
Mitophagy in Human Diseases



 , ,
, ,  and
and
Abstract
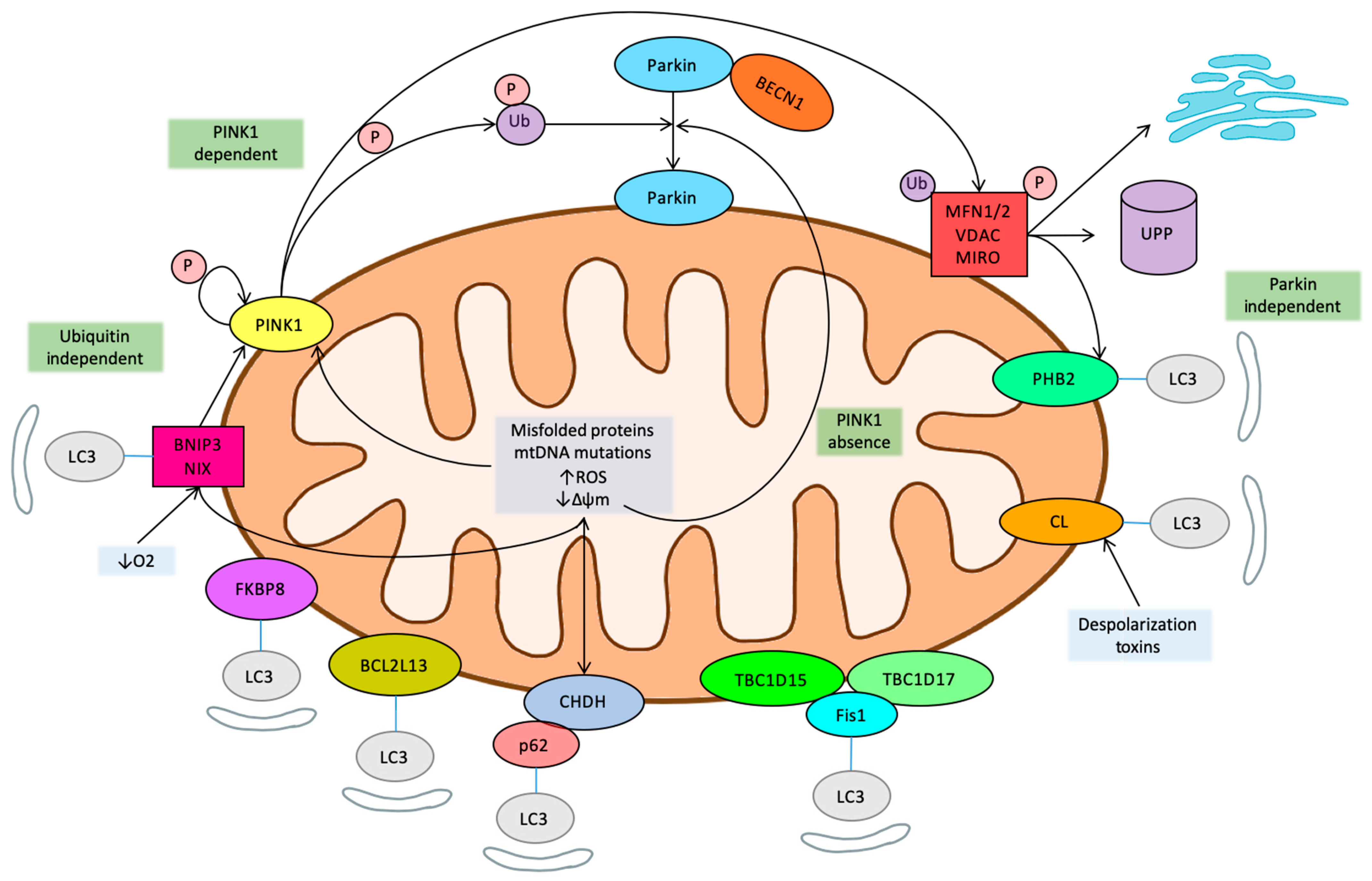
1. History and Pathways of Mitophagy
1.1. Mitophagy, a Type of Autophagy
1.2. PINK1
1.3. Mitochondrial Homeostasis-Related Pathways
1.4. LC3
1.5. Ubiquitin Independent Mitophagy
1.6. Novel Regulatory Pathways
2. Mitophagy in Neuropsychiatric and Neurodegenerative Diseases
2.1. Mitophagy in Neurodegenerative Diseases (Parkinson’s, Dementia, Alzheimer’s, Post-Stroke Cognitive Impairment)
2.1.1. Parkinson’s Disease
2.1.2. Dementia
2.1.3. Alzheimer’s Disease
2.1.4. Post-Stroke Cognitive Impairment (PSCI)
2.2. Mitophagy in Neurodegenerative Diseases: ALS and Huntington’s Disease
2.2.1. Amyotrophic Lateral Sclerosis
2.2.2. Huntington’s Disease
2.3. Mitophagy in Developmental Neurodegenerative Diseases (Autism and Epilepsy)
2.3.1. Autism
2.3.2. Epilepsy
2.4. Mitophagy in Psychiatric Diseases (Schizophrenia, Bipolar Disorder, and Depression)
2.4.1. Schizophrenia
2.4.2. Bipolar Disorder
2.4.3. Depression

{kind=link}
| Disease/Mutation | Mitophagy Defect | References |
|---|---|---|
| PD PINK1-deficient patients | Excessive mitophagosomes | [89,90,96,97] |
| PD PARKIN-deficient patients | Abnormal mitophagosomes; blockade mitochondrial turnover | [89,90,95] |
| Wild-type or PD A53T-Alpha-synuclein overexpression | Reduced mitophagy | [89] |
| PD L1444 GBA overexpression | Reduced mitochondrial dynamics | [89] |
| PD SREBF1 mutation | Reduced Parkin levels | [91,92] |
| PD FBX07 mutation | Impaired mitophagy | [101,102,103] |
| PD PARK7 mutation | Impaired mitochondria dynamics | [107] |
| PD LRKK2/PARK8 mutation | Decreased lysosomes, abnormal mitophagy | [116,117,118,119,120] |
| PD ATP13A2 mutation | Abnormal mitochondria function | [125,126] |
| PD VPS35 mutation | Reduced parkin activity | [128,129,130] |
| AD and Down syndrome dementia | Aberrant mitochondria | [137] |
| AD patients | Aberrant mitochondria Accumulation of autophagy intermediates | [145,146,147,148,149] |
| AD patients | Impaired Parkin mitochondrial translocation | [84,152] |
| AD DSC1 mutation | Reduced mitochondria transport | [90] |
| FTD p62 mutation | Reduced mitophagy Impaired lysosomes | [132,141,142] |
| ALS patients | Impaired mitophagy | [60,175] |
| ALS TBK1/OPTN mutations | Disrupted parkin activity | [177] |
| ALS p62 mutation | Damaged mitochondria Aberrant autophagic vacuoles | [181] |
| HD patients | Reduced mitochondriogenesis and mitophagy | [182,183,184,185,186,187,188,189] |
| HD Huntington mutation | Hyperactive mitophagy | [190] |
| Autism patients | Loss of mitophagy | [196] |
| Epilepsy HACE 1 mutations | Impaired mitophagy | [206,207,208] |
| Epilepsy CLN5 mutations | Impaired mitophagy | [210] |
| Schizophrenia DISC1 overexpression | Altered mitophagy Blocked mitochondrial transport | [219] |
| BD mutations | Reduced mitophagy Increased fission | [231,232,233,235] |
