

MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. What Are Mitochondrial Derived Peptides (MDPs)?
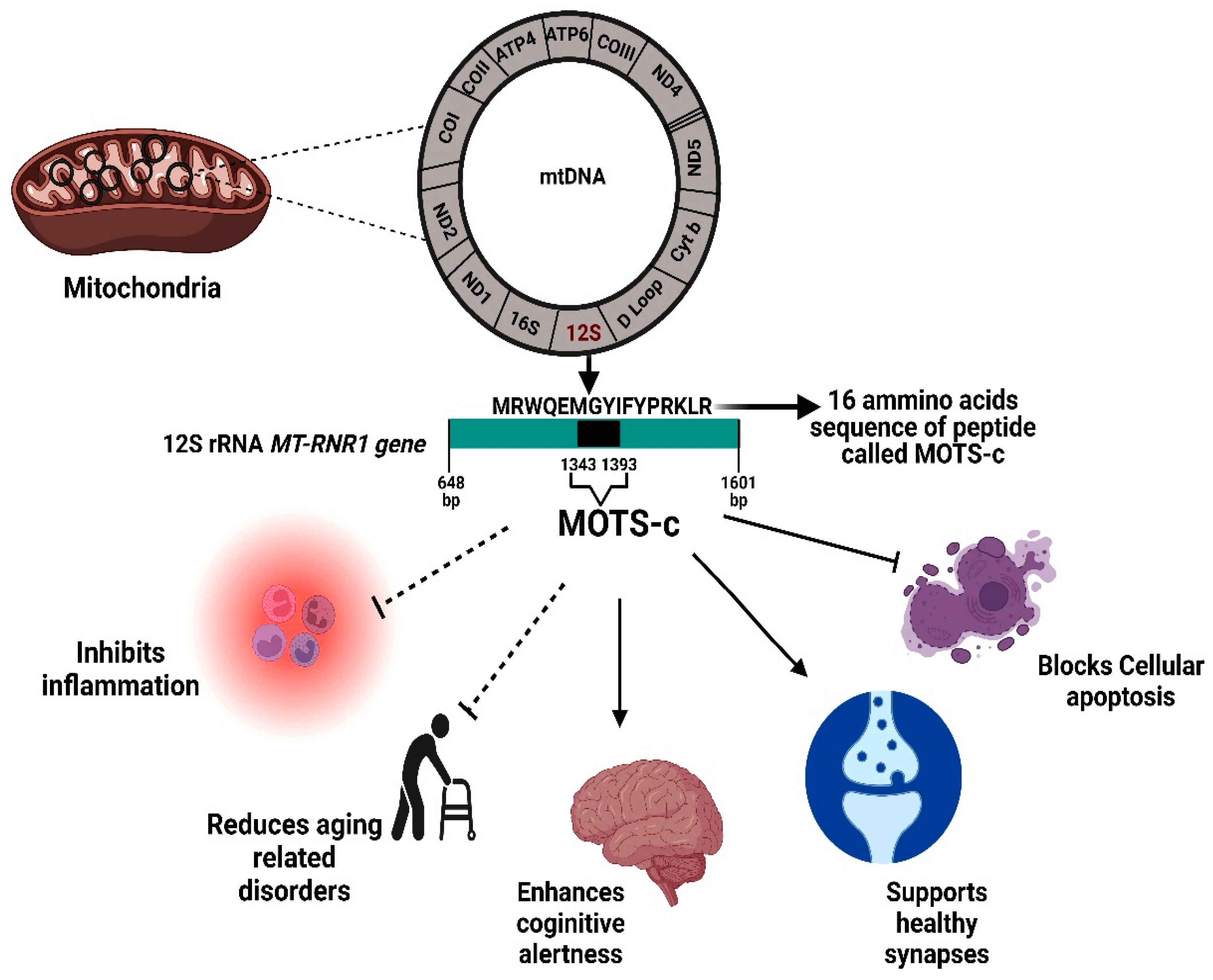
2.1. MOTS-c: Origin, History, and Structure
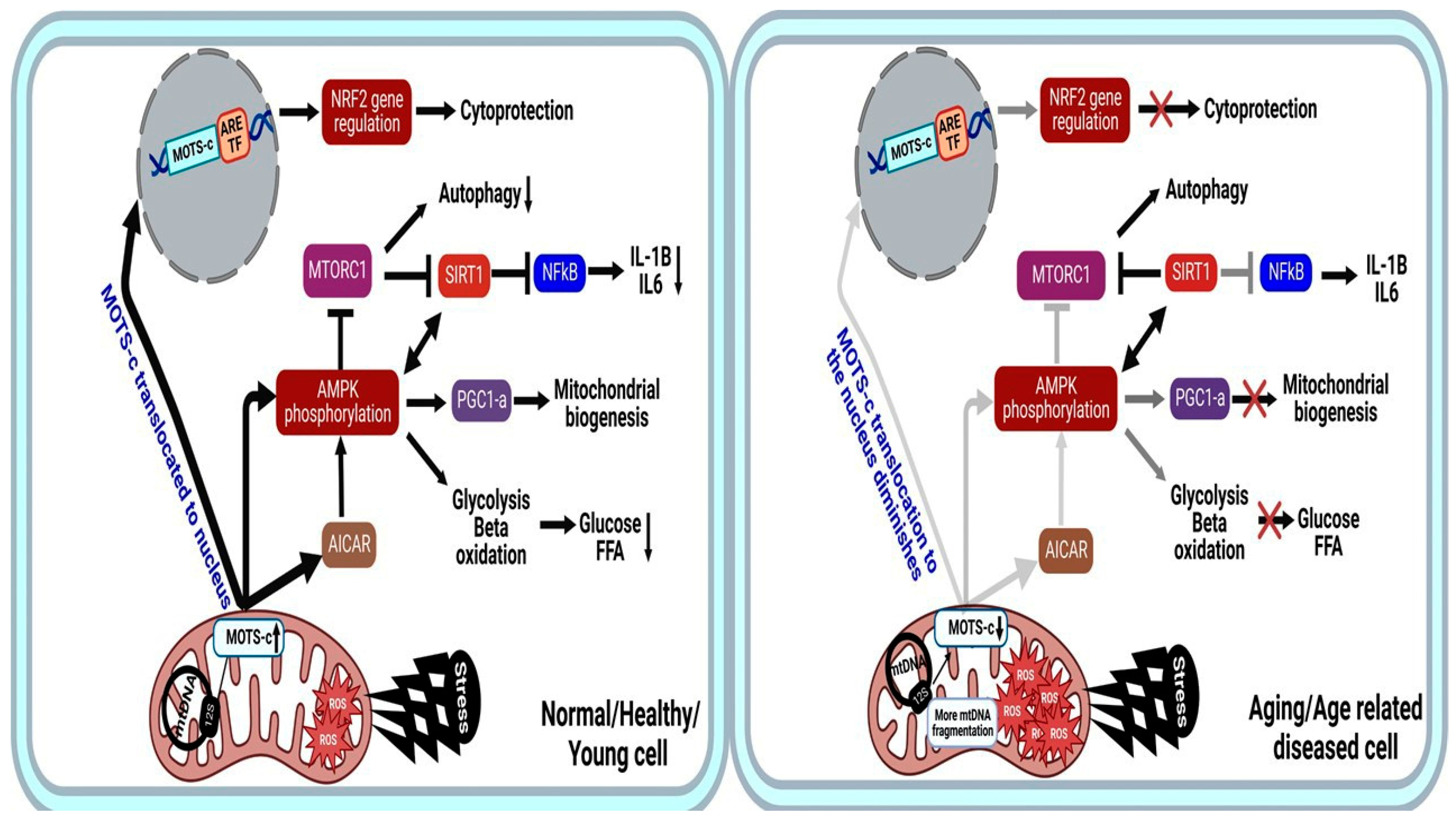
2.2. Molecular Mechanisms and Pathways of MOTS-C
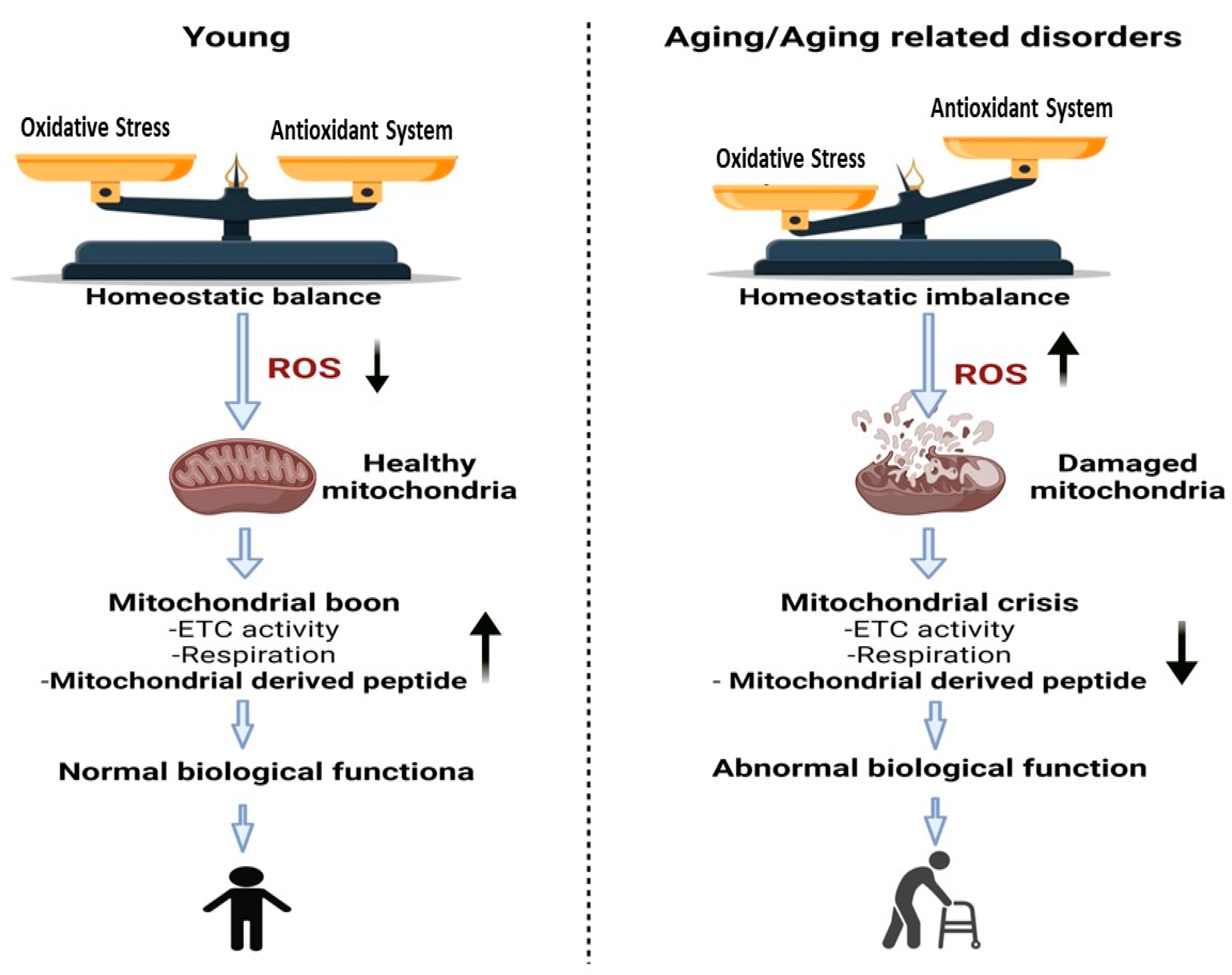
2.3. Aging and Longevity: MOTS-c
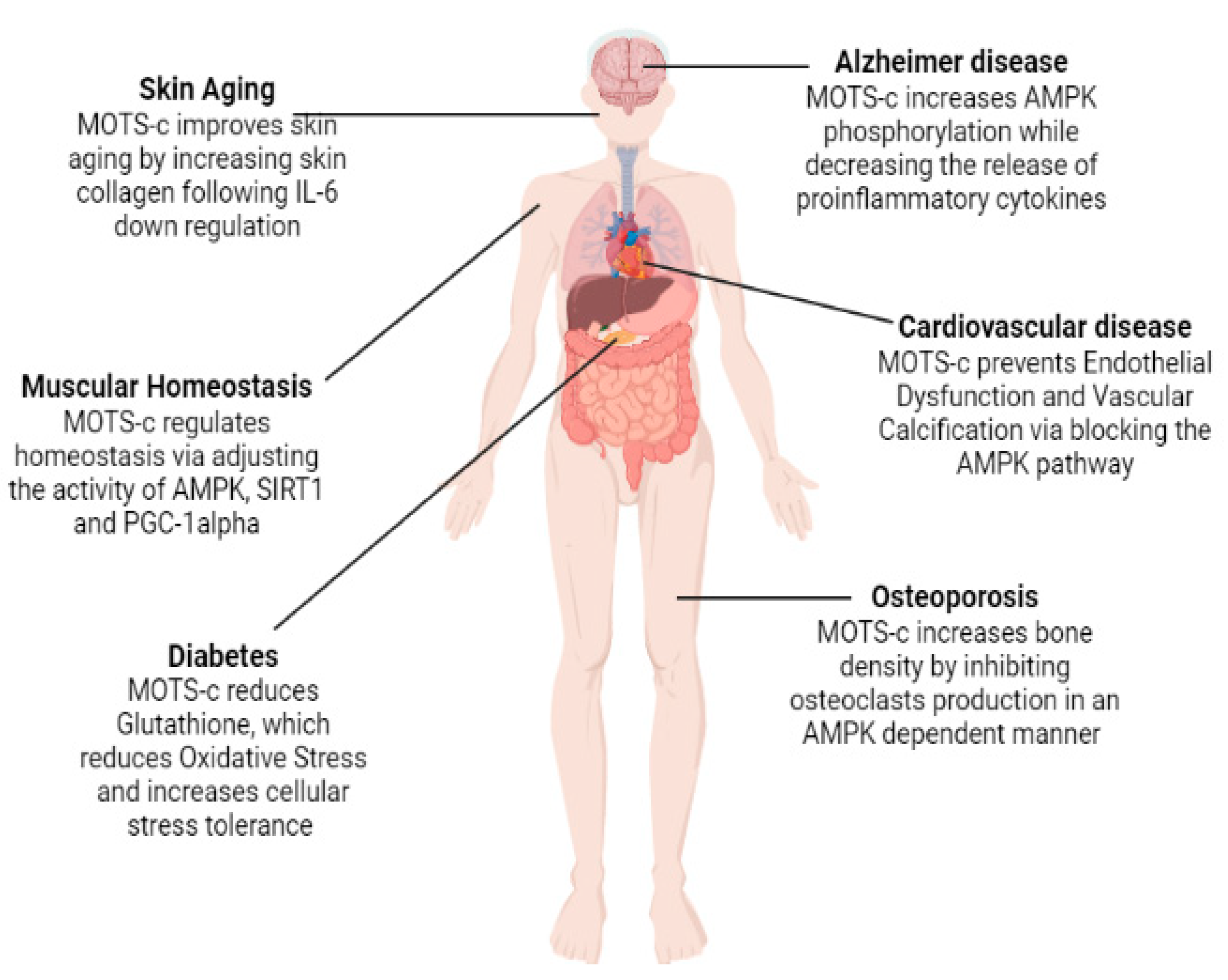
2.4. Age-Related Diseases: MOTS-c
2.4.1. Diabetes
2.4.2. Cardiovascular Diseases
2.4.3. Post-Menopausal Disorders
2.4.4. Osteoporosis
2.4.5. Alzheimer’s Disease
2.5. MOTS-c in Relation to Muscle Homeostasis and Physical Activity
3. Future Implications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Judge, A.; Dodd, M.S. Metabolism. Essays Biochem. 2020, 64, 607–647. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, J.R.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Hill, B.G.; Shiva, S.; Ballinger, S.; Zhang, J.; Darley-Usmar, V.M. Bioenergetics and translational metabolism: Implications for genetics, physiology and precision medicine. Biol. Chem. 2019, 401, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Zeng, J.; Drew, B.G.; Sallam, T.; Martin-Montalvo, A.; Wan, J.; Kim, S.J.; Mehta, H.; Hevener, A.L.; de Cabo, R.; et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015, 21, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621. [Google Scholar] [CrossRef]
- Yen, K.; Lee, C.; Mehta, H.; Cohen, P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J. Mol. Endocrinol. 2013, 50, R11–R19. [Google Scholar] [CrossRef]
- Mendelsohn, R.A.; Larrick, J.W. Mitochondrial-Derived Peptides Exacerbate Senescence. Rejuvenation Res. 2018, 21, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Miller, B.; Kumagai, H.; Silverstein, A.R.; Flores, M.; Yen, K. Mitochondrial-derived peptides in aging and age-related diseases. Geroscience 2021, 43, 1113–1121. [Google Scholar] [CrossRef]
- Gustafsson, M.C.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef]
- Galtier, N.; Enard, D.; Radondy, Y.; Bazin, E.; Belkhir, K. Mutation hot spots in mammalian mitochondrial DNA. Genome Res. 2006, 16, 215–222. [Google Scholar] [CrossRef]
- Sreekumar, G.P.; Kannan, R. Mechanisms of protection of retinal pigment epithelial cells from oxidant injury by humanin and other mitochondrial-derived peptides: Implications for age-related macular degeneration. Redox. Biol. 2020, 37, 101663. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Tsuzuki, T.; Nomiyama, H.; Setoyama, C.; Maeda, S.; Shimada, K. Presence of mitochondrial-DNA-like sequences in the human nuclear DNA. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Harhay, G.P.; Sonstegard, T.S.; Keele, J.W.; Heaton, M.P.; Clawson, M.L.; Snelling, W.M.; Wiedmann, R.T.; Van Tassell, C.P.; Smith, T.P. Characterization of 954 bovine full-CDS cDNA sequences. BMC Genom. 2005, 6, 166. [Google Scholar] [CrossRef]
