
Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies

 , and
, and
Abstract
:1. Introduction
2. Omics Studies in DM1
2.1. Omics Studies on Skeletal Muscle
2.2. Omics Studies in the Central Nervous System
2.3. Omics Studies in the Heart
2.4. Omics Studies in Other Affected Tissues in DM1: Lens and Thymus
2.5. Other Omics Studies in DM1
3. Omics Studies in Candidate Drug Validations
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.E.; Butterfield, R.J.; Mayne, K.; Newcomb, T.; Imburgia, C.; Dunn, D.; Duval, B.; Feldkamp, M.L.; Weiss, R.B. Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program. Neurology 2021, 96, e1045–e1053. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ranum, L.P.W. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr. Neurol. Neurosci. Rep. 2005, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Musova, Z.; Mazanec, R.; Krepelova, A.; Ehler, E.; Vales, J.; Jaklova, R.; Prochazka, T.; Koukal, P.; Marikova, T.; Kraus, J.; et al. Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. 2009, 149A, 1365–1374. [Google Scholar] [CrossRef]
- Ebralidze, A.; Wang, Y.; Petkova, V.; Ebralidse, K.; Junghans, R.P. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science 2004, 303, 383–387. [Google Scholar] [CrossRef]
- Huichalaf, C.; Sakai, K.; Jin, B.; Jones, K.; Wang, G.-L.; Schoser, B.; Schneider-Gold, C.; Sarkar, P.; Pereira-Smith, O.M.; Timchenko, N.; et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J. 2010, 24, 3706–3719. [Google Scholar] [CrossRef] [Green Version]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.-P.; Fischer, M.-C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef]
- López-Martínez, A.; Soblechero-Martín, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109. [Google Scholar] [CrossRef]
- Batra, R.; Charizanis, K.; Manchanda, M.; Mohan, A.; Li, M.; Finn, D.J.; Goodwin, M.; Zhang, C.; Sobczak, K.; Thornton, C.A.; et al. Loss of MBNL Leads to Disruption of Developmentally Regulated Alternative Polyadenylation in RNA-Mediated Disease. Mol. Cell 2014, 56, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, T.; Gagnon, C.; Groh, W.J.; Gutmann, L.; Johnson, N.E.; Meola, G.; Moxley, R.; Pandya, S.; Rogers, M.T.; Simpson, E.; et al. Consensus-based care recommendations for adults with myotonic dystrophy type 1. Neurol. Clin. Pract. 2018, 8, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Gilabert, M.; López-Castel, A.; Artero, R. Myotonic dystrophy type 1 drug development: A pipeline toward the market. Drug Discov. Today 2021, 26, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Fardaei, M.; Rogers, M.T.; Thorpe, H.M.; Larkin, K.; Hamshere, M.G.; Harper, P.S.; Brook, J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002, 11, 805–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-S.; Cao, Y.; Witwicka, H.E.; Tom, S.; Tapscott, S.J.; Wang, E.H. RNA-binding protein Muscleblind-like 3 (MBNL3) disrupts myocyte enhancer factor 2 (Mef2) {beta}-exon splicing. J. Biol. Chem. 2010, 285, 33779–33787. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, P.; Stepniak-Konieczna, E.; Sobczak, K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014, 42, 10873–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Dixon, D.M.; Dansithong, W.; Abdallah, W.F.; Roos, K.P.; Jordan, M.C.; Trac, B.; Lee, H.S.; Comai, L.; Reddy, S. Muscleblind-like 3 deficit results in a spectrum of age-associated pathologies observed in myotonic dystrophy. Sci. Rep. 2016, 6, 30999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, T.; Ladd, A.N. The importance of CELF control: Molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins: Importance of CELF control. WIREs RNA 2012, 3, 104–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasiri-Aghdam, M.; Garcia-Garduño, T.C.; Jave-Suárez, L.F. CELF Family Proteins in Cancer: Highlights on the RNA-Binding Protein/Noncoding RNA Regulatory Axis. Int. J. Mol. Sci. 2021, 22, 11056. [Google Scholar] [CrossRef]
- Greenberg, D.S.; Soreq, H. Alternative Splicing. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 97–98. ISBN 978-0-08-096156-9. [Google Scholar]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Yeh, H.-S.; Yong, J. Alternative Polyadenylation of mRNAs: 3′-Untranslated Region Matters in Gene Expression. Mol. Cells 2016, 39, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, N.J. Ending the message: Poly(A) signals then and now. Genes Dev. 2011, 25, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes-Pereira, M.; Foiry, L.; Nicole, A.; Huguet, A.; Junien, C.; Munnich, A.; Gourdon, G. CTG Trinucleotide Repeat “Big Jumps”: Large Expansions, Small Mice. PLoS Genet. 2007, 3, e52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.-Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, Physiological, and Motor Performance Defects in DMSXL Mice Carrying >1000 CTG Repeats from the Human DM1 Locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Hernández, O.; Guiraud-Dogan, C.; Sicot, G.; Huguet, A.; Luilier, S.; Steidl, E.; Saenger, S.; Marciniak, E.; Obriot, H.; Chevarin, C.; et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain 2013, 136, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arandel, L.; Polay Espinoza, M.; Matloka, M.; Bazinet, A.; De Dea Diniz, D.; Naouar, N.; Rau, F.; Jollet, A.; Edom-Vovard, F.; Mamchaoui, K.; et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Model Mech. 2017, 10, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Peña-Bautista, C.; Baquero, M.; Vento, M.; Cháfer-Pericás, C. Omics-based Biomarkers for the Early Alzheimer Disease Diagnosis and Reliable Therapeutic Targets Development. Curr. Neuropharmacol. 2019, 17, 630–647. [Google Scholar] [CrossRef]
- Heo, Y.J.; Hwa, C.; Lee, G.-H.; Park, J.-M.; An, J.-Y. Integrative Multi-Omics Approaches in Cancer Research: From Biological Networks to Clinical Subtypes. Mol. Cells 2021, 44, 433–443. [Google Scholar] [CrossRef]
- Morello, G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Cavallaro, S. From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 1151. [Google Scholar] [CrossRef]