| Depression DISC1/p62 mutations | Reduced mitophagy Impaired mitochondrial function | [9,241,242,243,244] |
3. Mitophagy in Liver Diseases
3.1. Non-Alcoholic Fatty Liver Disease
3.2. Alcoholic Liver Disease
4. Mitophagy in Type 2 Diabetes and Obesity
4.1. Type 2 Diabetes
4.2. Obesity
5. The Role of Mitophagy in Cardiovascular Diseases (CVD)
5.1. Mitophagy in Macrovascular Diseases: Atherosclerosis
5.2. Mitophagy in Heart-Related Diseases: Heart Failure, Ischemia-Reperfusion Injury, and Cardiac Hypertrophy
5.2.1. Heart Failure
5.2.2. Ischemia-Reperfusion Injury
5.2.3. Cardiac Hypertrophy
5.3. Mitophagy in Microvascular Diseases: The Case of Diabetic Retinopathy
6. Mitophagy in Skeletal Muscle Diseases
7. Mitophagy and Lifestyle Impact on Aging
8. Mitophagy in Cancer
9. Mitophagy in Inflammation
10. Targeting Mitophagy
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| ABAD | aβ-binding alcohol dehydrogenase |
| AβPP | Aβ precursor protein |
| AD | Alzheimer’s disease |
| AGEs | Advanced glycation end products |
| ALCAT1 | Acyl-CoA lysocardiolipin acyltransferase-1 |
| ALD | Alcoholic liver disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | Adenosine monophosphate activated protein kinase |
| ApoE4 | Apolipoprotein e4 |
| AS | Atherosclerosis |
| ATG32 | Autophagy-related protein 32 |
| ATP+ | Adenosine triphosphate |
| AUTAC | Autophagy targeting chimera |
| AV | autophagic vacuoles |
| α-syn | 𝛼-synuclein |
| BAT | Brown adipose tissue |
| BCL2 | B-Cell CLL/Lymphoma 2 |
| BCL2L13 | Bcl2 like 13 |
| BECN1 | Coiled-coil myosin-like BCL2-interacting protein |
| BNIP3 | BCL2/adenovirus E1B 19 kDa interacting protein 3 |
| BNIP3L/NIX | BCL2/adenovirus E1B 19 kDa inter-acting protein 3 like |
| BP | Bipolar Disorder |
| BRCA1 | Breast Cancer 1 |
| CACNA1C | Calcium voltage-gated channel subunit alpha1 C |
| CaMKK-β | calcium-dependent protein kinase kinase-β |
| CHDH | Choline dehydrogenase |
| CHMP2B | Charged multivesicular body protein 2b |
| CK2α | casein Kinase 2 |
| CL | Cardiolipin |
| CLN5 | Ceroid-Lipofuscinosis, Neuronal 5 |
| CMA | Chaperone mediated autophagy |
| CVD | Cardiovascular disease |
| CoQ | Coenzyme Q10 |
| COX | Cytochrome c oxidase |
| cypD | Cyclophilin D |
| DCT-1 | Daf-16/FOXO controlled germline tumor affecting-1 |
| DISC1 | Disrupted in schizophrenia 1 |
| DJ-1 | Parkinsonism Associated Deglycase |
| DPR | Dipeptide repeat |
| DR | Diabetic retinopathy |
| Drp1 | Dynamin-related protein 1 |
| ER | Endoplasmic reticulum |
| ETC | Electron transport chain |
| FBXO7 | F-box only protein 7 |
| FBXW7 | F-box and WD40 domain protein only protein 7 |
| FGF21 | Fibroblast Growth Factor 21 |
| FIS1 | Mitochondrial fission 1 protein |
| FKBP8 | FK506-binding protein 8 |
| FTD | Frontotemporal Dementia |
| FUNDC1 | FUN14 domain containing protein 1 |
| GABA | γ-aminobutyric acid |