- Brown, M.W.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.J.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Yang, B.; Yu, Q.; Chang, B.; Guo, Q.; Xu, S.; Yi, X.; Cao, S. MOTS-c interacts synergistically with exercise intervention to regulate PGC-1α expression, attenuate insulin resistance and enhance glucose metabolism in mice via AMPK signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 16612–16616. [Google Scholar] [CrossRef] [PubMed]
- Aspden, J.L.; Eyre-Walker, Y.C.; Phillips, R.J.; Amin, U.; Mumtaz, M.A.; Brocard, M.; Couso, J.P. Extensive translation of small open reading frames revealed by Poly-Ribo-Seq. eLife 2014, 3, e03528. [Google Scholar] [CrossRef]
- Durieux, J.; Wolff, S.; Dillin, A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 2011, 144, 79–91. [Google Scholar] [CrossRef]
- Woo, K.D.; Shadel, G.S. Mitochondrial stress signals revise an old aging theory. Cell 2011, 144, 11–12. [Google Scholar] [CrossRef]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018, 28, 516–524.e7. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kurita, M.; Aiso, S.; Nishimoto, I.; Matsuoka, M. Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor α/WSX-1/gp130. Mol. Biol. Cell 2009, 20, 2864–2873. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Kim, K.H.; Cohen, P. MOTS-c: A novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic. Biol. Med. 2016, 100, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.C.; Lai, R.W.; Woodhead, J.S.; Joly, J.H.; Mitchell, C.J.; Cameron-Smith, D.; Lu, R.; Cohen, P.; Graham, N.A.; Benayoun, B.A.; et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zarse, K.; Ristow, M. A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell Metab. 2015, 21, 355–356. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef] [PubMed]
- Crozet, P.; Margalha, L.; Confraria, A.; Rodrigues, A.; Martinho, C.; Adamo, M.; Elias, C.A.; Baena-González, E. Mechanisms of regulation of SNF1/AMPK/SnRK1 protein kinases. Front. Plant Sci. 2014, 5, 190. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Schnyder, S.; Handschin, C. Skeletal muscle as an endocrine organ: PGC-1α, myokines and exercise. Bone 2015, 80, 115–125. [Google Scholar] [CrossRef]
- Li, Q.; Lu, H.; Hu, G.; Ye, Z.; Zhai, D.; Yan, Z.; Wang, L.; Xiang, A.; Lu, Z. Earlier changes in mice after D-galactose treatment were improved by mitochondria derived small peptide MOTS-c. Biochem. Biophys. Res Commun. 2019, 513, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Fuku, N.; Pareja-Galeano, H.; Zempo, H.; Alis, R.; Arai, Y.; Lucia, A.; Hirose, N. The mitochondrial-derived peptide MOTS-c: A player in exceptional longevity? Aging Cell 2015, 14, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Guo, X.; Wang, Z.; Wang, P.; Zhang, Z.; Dong, J.; Zhuang, R.; Zhou, Y.; Ma, G.; Cai, W. Alphalipoic acid prevents oxidative stress and peripheral neuropathy in nab-paclitaxel-treated rats through the Nrf2 signalling pathway. Oxid. Med. Cell Longev. 