- Bonder, M.J.; Smail, C.; Gloudemans, M.J.; Frésard, L.; Jakubosky, D.; D’Antonio, M.; Li, X.; Ferraro, N.M.; Carcamo-Orive, I.; Mirauta, B.; et al. Identification of rare and common regulatory variants in pluripotent cells using population-scale transcriptomics. Nat. Genet. 2021, 53, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of New Sequencing Technologies for Transcriptome Analysis. Annu. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Cline, M.S.; Osborne, R.J.; Tuttle, D.L.; Clark, T.A.; Donohue, J.P.; Hall, M.P.; Shiue, L.; Swanson, M.S.; Thornton, C.A.; et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010, 17, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Llorian, M.; Smith, C.W. Decoding muscle alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 380–387. [Google Scholar] [CrossRef]
- Wang, E.T.; Cody, N.A.L.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide Regulation of Pre-mRNA Splicing and mRNA Localization by Muscleblind Proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [Green Version]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.K.; Tang, Z.; Thornton, C.A. Targeted splice sequencing reveals RNA toxicity and therapeutic response in myotonic dystrophy. Nucleic Acids Res. 2021, 49, 2240–2254. [Google Scholar] [CrossRef]
- Franck, S.; Couvreu De Deckersberg, E.; Bubenik, J.L.; Markouli, C.; Barbé, L.; Allemeersch, J.; Hilven, P.; Duqué, G.; Swanson, M.S.; Gheldof, A.; et al. Myotonic dystrophy type 1 embryonic stem cells show decreased myogenic potential, increased CpG methylation at the DMPK locus and RNA mis-splicing. Biol. Open 2022, 11, bio058978. [Google Scholar] [CrossRef]
- Wang, E.T.; Treacy, D.; Eichinger, K.; Struck, A.; Estabrook, J.; Olafson, H.; Wang, T.T.; Bhatt, K.; Westbrook, T.; Sedehizadeh, S.; et al. Transcriptome alterations in myotonic dystrophy skeletal muscle and heart. Hum. Mol. Genet. 2019, 28, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.D.; Struck, A.J.; Gupta, R.; Farnsworth, D.R.; Mahady, A.E.; Eichinger, K.; Thornton, C.A.; Wang, E.T.; Berglund, J.A. Dose-Dependent Regulation of Alternative Splicing by MBNL Proteins Reveals Biomarkers for Myotonic Dystrophy. PLoS Genet. 2016, 12, e1006316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamori, M.; Sobczak, K.; Puwanant, A.; Welle, S.; Eichinger, K.; Pandya, S.; Dekdebrun, J.; Heatwole, C.R.; McDermott, M.P.; Chen, T.; et al. Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 2013, 74, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Oliveira, R.; Sznajder, Ł.J.; Swanson, M.S. Myotonic Dystrophy and Developmental Regulation of RNA Processing. Compr. Physiol. 2018, 8, 509–553. [Google Scholar] [CrossRef] [PubMed]
- Chau, A.; Kalsotra, A. Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics: DM1 disease mechanisms and therapeutics. Dev. Dyn. 2015, 244, 377–390. [Google Scholar] [CrossRef]
- Bachinski, L.L.; Baggerly, K.A.; Neubauer, V.L.; Nixon, T.J.; Raheem, O.; Sirito, M.; Unruh, A.K.; Zhang, J.; Nagarajan, L.; Timchenko, L.T.; et al. Most expression and splicing changes in myotonic dystrophy type 1 and type 2 skeletal muscle are shared with other muscular dystrophies. Neuromuscul. Disord. 2014, 24, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef]
- André, L.M.; van Cruchten, R.T.P.; Willemse, M.; Bezstarosti, K.; Demmers, J.A.A.; van Agtmaal, E.L.; Wansink, D.G.; Wieringa, B. Recovery in the Myogenic Program of Congenital Myotonic Dystrophy Myoblasts after Excision of the Expanded (CTG)n Repeat. Int. J. Mol. Sci. 2019, 20, 5685. [Google Scholar] [CrossRef] [Green Version]