| GSK3β | Glycogen Synthase Kinase 3 Beta |
| GTPases | Guanosine triphosphate hydrolases |
| GPER | G protein-coupled estrogen receptor 1 |
| HACE1 | HECT Domain And Ankyrin Repeat Containing E3 Ubiquitin Protein Ligase 1 |
| HD | Huntington’s disease |
| HIF1α | Hypoxia-inducible factor 1 Subunit Alpha |
| HIV | Human immunodeficiency virus |
| HF | Heart failure |
| HG | High glucose |
| IL-1β | Interleukin 1 beta |
| IMM | Inner mitochondrial membrane |
| IPC | Ischemic preconditioning |
| I/R | Ischemia-reperfusion |
| IRI | Ischemia/reperfusion injury |
| KA | Kainic acid |
| KO | Knock out |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| LIR | LC3-interacting region |
| L-OPA1 | Full-length OPA1 |
| LRRK2 | Leucin-rich repeat serine/threonine kinase2 |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
| MERRF | Myoclonic epilepsy with ragged red fibers |
| Mff | Mitochondrial fission factor |
| MFN | Mitofusin |
| mHtt | Mutant Htt |
| MI | Myocardial infarction |
| MIDs | Mitochondrial Disorders |
| MiD49 | Mitochondrial dynamic protein 49 kDa |
| MiD51 | Mitochondrial dynamic protein 51 kDa |
| MIM | Mitochondrial inner membrane |
| MIRAS | Myoclonic mitochondrial recessive ataxia syndrome |
| MIRO | Mitochondrial Rho |
| MitoPLD | Mitochondrial Phospholipase D |
| MSN | Medium spiny neurons |
| mTOR | Mechanistic Target of Rapamycin Kinase |
| MEMSA | Myoclonus, epilepsy, myOPAthy, sensory ataxia |
| MPP | Mitochondrial processing peptidase |
| MPP+ | 1methyl-4-phenylpyridinium |
| mPTP | mitochondrial permeability transition pore |
| MSNs | Striatal medium spiny GABA neurons |
| Mst1 | Macrophage stimulating 1 |
| mtDNA | Mitochondrial DNA |
| MT-ND5 | Mitochondrially Encoded NADH:Ubiquinone Oxidoreductase Core Subunit 5 |
| MT-ND2 | Mitochondrially Encoded NADH:Ubiquinone Oxidoreductase Core Subunit 2 |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH+ | Nicotinamide adenine dinucleotide hydride |
| NAFLD | Non-alcoholic fatty liver disease |
| NCAN | Neurocan |
| NBR1 | Neighbor of BRCA1 gene 1 |
| NDP52 | Nuclear domain 10 protein 52 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRP3 | NLR Family Pyrin Domain Containing 3 |
| NO | Nitric oxide |
| 6-OHDA | 6-hydroxydopamine |
| OMM | Outer mitochondrial membrane |
| ONOO- | Peroxynitrite |
| OPA1 | Dynamin-like GTPase optic atrophy 1 |
| OPTN | Optineurin |
| oxLDL | Oxidized LDL |
| OXPHOS | Oxidative phosphorylation |
| PARL | Presenilin-associated rhomboid-like protein |
| PB1 | Phox and Bem1 domain |
| PD | Parkinson’s disease |
| PDE4 | Phosphodiesterase 4A |
| PGC-1𝛼 | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PHB | Prohibitin 2 |
| PINK1 | PTEN-induced putative kinase 1 |
| PKC | Protein Kinase C |
| PSCI | Post-stroke cognitive impairment |
| pTau | Tau protein |
| p62 | Sequestosome-1 |
| RBX1 | Ring-Box 1 |
| ROS | Reactive oxygen species |