2019, 2019, 3142732. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- D’Souza, R.F.; Woodhead, J.S.; Hedges, C.P.; Zeng, N.; Wan, J.; Kumagai, H.; Lee, C.; Cohen, P.; Cameron-Smith, D.; Mitchell, C.J.; et al. Increased expression of the mitochondrial derived peptide, MOTS-c, in skeletal muscle of healthy aging men is associated with myofiber composition. Aging 2020, 12, 5244–5258. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, S.M.; Sinclair, D.A. Slowing ageing by design: The rise of NAD (+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Haigis, C.M.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef]
- Zimmerman, J.A.; Malloy, V.; Krajcik, R.; Orentreich, N. Nutritional control of aging. Exp. Gerontol. 2003, 38, 47–52. [Google Scholar] [CrossRef]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Crimmins, E.M. Lifespan and Healthspan: Past, Present, and Promise. Gerontologist 2015, 55, 901–911. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Zouboulis, C.C. Molecular mechanisms of skin aging: State of the art. Ann. N. Y. Acad. Sci. 2007, 1119, 40–50. [Google Scholar] [CrossRef]
- Luckett, R.L.; Gallucci, R.M. Interleukin-6 (IL-6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL-6KO mice. Br. J. Dermatol. 2007, 156, 1163–1171. [Google Scholar] [CrossRef]
- Shadyab, H.A.; LaCroix, A.Z. Genetic factors associated with longevity: A review of recent findings. Ageing Res. Rev. 2015, 19, 1–7. [Google Scholar] [CrossRef]
- Sergiev, V.P.; Dontsova, O.A.; Berezkin, G.V. Theories of aging: An ever-evolving field. Acta Nat. 2015, 7, 9–18. [Google Scholar] [CrossRef]
- Dai, D.F.; Chiao, Y.A.; Martin, G.M.; Marcinek, D.J.; Basisty, N.; Quarles, E.K.; Rabinovitch, P.S. Mitochondrial-targeted catalase: Extended longevity and the roles in various disease models. Prog. Mol. Biol. Transl. Sci. 2017, 146, 203–241. [Google Scholar]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, L.; Zhuang, Z.; Hu, X.; Dong, D. Mitochondrial-Derived Peptides in Diabetes and Its Complications. Front. Endocrinol. 2021, 12, 808120. [Google Scholar] [CrossRef] [PubMed]
- Sada, K.; Nishikawa, T.; Kukidome, D.; Yoshinaga, T.; Kajihara, N.; Sonoda, K.; Senokuchi, T.; Motoshima, H.; Matsumura, T.; Araki, E. Hyperglycemia induces cellular hypoxia through production of mitochondrial ROS followed by suppression of aquaporin-1. PLoS ONE 2016, 11, e0158619. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Gudbjörnsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, Å. Type 1 diabetes mellitus. Nat. Rev. Dis. Primers 2017, 3, 17016. [Google Scholar] [CrossRef]
- Pugliese, A. Autoreactive T cells in type 1 diabetes. J. Clin. Investig. 2017, 127, 2881–2891. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Bailis, W.; Shyer, J.A.; Zhao, J.; Canaveras, J.C.; Al Khazal, F.J.; Qu, R.; Steach, H.R.; Bielecki, P.; Khan, O.; Jackson, R.; et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571, 403–407. [Google Scholar] [CrossRef]