- Meola, G.; Sansone, V. Cerebral involvement in myotonic dystrophies. Muscle Nerve 2007, 36, 294–306. [Google Scholar] [CrossRef]
- Sicot, G.; Servais, L.; Dinca, D.M.; Leroy, A.; Prigogine, C.; Medja, F.; Braz, S.O.; Huguet-Lachon, A.; Chhuon, C.; Nicole, A.; et al. Downregulation of the Glial GLT1 Glutamate Transporter and Purkinje Cell Dysfunction in a Mouse Model of Myotonic Dystrophy. Cell Rep. 2017, 19, 2718–2729. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Mankodi, A.; Swanson, M.S.; Moxley, R.T.; Thornton, C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004, 13, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourdon, G.; Meola, G. Myotonic Dystrophies: State of the Art of New Therapeutic Developments for the CNS. Front Cell Neurosci. 2017, 11, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, M.; Mohan, A.; Batra, R.; Lee, K.-Y.; Charizanis, K.; Gómez, F.J.F.; Eddarkaoui, S.; Sergeant, N.; Buée, L.; Kimura, T.; et al. MBNL Sequestration by Toxic RNAs and RNA Mis-Processing in the Myotonic Dystrophy Brain. Cell Rep. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azotla-Vilchis, C.N.; Sanchez-Celis, D.; Agonizantes-Juárez, L.E.; Suárez-Sánchez, R.; Hernández-Hernández, J.M.; Peña, J.; Vázquez-Santillán, K.; Leyva-García, N.; Ortega, A.; Maldonado, V.; et al. Transcriptome Analysis Reveals Altered Inflammatory Pathway in an Inducible Glial Cell Model of Myotonic Dystrophy Type 1. Biomolecules 2021, 11, 159. [Google Scholar] [CrossRef]
- González-Barriga, A.; Lallemant, L.; Dincã, D.M.; Braz, S.O.; Polvèche, H.; Magneron, P.; Pionneau, C.; Huguet-Lachon, A.; Claude, J.-B.; Chhuon, C.; et al. Integrative Cell Type-Specific Multi-Omics Approaches Reveal Impaired Programs of Glial Cell Differentiation in Mouse Culture Models of DM1. Front. Cell. Neurosci. 2021, 15, 662035. [Google Scholar] [CrossRef]
- Otero, B.A.; Poukalov, K.; Hildebrandt, R.P.; Thornton, C.A.; Jinnai, K.; Fujimura, H.; Kimura, T.; Hagerman, K.A.; Sampson, J.B.; Day, J.W.; et al. Transcriptome alterations in myotonic dystrophy frontal cortex. Cell Rep. 2021, 34, 108634. [Google Scholar] [CrossRef]
- Blech-Hermoni, Y.; Dasgupta, T.; Coram, R.J.; Ladd, A.N. Identification of Targets of CUG-BP, Elav-Like Family Member 1 (CELF1) Regulation in Embryonic Heart Muscle. PLoS ONE 2016, 11, e0149061. [Google Scholar] [CrossRef]
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Singh, R.K.; Gurha, P.; Ward, A.J.; Creighton, C.J.; Cooper, T.A. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue dramatically altering miRNA and mRNA expression. Cell Rep. 2014, 6, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Misra, C.; Bangru, S.; Lin, F.; Lam, K.; Koenig, S.N.; Lubbers, E.R.; Hedhli, J.; Murphy, N.P.; Parker, D.J.; Dobrucki, L.W.; et al. Aberrant Expression of a Non-muscle RBFOX2 Isoform Triggers Cardiac Conduction Defects in Myotonic Dystrophy. Dev. Cell 2020, 52, 748–763.e6. [Google Scholar] [CrossRef]
- Rao, A.N.; Campbell, H.M.; Guan, X.; Word, T.A.; Wehrens, X.H.T.; Xia, Z.; Cooper, T.A. Reversible cardiac disease features in an inducible CUG repeat RNA–expressing mouse model of myotonic dystrophy. JCI Insight 2021, 6, e143465. [Google Scholar] [CrossRef] [PubMed]