| SAMP8 | Senescence accelerated mice P8 |
| SCAE | Spinocerebellar ataxia with epilepsy |
| SCF complex | Skp-Cullin-F-box ubiquitin E3 ligase complex |
| SE | Status epilepticus |
| Serine 65 | Ser65 |
| SIRT1 | Silent Mating Type Information Regulation 2, S. Cerevisiae, Homolog 1 |
| SLP-2 | Stomatin-like protein 2 |
| SMURF1 | SMAD Specific E3 Ubiquitin Protein Ligase 1 |
| Sod1 | Superoxide dismutase 1 |
| SREBF1 | Sterol regulatory element binding transcription factor 1 |
| STEMI | ST-Elevation Myocardial Infarction |
| TAX1 | Trans-activating transcriptional regulatory protein of HTLV-1 |
| TAX1BP1 | TAX1 binding protein 1 |
| TBC1D | TBC1 domain family member |
| TBK1 | Tank-binding kinase 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| TENM4 | Teneurin Transmembrane Protein 4 |
| TFEB | Transcription Factor EB |
| TIMM | Translocase of the inner mitochondrial membrane |
| TNIK | TRAF2 And NCK Interacting Kinase |
| TLR | Toll-like receptor |
| FUS/TLS | Translocated in Liposarcoma |
| TOMM | Translocase of the outer mitochondrial membrane |
| TSPO | Translocator protein |
| Trx | Thioredoxin |
| TXNIP | Thioredoxin-Interacting Protein |
| T2D | Type 2 diabetes |
| UPS | Ubiquitin proteasome system |
| USP30 | Ubiquitin carboxyl-terminal hydrolase 30 |
| VDAC1 | Voltage-dependent anion channel 1 |
| VCP | Valosin-containing protein |
| VSMCs | Vascular smooth muscle cell |
| Rab-GAP | Rab GTPase activating protein |
| RGCs | Retinal ganglion cells |
| VCI | Vascular cognitive impairment |
| WAT | White adipose tissue |
| WDFY3/Alfy | WD Repeat And FYVE Domain Containing 3 |
| WT | Wild type |
References
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Lemasters, J.J. Selective Mitochondrial Autophagy, or Mitophagy, as a Targeted Defense Against Oxidative Stress, Mitochondrial Dysfunction, and Aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective Degradation of Mitochondria by Mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Bakula, D.; Scheibye-Knudsen, M. Mitoph Aging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 2020, 8. [Google Scholar]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a Decisive Process for Cell Death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Kanki, T.; Klionsky, D.J. Mitophagy in yeast occurs through a selective mechanism. J. Biol. Chem. 2008, 283, 32386–32393. [Google Scholar] [CrossRef]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.-S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of Mitophagy in Cellular Homeostasis, Physiology and Pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Zachari, M.; Ktistakis, N.T. Mammalian Mitophagosome Formation: A Focus on the Early Signals and Steps. Front. Cell Dev. Biol. 2020, 8, 171. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Variants of Mitochondrial Autophagy: Types 1 and 2 Mitophagy and Micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Um, J.-H.; Kim, Y.Y.; Finkel, T.; Yun, J. Sensitive Measurement of Mitophagy by Flow Cytometry Using the pH-Dependent Fluorescent Reporter Mt-Keima. J. Vis. Exp. 2018, 138, 58099. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef]