- Weinberg, E.S.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Kong, B.S.; Min, S.H.; Lee, C.; Cho, Y.M. Mitochondrial-encoded MOTS-c prevents pancreatic islet destruction in autoimmune diabetes. Cell Rep. 2021, 36, 109447. [Google Scholar] [CrossRef]
- Kim, S.J.; Miller, B.; Mehta, H.H.; Xiao, J.; Wan, J.; Arpawong, T.E.; Yen, K.; Cohen, P. The mitochondrial-derived peptide MOTS-c is a regulator of plasma metabolites and enhances insulin sensitivity. Physiol. Rep. 2019, 7, e14171. [Google Scholar] [CrossRef]
- Foster-Schubert, K.E.; Alfano, C.M.; Duggan, C.R.; Xiao, L.; Campbell, K.L.; Kong, A.; Bain, C.E.; Wang, C.Y.; Blackburn, G.L.; McTiernan, A. Effect of diet and exercise, alone or combined, on weight and body composition in overweight-to-obese postmenopausal women. Obesity 2012, 20, 1628–1638. [Google Scholar] [CrossRef]
- Sivitz, I.W.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox. Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef]
- Ramanjaneya, M.; Bettahi, I.; Jerobin, J.; Chandra, P.; Abi Khalil, C.; Skarulis, M.; Atkin, S.L.; Abou-Samra, A.B. Mitochondrial-derived peptides are down regulated in diabetes subjects. Front Endocrinol. 2019, 10, 331. [Google Scholar] [CrossRef] [PubMed]
- Zempo, H.; Kim, S.J.; Fuku, N.; Nishida, Y.; Higaki, Y.; Wan, J.; Yen, K.; Miller, B.; Vicinanza, R.; Miyamoto-Mikami, E.; et al. A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c. Aging 2021, 13, 1692–1717. [Google Scholar] [CrossRef]
- Johns, E.C.; Denison, F.C.; Norman, J.E.; Reynolds, R.M. Gestational diabetes mellitus: Mechanisms, treatment, and complications. Trends Endocrinol. Metab. 2018, 29, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.R.; Reynolds, R.M. Short- and long-term outcomes of gestational diabetes and its treatment on fetal development. Prenat. Diagn. 2020, 40, 1085–1091. [Google Scholar] [CrossRef]
- Melchior, H.; Kurch-Bek, D.; Mund, M. The Prevalence of Gestational Diabetes. Dtsch. Arztebl. Int. 2017, 114, 412–418. [Google Scholar] [CrossRef]
- England, L.J.; Dietz, P.M.; Njoroge, T.; Callaghan, W.M.; Bruce, C.; Buus, R.M.; Williamson, D.F. Preventing type 2 diabetes: Public health implications for women with a history of gestational diabetes mellitus. Am. J. Obstet. Gynecol. 2009, 200, 365.e1–365.e8. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Pan, Y.; He, J.; Zhong, H.; Wu, Y.; Ji, C.; Liu, L.; Cui, X. The mitochondrial-derived peptide MOTS-c relieves hyperglycemia and insulin resistance in gestational diabetes mellitus. Pharmacol. Res. 2022, 175, 105987. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Nikiforov, N.G.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. The role of mitochondria-derived peptides in cardiovascular diseases and their potential as therapeutic targets. Int. J. Mol. Sci. 2021, 22, 8770. [Google Scholar] [CrossRef]