- Klesert, T.R.; Cho, D.H.; Clark, J.I.; Maylie, J.; Adelman, J.; Snider, L.; Yuen, E.C.; Soriano, P.; Tapscott, S.J. Mice deficient in Six5 develop cataracts: Implications for myotonic dystrophy. Nat. Genet. 2000, 25, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, J.D.; Lott, M.C.; Russell, S.L.; Moulton, V.; Sanderson, J.; Wormstone, I.M.; Broadway, D.C. Activation of the innate immune response and interferon signalling in myotonic dystrophy type 1 and type 2 cataracts. Hum. Mol. Genet. 2012, 21, 852–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanadia, R.N.; Urbinati, C.R.; Crusselle, V.J.; Luo, D.; Lee, Y.-J.; Harrison, J.K.; Oh, S.P.; Swanson, M.S. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr. Patterns 2003, 3, 459–462. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Scotti, M.M.; Shin, J.; Taylor, K.; Ivankovic, F.; Nutter, C.A.; Aslam, F.N.; Subramony, S.H.; Ranum, L.P.W.; Swanson, M.S. Loss of MBNL1 induces RNA misprocessing in the thymus and peripheral blood. Nat. Commun. 2020, 11, 2022. [Google Scholar] [CrossRef] [Green Version]
- Kernfeld, E.M.; Genga, R.M.J.; Neherin, K.; Magaletta, M.E.; Xu, P.; Maehr, R. A Single-Cell Transcriptomic Atlas of Thymus Organogenesis Resolves Cell Types and Developmental Maturation. Immunity 2018, 48, 1258–1270.e6. [Google Scholar] [CrossRef] [Green Version]
- Weber, B.N.; Chi, A.W.-S.; Chavez, A.; Yashiro-Ohtani, Y.; Yang, Q.; Shestova, O.; Bhandoola, A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature 2011, 476, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visconti, V.V.; Centofanti, F.; Fittipaldi, S.; Macrì, E.; Novelli, G.; Botta, A. Epigenetics of Myotonic Dystrophies: A Minireview. Int. J. Mol. Sci. 2021, 22, 12594. [Google Scholar] [CrossRef]
- Hamzeiy, H.; Savaş, D.; Tunca, C.; Şen, N.E.; Gündoğdu Eken, A.; Şahbaz, I.; Calini, D.; Tiloca, C.; Ticozzi, N.; Ratti, A.; et al. Elevated Global DNA Methylation Is Not Exclusive to Amyotrophic Lateral Sclerosis and Is Also Observed in Spinocerebellar Ataxia Types 1 and 2. NDD 2018, 18, 38–48. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- López Castel, A.; Overby, S.J.; Artero, R. MicroRNA-Based Therapeutic Perspectives in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 5600. [Google Scholar] [CrossRef] [PubMed]
- Cappella, M.; Perfetti, A.; Cardinali, B.; Garcia-Manteiga, J.M.; Carrara, M.; Provenzano, C.; Fuschi, P.; Cardani, R.; Renna, L.V.; Meola, G.; et al. High-throughput analysis of the RNA-induced silencing complex in myotonic dystrophy type 1 patients identifies the dysregulation of miR-29c and its target ASB2. Cell Death Dis. 2018, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Koutalianos, D.; Koutsoulidou, A.; Mytidou, C.; Kakouri, A.C.; Oulas, A.; Tomazou, M.; Kyriakides, T.C.; Prokopi, M.; Kapnisis, K.; Nikolenko, N.; et al. miR-223-3p and miR-24-3p as novel serum-based biomarkers for myotonic dystrophy type 1. Mol. Ther. -Methods Clin. Dev. 2021, 23, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Mateus, T.; Martins, F.; Nunes, A.; Herdeiro, M.T.; Rebelo, S. Metabolic Alterations in Myotonic Dystrophy Type 1 and Their Correlation with Lipin. Int. J. Env. Res. Public Health 2021, 18, 1794. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, N.K.; Annarao, S.; Sinha, N. Metabolic status of patients with muscular dystrophy in early phase of the disease: In vitro, high resolution NMR spectroscopy based metabolomics analysis of serum. Life Sci. 2016, 151, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Spitali, P.; Hettne, K.; Tsonaka, R.; Sabir, E.; Seyer, A.; Hemerik, J.B.A.; Goeman, J.J.; Picillo, E.; Ergoli, M.; Politano, L.; et al. Cross-sectional serum metabolomic study of multiple forms of muscular dystrophy. J. Cell Mol. Med. 2018, 22, 2442–2448. [Google Scholar] [CrossRef] [Green Version]
- Overby, S.J.; Cerro-Herreros, E.; Llamusi, B.; Artero, R. RNA-mediated therapies in myotonic dystrophy. Drug Discov. Today 2018, 23, 2013–2022. [Google Scholar] [CrossRef]
- Bargiela, A.; Cerro-Herreros, E.; Fernandez-Costa, J.M.; Vilchez, J.J.; Llamusi, B.; Artero, R. Increased autophagy and apoptosis contribute to muscle atrophy in a myotonic dystrophy type 1 Drosophila model. Dis. Model Mech. 2015, 8, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Sabater-Arcis, M.; Bargiela, A.; Furling, D.; Artero, R. miR-7 Restores Phenotypes in Myotonic Dystrophy Muscle Cells by Repressing Hyperactivated Autophagy. Mol. Ther. Nucleic Acids 2020, 19, 278–292. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Bargiela, A.; Sabater-Arcis, M.; Espinosa-Espinosa, J.; Zulaica, M.; de Munain, A.L.; Artero, R. Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. USA 2019, 116, 25203–25213. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, M.; Taylor, K.; Mochizuki, H.; Sobczak, K.; Takahashi, M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 2015, 3, 42–54. [Google Scholar] [CrossRef]
- Jenquin, J.R.; Yang, H.; Huigens III, R.W.; Nakamori, M.; Berglund, J.A. Combination Treatment of Erythromycin and Furamidine Provides Additive and Synergistic Rescue of Mis-splicing in Myotonic Dystrophy Type 1 Models. ACS Pharmacol. Transl. Sci. 2019, 2, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Laustriat, D.; Gide, J.; Barrault, L.; Chautard, E.; Benoit, C.; Auboeuf, D.; Boland, A.; Battail, C.; Artiguenave, F.; Deleuze, J.-F.; et al. In Vitro and In Vivo Modulation of Alternative Splicing by the Biguanide Metformin. Mol. Ther. Nucleic Acids 2015, 4, e262. [Google Scholar] [CrossRef] [PubMed]
- Jauvin, D.; Chrétien, J.; Pandey, S.K.; Martineau, L.; Revillod, L.; Bassez, G.; Lachon, A.; McLeod, A.R.; Gourdon, G.; Wheeler, T.M.; et al. Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol. Ther. Nucleic Acids 2017, 7, 465–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerro-Herreros, E.; Sabater-Arcis, M.; Fernandez-Costa, J.M.; Moreno, N.; Perez-Alonso, M.; Llamusi, B.; Artero, R. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018, 9, 2482. [Google Scholar] [CrossRef]
- Cerro-Herreros, E.; González-Martínez, I.; Moreno, N.; Espinosa-Espinosa, J.; Fernández-Costa, J.M.; Colom-Rodrigo, A.; Overby, S.J.; Seoane-Miraz, D.; Poyatos-García, J.; Vilchez, J.J.; et al. Preclinical characterization of antagomiR-218 as a potential treatment for myotonic dystrophy. Mol. Ther.-Nucleic Acids 2021, 26, 174–191. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Valanejad, L.; Zalewski, Z.A.; Karns, R.; Puymirat, J.; Nelson, D.; Witte, D.; Woodgett, J.; Timchenko, N.A.; et al. Correction of GSK3β at young age prevents muscle pathology in mice with myotonic dystrophy type 1. FASEB J. 2018, 32, 2073–2085. [Google Scholar] [CrossRef] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [Green Version]