- Cummins, N.; Götz, J. Shedding light on mitophagy in neurons: What is the evidence for PINK1/Parkin mitophagy in vivo? Cell Mol. Life Sci. 2018, 75, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Zhu, W.; Yu, J.; Wang, Q.; Zhang, J.; Cui, Y.; Pan, X.; Gao, X.; Sun, H. Succinate Accumulation Induces Mitochondrial Reactive Oxygen Species Generation and Promotes Status Epilepticus in the Kainic Acid Rat Model. Redox Biol. 2020, 28, 101365. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and Mitophagy in cANCER. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Regulation and Roles of Mitophagy at Synapses. Mech. Ageing Dev. 2020, 187, 111216. [Google Scholar] [CrossRef]
- Schiavi, A.; Strappazzon, F.; Ventura, N. Mitophagy and Iron: Two Actors Sharing the Stage in Age-Associated Neuronal Pathologies. Mech. Ageing Dev. 2020, 188, 111252. [Google Scholar] [CrossRef] [PubMed]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S. PINK1 import regulation at a crossroad of mitochondrial fate: The molecular mechanisms of PINK1 import. J. Biochem. 2020, 167, 217–224. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N. Phospho-ubiquitin: Upending the PINK–Parkin–ubiquitin cascade. J. Biochem. 2016, 159, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.J.; Hess, S.; Chan, D.C. Broad Activation of the Ubiquitin–Proteasome System by Parkin is Critical for Mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2014, 4, 6–13. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.Y.; Renton, A.E.M.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Yamano, K.; Youle, R.J. PINK1 is Degraded Through the N-end Rule Pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Nat. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During Autophagy Mitochondria Elongate, are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Westrate, L.M.; Drocco, J.A.; Martin, K.R.; Hlavacek, W.S.; MacKeigan, J.P. Mitochondrial Morphological Features are Associated with Fission and Fusion Events. PLoS ONE 2014, 9, e95265. [Google Scholar] [CrossRef]
- Zhou, Y.; Long, Q.; Liu, X. A new sight: Topology-dependent mitophagy. Cell Biol. Toxicol. 2020, 36, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.E.; Arias-Durán, C.; Ávalos-Guajardo, Y.; Aedo, G.; Verdejo, H.E.; Parra, V.; Lavandero, S. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol. Asp. Med. 2020, 71, 100822. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.S.; Zhou, Y.; Wen, P.-C.; Tajkhorshid, E.; Kwok, W.-M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitiNation. Proc. Nat. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Calì, T. PINK1/Parkin Mediated Mitophagy, Ca2+ Signalling, and ER–Mitochondria Contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef]