- Vancheri, F.; Longo, G.; Vancheri, S.; Henein, M. Coronary microvascular dysfunction. J. Clin. Med. 2020, 9, 2880. [Google Scholar] [CrossRef] [PubMed]
- North, J.B.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.; Huang, Q.; Li, S.; Zhou, Y.; Li, Z. Cardioprotection of CAPE-oNO2 against myocardial ischemia/reperfusion induced ROS generation via regulating the SIRT1/eNOS/NF-κB pathway in vivo and in vitro. Redox. Biol. 2018, 15, 62–73. [Google Scholar] [CrossRef]
- Yoon, M.H.; Reriani, M.; Mario, G.; Rihal, C.; Gulati, R.; Lennon, R.; Tilford, J.M.; Lerman, L.O.; Lerman, A. Long-term endothelin receptor antagonism attenuates coronary plaque progression in patients with early atherosclerosis. Int. J. Cardiol. 2013, 168, 1316–1321. [Google Scholar] [CrossRef]
- Choi, B.J.; Matsuo, Y.; Aoki, T.; Kwon, T.G.; Prasad, A.; Gulati, R.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Coronary endothelial dysfunction is associated with inflammation and vasa vasorum proliferation in patients with early atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2473–2477. [Google Scholar] [CrossRef]
- Qin, Q.; Delrio, S.; Wan, J.; Widmer, R.J.; Cohen, P.; Lerman, L.O.; Lerman, A. Downregulation of circulating MOTS-c levels in patients with coronary endothelial dysfunction. Int. J. Cardiol. 2018, 254, 23–27. [Google Scholar] [CrossRef]
- Zhai, D.; Ye, Z.; Jiang, Y.; Xu, C.; Ruan, B.; Yang, Y.; Lei, X.; Xiang, A.; Lu, H.; Zhu, Z.; et al. MOTS-c peptide increases survival and decreases bacterial load in mice infected with MRSA. Mol. Immunol. 2017, 92, 151–160. [Google Scholar] [CrossRef]
- Li, H.; Ren, K.; Jiang, T.; Zhao, G.J. MOTS-c attenuates endothelial dysfunction via suppressing the MAPK/NF-κB pathway. Int. J. Cardiol. 2018, 268, 40. [Google Scholar] [CrossRef]
- Yan, Z.; Zhu, S.; Wang, H.; Wang, L.; Du, T.; Ye, Z.; Zhai, D.; Zhu, Z.; Tian, X.; Lu, Z.; et al. MOTS-c inhibits Osteolysis in the Mouse Calvaria by affecting osteocyte-osteoclast crosstalk and inhibiting inflammation. Pharmacol. Res. 2019, 147, 104381. [Google Scholar] [CrossRef]
- Marulanda, J.; Alqarni, S.; Murshed, M. Mechanisms of vascular calcification and associated diseases. Curr. Pharm. Des. 2014, 20, 5801–5810. [Google Scholar] [CrossRef]
- Wei, M.; Gan, L.; Liu, Z.; Liu, L.; Chang, J.R.; Yin, D.C.; Cao, H.L.; Su, X.L.; Smith, W.W. Mitochondrial-derived peptide MOTS-c attenuates vascular calcification and secondary myocardial remodeling via adenosine monophosphate-activated protein kinase signaling pathway. Cardiorenal. Med. 2020, 10, 42–50. [Google Scholar] [CrossRef]
- Boccellino, M.; Di Domenico, M.; Donniacuo, M.; Bitti, G.; Gritti, G.; Ambrosio, P.; Quagliuolo, L.; Rinaldi, B. AT1-receptor blockade: Protective effects of irbesartan in cardiomyocytes under hypoxic stress. PLoS ONE 2018, 13, e0202297. [Google Scholar] [CrossRef]