- Dastidar, S.; Majumdar, D.; Tipanee, J.; Singh, K.; Klein, A.F.; Furling, D.; Chuah, M.K.; VandenDriessche, T. Comprehensive transcriptome-wide analysis of spliceopathy correction of myotonic dystrophy using CRISPR-Cas9 in iPSCs-derived cardiomyocytes. Mol. Ther. 2021, 30, 75–91. [Google Scholar] [CrossRef]
- Morales, F.; Couto, J.M.; Higham, C.F.; Hogg, G.; Cuenca, P.; Braida, C.; Wilson, R.H.; Adam, B.; del Valle, G.; Brian, R.; et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum. Mol. Genet. 2012, 21, 3558–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merker, J.D.; Wenger, A.M.; Sneddon, T.; Grove, M.; Zappala, Z.; Fresard, L.; Waggott, D.; Utiramerur, S.; Hou, Y.; Smith, K.S.; et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet. Med. 2018, 20, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flower, M.; Lomeikaite, V.; Ciosi, M.; Cumming, S.; Morales, F.; Lo, K.; Hensman Moss, D.; Jones, L.; Holmans, P.; Monckton, D.G.; et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain 2019, 142, 1876–1886. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, S.; Matsumoto, N. Long-read sequencing for rare human genetic diseases. J. Hum. Genet. 2020, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chintalaphani, S.R.; Pineda, S.S.; Deveson, I.W.; Kumar, K.R. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol. Commun. 2021, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Czubak, K.; Taylor, K.; Piasecka, A.; Sobczak, K.; Kozlowska, K.; Philips, A.; Sedehizadeh, S.; Brook, J.D.; Wojciechowska, M.; Kozlowski, P. Global Increase in Circular RNA Levels in Myotonic Dystrophy. Front. Genet. 2019, 10, 649. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
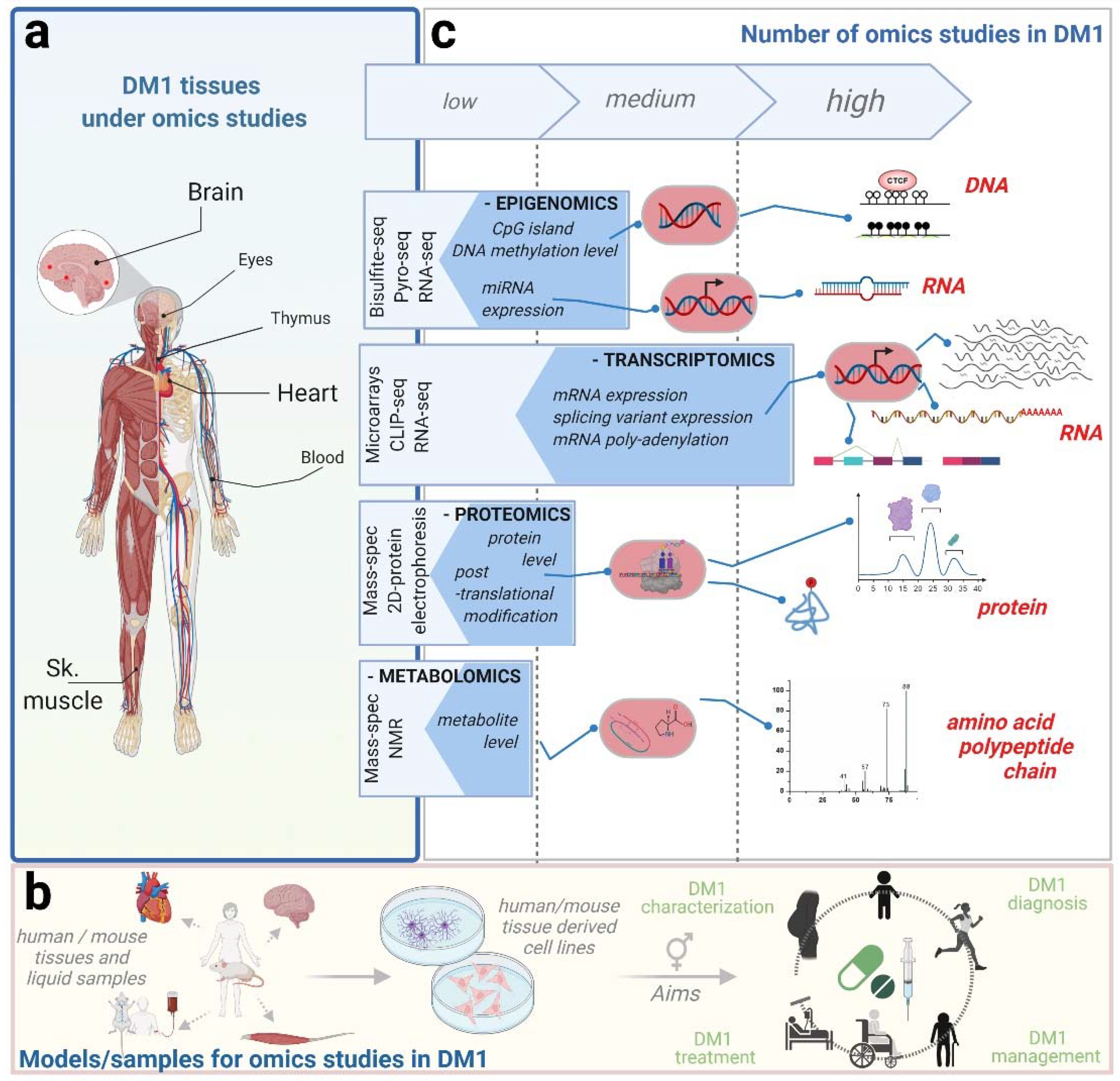

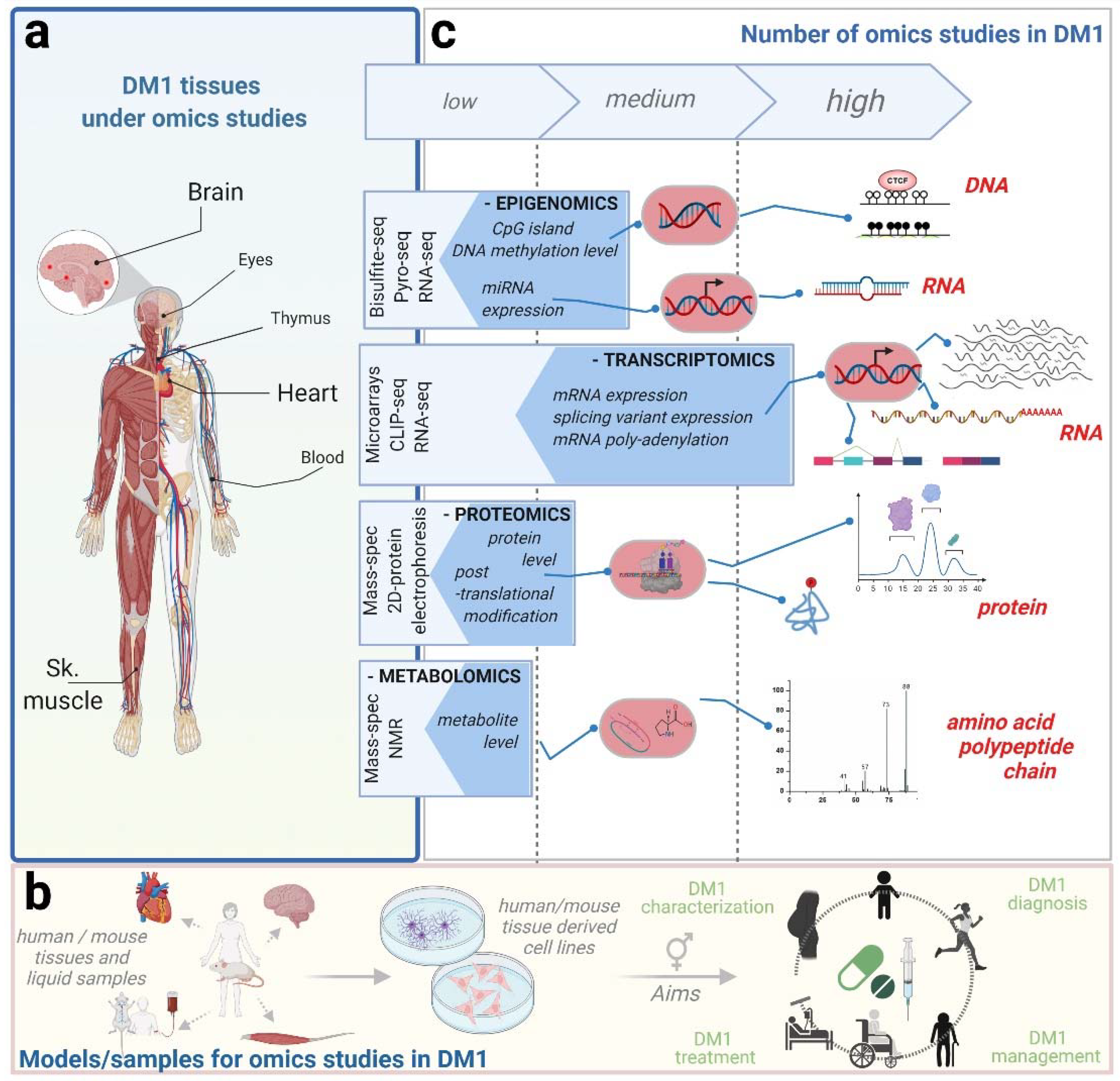{kind=link}
| Technique | Type | Sample | Species | Objective | Reference | Cite |
|---|---|---|---|---|---|---|
| CLIP-Seq | Illumina Genome Analyzer II | Embryonic fibroblasts | Human, mouse | Alternative polyadenylation | GSE60487 | [9] |
| 2D WB and nano LC-MS/MS | MALDI-TOF Voyager -DE-STR and Q-TOF MS | Frontal cortex and hippocampus | Mouse | Proteome and phosphoproteome analysis | Not provided | [26] |
| Microarray | ares_ucsc_mouse_59198_affyMouseA | Skeletal muscle biopsies | Mouse | Alternative splicing | GSE17986 | [36] |
| RNA-Seq | Illumina HiSeq 2000 | Brain, heart, skeletal muscle, and cell cultures | Mouse | Transcriptome analysis | GSE39911 | [38] |
| CLIP-Seq | Illumina Genome Analyzer II | Brain, heart, skeletal muscle, and cell cultures | Mouse | RBP binding sites | GSE39911 | [38] |
| RNA-Seq | Illumina HiSeq 2500 | Quadriceps | Mouse | Transcriptome analysis | PRJNA625451 | [40] |
| RNA-Seq | Illumina NextSeq 500 | Embryonic stem cells, myoblast, myotubes | Human | Transcriptome analysis | GSE160916 | [41] |
| RNA-Seq | Illumina HiSeq 2000 | Skeletal muscle and heart biopsies and autopsies | Human | Transcriptome analysis | GSE86356 | [42] |
| Microarray | Human Exon 1.0 ST array | Skeletal muscle biopsies | Human | Alternative splicing | GSE48828 | [47] |
