Scholar] [CrossRef]
- Herring, B.E.; Nicoll, R.A. Long-Term Potentiation: From CaMKII to AMPA Receptor Trafficking. Annu. Rev. Physiol. 2016, 78, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Chiu, S.L.; Zhu, Q.; Huganir, R.L. GRIP1 regulates synaptic plasticity and learning and memory. Proc. Natl. Acad. Sci. USA 2020, 117, 25085–25091. [Google Scholar] [CrossRef] [PubMed]
- Sloutsky, R.; Stratton, M.M. Functional implications of CaMKII alternative splicing. Eur. J. Neurosci. 2021, 54, 6780–6794. [Google Scholar] [CrossRef] [PubMed]
- Hallock, P.T.; Chin, S.; Blais, S.; Neubert, T.A.; Glass, D.J. Sorbs1 and 2 interact with Crk-L and are required for Acetylcholine receptor cluster formation. Mol. Cell. Biol. 2015, 36, 262–270. [Google Scholar] [CrossRef]
- Boileau, A.J.; Pearce, R.A.; Czajkowski, C. The Short Splice Variant of the γ2 Subunit Acts as an External Modulator of GABAA Receptor Function. J. Neurosci. 2010, 30, 4895–4903. [Google Scholar] [CrossRef] [PubMed]
- Eysert, F.; Coulon, A.; Boscher, E.; Vreulx, A.C.; Flaig, A.; Mendes, T.; Hughes, S.; Grenier-Boley, B.; Hanoulle, X.; Demiautte, F.; et al. Alzheimer’s genetic risk factor FERMT2 (Kindlin-2) controls axonal growth and synaptic plasticity in an APP-dependent manner. Mol. Psychiatry 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- Dixon, D.M.; Choi, J.; El-Ghazali, A.; Park, S.Y.; Roos, K.P.; Jordan, M.C.; Fishbein, M.C.; Comai, L.; Reddy, S. Loss of muscleblind-like 1 results in cardiac pathology and persistence of embryonic splice isoforms. Sci. Rep. 2015, 5, 9042. [Google Scholar] [CrossRef] [PubMed]
- Klinck, R.; Fourrier, A.; Thibault, P.; Toutant, J.; Durand, M.; Lapointe, E.; Caillet-Boudin, M.L.; Sergeant, N.; Gourdon, G.; Meola, G.; et al. RBFOX1 Cooperates with MBNL1 to Control Splicing in Muscle, Including Events Altered in Myotonic Dystrophy Type 1. PLoS ONE 2014, 9, e107324. [Google Scholar] [CrossRef]
- Wang, Q.; Jie, W.; Liu, J.H.; Yang, J.M.; Gao, T.M. An astroglial basis of major depressive disorder? An overview. Glia 2017, 65, 1227–1250. [Google Scholar] [CrossRef]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549.e9. [Google Scholar] [CrossRef]
- Thomas, J.D.; Sznajder, Ł.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Vivar, C.; van Praag, H. Functional circuits of new neurons in the dentate gyrus. Front. Neural Circuits 2013, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Bacigaluppi, M.; Sferruzza, G.; Butti, E.; Ottoboni, L.; Martino, G. Endogenous neural precursor cells in health and disease. Brain Res. 2020, 1730, 146619. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Lia-Baldini, A.S.; Duros, C.; Fouquet, C.; Lacroix, C.; Hofmann-Radvanyi, H.; Junien, C.; Gourdon, G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum. Mol. Genet. 2000, 9, 1185–1194. [Google Scholar] [CrossRef]
- Hagihara, H.; Toyama, K.; Yamasaki, N.; Miyakawa, T. Dissection of Hippocampal Dentate Gyrus from Adult Mouse. JoVE J. Vis. Exp. 2009, 33, e1543. [Google Scholar] [CrossRef]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.Y.; Charizanis, K.; Fernández Gómez, F.J.; Eddarkaoui, S.; Sergeant, N.; Buée, L.; Kimura, T.; et al. MBNL Sequestration by Toxic RNAs and RNA Misprocessing in the Myotonic Dystrophy Brain. Cell Rep. 2015, 12, 1159–1168. [Google Scholar] [CrossRef]
- Anderson, W.W.; Collingridge, G.L. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J. Neurosci. Methods 2007, 162, 346–356. [Google Scholar] [CrossRef]
- Paxinos, G.; Frankin, K.B.J. The Mouse Brain Steriotaxic Coordinates; Elsevier Science & Technology Books; Academic Press: Cambridge, MA, USA, 2001; ISBN 9780125476379. [Google Scholar]
- Sauvinet, V.; Parrot, S.; Benturquia, N.; Bravo-Moratón, E.; Renaud, B.; Denoroy, L. In vivo simultaneous monitoring of γ-aminobutyric acid, glutamate, and L-aspartate using brain microdialysis and capillary electrophoresis with laser-induced fluorescence detection: Analytical developments and in vitro/in vivo validations. Electrophoresis 2003, 24, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Burchett, J.M.; Restivo, J.L.; Schuler, D.R.; Verghese, P.B.; Mahan, T.E.; Landreth, G.E.; Castellano, J.M.; Jiang, H.; Cirrito, J.R.; et al. In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol. Neurodegener. 2013, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.; Walt, H.; Joseph, B.J. In vivo calibration of microdialysis probes for exogenous compounds. Anal. Chem. 2002, 64, 577–583. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potier, B.; Lallemant, L.; Parrot, S.; Huguet-Lachon, A.; Gourdon, G.; Dutar, P.; Gomes-Pereira, M. DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus. Int. J. Mol. Sci. 2022, 23, 592. https://doi.org/10.3390/ijms23020592
Potier B, Lallemant L, Parrot S, Huguet-Lachon A, Gourdon G, Dutar P, Gomes-Pereira M. DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus. International Journal of Molecular Sciences. 2022; 23(2):592. https://doi.org/10.3390/ijms23020592
Chicago/Turabian StylePotier, Brigitte, Louison Lallemant, Sandrine Parrot, Aline Huguet-Lachon, Geneviève Gourdon, Patrick Dutar, and Mário Gomes-Pereira. 2022. "DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus" International Journal of Molecular Sciences 23, no. 2: 592. https://doi.org/10.3390/ijms23020592
APA StylePotier, B., Lallemant, L., Parrot, S., Huguet-Lachon, A., Gourdon, G., Dutar, P., & Gomes-Pereira, M. (2022). DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus. International Journal of Molecular Sciences, 23(2), 592. https://doi.org/10.3390/ijms23020592