- Honda, J.; Kimura, T.; Sakai, S.; Maruyama, H.; Tajiri, K.; Murakoshi, N.; Homma, S.; Miyauchi, T.; Aonuma, K. The Glucagon-Like Peptide-1 Receptor Agonist Liraglutide Improves Hypoxia-Induced Pulmonary Hypertension in Mice Partly via Normalization of Reduced ET B Receptor Expression. Physiol. Res. 2018, 67 (Suppl. 1), S175–S184. [Google Scholar] [CrossRef]
- Eapen, M.S.; Hansbro, P.M.; McAlinden, K.; Kim, R.Y.; Ward, C.; Hackett, T.L.; Walters, E.H.; Sohal, S.S. Abnormal M1/M2 macrophage phenotype profiles in the small airway wall and lumen in smokers and chronic obstructive pulmonary disease (COPD). Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Liu, H.; Li, K.; Su, X. Homocysteine up-regulates ETB receptors via suppression of autophagy in vascular smooth muscle cells. Microvasc. Res. 2018, 119, 13–21. [Google Scholar] [CrossRef]
- Rochette, L.; Rigal, E.; Dogon, G.; Malka, G.; Zeller, M.; Vergely, C.; Cottin, Y. Mitochondrial-derived peptides: New markers for cardiometabolic dysfunction. Arch. Cardiovasc. Dis. 2022, 115, 48–56. [Google Scholar] [CrossRef]
- Sharma, K.; Bansal, M. Association of age at menopause with post-menopausal symptoms, menarche age and other reproductive factors among rural females in Shimla, Himachal pradesh. J. Biosoc. Sci. 2018, 50, 19–25. [Google Scholar] [CrossRef]
- Pei, J.; Harakalova, M.; den Ruijter, H.; Pasterkamp, G.; Duncker, D.J.; Verhaar, M.C.; Asselbergs, F.W.; Cheng, C. Cardiorenal disease connection during post-menopause: The protective role of estrogen in uremic toxins induced microvascular dysfunction. Int. J. Cardiol. 2017, 238, 22–30. [Google Scholar] [CrossRef]
- El Maghraoui, A.; Rezqi, A.; El Mrahi, S.; Sadni, S.; Ghozlani, I.; Mounach, A. Osteoporosis, vertebral fractures and metabolic syndrome in postmenopausal women. BMC Endocr. Disord. 2014, 14, 93. [Google Scholar] [CrossRef]
- Lu, H.; Tang, S.; Xue, C.; Liu, Y.; Wang, J.; Zhang, W.; Luo, W.; Chen, J. Mitochondrial-derived peptide MOTS-c increases adipose thermogenic activation to promote cold adaptation. Int. J. Mol. Sci. 2019, 20, 2456. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, G.H.; Zhang, Y.K.; Shen, G.S.; Xu, Y.J.; Xu, N.W. Research on the correlation of diabetes mellitus complicated with osteoporosis with lipid metabolism, adipokines and inflammatory factors and its regression analysis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3900–3905. [Google Scholar]
- Seeman, E.; Delmas, P.D. Bone quality—The material and structural basis of bone strength and fragility. N. Engl. J. Med. 2006, 354, 2250–2261. [Google Scholar] [CrossRef]