| LC-MS/MS | EASY-nLC 1200-rbitrap Tribid MS | Myoblasts | Human | Global proteome analysis | PXD016056 | [49] |
| iTraq Nano-LC-MS/MS | Ultimate 3000 RSLC LTQ-Orbitrap Velos MS | Cerebellum | Mouse | Global proteome analysis | Not provided | [51] |
| CLIP-Seq | Illumina Genome Analyzer II | Brain | Human, mouse | RBP binding sites, alternative polyadenylation | GSE68890 | [54] |
| Microarray | Clariom D Arrays | Cell culture | Human | Transcriptome analysis | GSE164057 | [55] |
| RNA-Seq | Illumina HiSeq 2500 | Astrocytes, oligodendrocytes, and neurons | Mouse | Transcriptome analysis | GSE162093 | [56] |
| 2D WB and nano LC-MS/MS | Nano RSLC-Q | Astrocytes | Mouse | Phosphoproteome analysis | PXD025011 | [56] |
| RNA-Seq | Illumina NextSeq 500 | Frontal cortex biopsies | Human | Transcriptome analysis | GSE157428 | [57] |
| CLIP-Seq | Illumina Genome Analyzer II | Heart | Chicken | RBP binding sites | GSE67360 | [58] |
| RNA-Seq | Illumina HiSeq 2000 | Muscle, heart | Mouse | Transcriptome analysis | GSE61893 | [59] |
| CLIP-Seq | Illumina Genome Analyzer II | Muscle, heart | Mouse | RBP binding sites | GSE61893 | [59] |
| Microarray | Illumina MouseWG-6 v2.0 expression beadchip | Heart | Mouse | Transcriptome analysis | GSE48991 | [60] |
| RNA-Seq | Illumina HiSeq 4000 | Heart | Mouse | Transcriptome analysis | GSE126771 | [61] |
| RNA-Seq | Illumina NovaSeq 6000 | Heart | Mouse | Transcriptome analysis | GSE164825 | [62] |
| Microarray | HumanHT-12 v3 Expression BeadChip | Lens epithelial | Human | Transcriptome analysis | E-MEXP-3365 | [64] |
| RNA-Seq | Illumina HiSeq 2500, Illumina NextSeq 500 | Thymus | Human, mouse | Transcriptome analysis | GSE138691 | [66] |
| RNA-Seq | Illumina HiSeq 2500 | Biceps brachii | Human | miRNA/mRNA interactions | GSE108592 | [73] |
| RNA-Seq | Illumina NextSeq 500 | Blood Muscles, heart, and brain | Human, mouse | miRNA analysis | PRJEB46413 | [74] |
| RNA-Seq | Illumina NextSeq 550 | Cell culture | Human | Transcriptome analysis | GSE128844 | [82] |
| RNA-Seq | Illumina NextSeq 500 | Quadriceps | Mouse | Transcriptome analysis | PRJNA555349 | [84] |
| RNA-Seq | Illumina HiSeq 2000 | Cell culture | Human | Transcriptome analysis | GSE138789 | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espinosa-Espinosa, J.; González-Barriga, A.; López-Castel, A.; Artero, R. Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies. Int. J. Mol. Sci. 2022, 23, 1441. https://doi.org/10.3390/ijms23031441
Espinosa-Espinosa J, González-Barriga A, López-Castel A, Artero R. Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies. International Journal of Molecular Sciences. 2022; 23(3):1441. https://doi.org/10.3390/ijms23031441
Chicago/Turabian StyleEspinosa-Espinosa, Jorge, Anchel González-Barriga, Arturo López-Castel, and Rubén Artero. 2022. "Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies" International Journal of Molecular Sciences 23, no. 3: 1441. https://doi.org/10.3390/ijms23031441
APA StyleEspinosa-Espinosa, J., González-Barriga, A., López-Castel, A., & Artero, R. (2022). Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies. International Journal of Molecular Sciences, 23(3), 1441. https://doi.org/10.3390/ijms23031441