- Kwan, P. Osteoporosis: From osteoscience to neuroscience and beyond. Mech. Ageing Dev. 2015, 145, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.S.; Schwabe, G.C.; Ehlers, C.; Marschall, C.; Reis, A.; Thiel, C.; Graul-Neumann, L. Two novel distinct COL1A2 mutations highlight the complexity of genotype–phenotype correlations in osteogenesis imperfecta and related connective tissue disorders. Eur. J. Med. Genet. 2013, 56, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Quint, P.; Ruan, M.; Pederson, L.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. TGF-β induces Wnt10b in osteoclasts from female mice to enhance coupling to osteoblasts. Endocrinology 2013, 154, 3745–3752. [Google Scholar] [CrossRef] [PubMed]
- Che, N.; Qiu, W.; Wang, J.K.; Sun, X.X.; Xu, L.X.; Liu, R.; Gu, L. MOTS-c improves osteoporosis by promoting the synthesis of type I collagen in osteoblasts via TGF-β/SMAD signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3183–3189. [Google Scholar]
- Hu, T.B.; Chen, W.Z. MOTS-c improves osteoporosis by promoting osteogenic differentiation of bone marrow mesenchymal stem cells via TGF-β/Smad pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7156–7163. [Google Scholar] [PubMed]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef]
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99 (Pt A), 11–15. [Google Scholar] [CrossRef]
- Ikeda, K.; Takeshita, S. The role of osteoclast differentiation and function in skeletal homeostasis. J. Biochem. 2016, 159, 1–8. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xiu, Y.; Li, J.; Xing, L.; Yao, Z. NF-κB-mediated regulation of osteoclastogenesis. Endocrinol. Metab. 2015, 30, 35–44. [Google Scholar] [CrossRef]
- Jeyabalan, J.; Shah, M.; Viollet, B.; Chenu, C. AMP-activated protein kinase pathway and bone metabolism. J. Endocrinol. 2012, 212, 277–290. [Google Scholar] [CrossRef]
- Kim, J.Y.; Min, J.Y.; Baek, J.M.; Ahn, S.J.; Jun, H.Y.; Yoon, K.H.; Choi, M.K.; Lee, M.S.; Oh, J. CTRP3 acts as a negative regulator of osteoclastogenesis through AMPK-c-Fos-NFATc1 signaling in vitro and RANKL-induced calvarial bone destruction in vivo. Bone 2015, 79, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Kola, B.; Bataveljic, A.; Arnett, T.R.; Viollet, B.; Saxon, L.; Korbonits, M.; Chenu, C. AMP-activated protein kinase (AMPK) activation regulates in vitro bone formation and bone mass. Bone 2010, 47, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Ming, W.; Lu, G.; Xin, S.; Huanyu, L.; Yinghao, J.; Xiaoying, L.; Chengming, X.; Banjun, R.; Li, W.; Zifan, L. Mitochondria related peptide MOTS-c suppresses ovariectomy-induced bone loss via AMPK activation. Biochem. Biophys. Res. Commun. 2016, 476, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Zaveri, T.D.; Dolgova, N.V.; Lewis, J.S.; Hamaker, K.; Clare-Salzler, M.J.; Keselowsky, B.G. Macrophage integrins modulate response to ultra-high molecular weight polyethylene particles and direct particle-induced osteolysis. Biomaterials 2017, 115, 128–140. [Google Scholar] [CrossRef]
- Goodman, S.B.; Gibon, E.; Yao, Z. The basic science of periprosthetic osteolysis. Instr. Course Lect. 2013, 62, 201–206. [Google Scholar]
- Sartori, M.; Vincenzi, F.; Ravani, A.; Cepollaro, S.; Martini, L.; Varani, K.; Fini, M.; Tschon, M. RAW 264.7 co-cultured with ultra-high molecular weight polyethylene particles spontaneously differentiate into osteoclasts: An in vitro model of periprosthetic osteolysis. J. Biomed. Mater. Res A 2017, 105, 510–520. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Cedazo-Minguez, A.; Kenigsberg, P.A.; Page, G.; Duarte, A.I.; Giusti, P.; Zusso, M.; Robert, P.; Frisoni, G.B.; Cattaneo, A.; et al. Current and emerging avenues for Alzheimer’s disease drug targets. J. Intern. Med. 2019, 286, 398–437. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Jiang, J.; Chang, X.; Nie, Y.; Shen, Y.; Liang, X.; Peng, Y.; Chang, M. Peripheral Administration of a Cell-Penetrating MOTS-c Analogue Enhances Memory and Attenuates Aβ1–42-or LPS-Induced Memory Impairment through Inhibiting Neuroinflammation. ACS Chem. Neurosci. 2021, 12, 1506–1518. [Google Scholar] [CrossRef]
- Lancaster, I.G.; Febbraio, M.A. The immunomodulating role of exercise in metabolic disease. Trends Immunol. 2014, 35, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Zanuso, S.; Jimenez, A.; Pugliese, G.; Corigliano, G.; Balducci, S. Exercise for the management of type 2 diabetes: A review of the evidence. Acta Diabetol. 2010, 47, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, M.; Ma, J.; Pang, X.; Yuan, J.; Pan, Y.; Fu, Y.; Laher, I. MOTS-c and Exercise Restore Cardiac Function by Activating of NRG1-ErbB Signaling in Diabetic Rats. Front Endocrinol. 2022, 13, 812032. [Google Scholar] [CrossRef] [PubMed]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef]
- Wallace, D.C. Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 1–16. [Google Scholar] [CrossRef]
- Verdin, E. NAD⁺ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Richie, J.P., Jr.; Leutzinger, Y.; Parthasarathy, S.; Maixoy, V.; Orentreich, N.; Zimmerman, J.A. Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB J. 1994, 8, 1302–1307. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohtashami, Z.; Singh, M.K.; Salimiaghdam, N.; Ozgul, M.; Kenney, M.C. MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 11991. https://doi.org/10.3390/ijms231911991
Mohtashami Z, Singh MK, Salimiaghdam N, Ozgul M, Kenney MC. MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases. International Journal of Molecular Sciences. 2022; 23(19):11991. https://doi.org/10.3390/ijms231911991
Chicago/Turabian StyleMohtashami, Zahra, Mithalesh K. Singh, Nasim Salimiaghdam, Mustafa Ozgul, and M. Cristina Kenney. 2022. "MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases" International Journal of Molecular Sciences 23, no. 19: 11991. https://doi.org/10.3390/ijms231911991
APA StyleMohtashami, Z., Singh, M. K., Salimiaghdam, N., Ozgul, M., & Kenney, M. C. (2022). MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases. International Journal of Molecular Sciences, 23(19), 11991. https://doi.org/10.3390/ijms231911991