- Safiulina, D.; Kuum, M.; Choubey, V.; Gogichaishvili, N.; Liiv, J.; Hickey, M.A.; Cagalinec, M.; Mandel, M.; Zeb, A.; Liiv, M.; et al. Miro proteins prime mitochondria for Parkin translocation and mitophagy. EMBO J. 2019, 38, e99384. [Google Scholar] [CrossRef] [PubMed]
- Ney, P.A. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 2015, 1853, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Autophagy machinery in the context of mammalian mitophagy. Biochim. Biophys. Acta 2015, 1853, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L.F. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Sánchez, R.; Yakhine-Diop, S.M.S.; Bravo-San Pedro, J.M.; Pizarro-Estrella, E.; Rodríguez-Arribas, M.; Climent, V.; Martin-Cano, F.E.; González-Soltero, M.E.; Tandon, A.; Fuentes, J.M.; et al. PINK1 Deficiency Enhances Autophagy and Mitophagy Induction. Mol. Cell Oncol. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in Cell Death, Autophagy, and Mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Park, S.; Choi, S.-G.; Yoo, S.-M.; Son, J.H.; Jung, Y.-K. Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 2014, 10, 1906–1920. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 2014, 3, e01612. [Google Scholar] [CrossRef]
- Xia, X.; Katzenell, S.; Reinhart, E.F.; Bauer, K.M.; Pellegrini, M.; Ragusa, M.J. A pseudo-receiver domain in Atg32 is required for mitophagy. Autophagy 2018, 14, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 Recruits LC3A to Mediate Parkin-Independent Mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chiang, W.-C.; Sumpter, R.; Mishra, P.; Levine, B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 2017, 168, 224–238.e10. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination signal for Mitophagy in Neuronal Cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef]
- Saito, T.; Nah, J.; Oka, S.-I.; Mukai, R.; Monden, Y.; Maejima, Y.; Ikeda, Y.; Sciarretta, S.; Liu, T.; Li, H.; et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J. Clin. Investig. 2019, 129, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Qi, J.; Ronai, Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends. Biochem. Sci. 2013, 38, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.-L.; Kaasik, A. BECN1 is Involved in the Initiation of Mitophagy. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Cugliandolo, F.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef]
- Heeman, B.; den Haute, C.V.; Aelvoet, S.-A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.H.; Willems, P.H.G.M.; et al. Depletion of PINK1 Affects Mitochondrial Metabolism, Calcium Homeostasis and Energy Maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef]
- Gómez-Sánchez, R.; Gegg, M.E.; Bravo-San Pedro, J.M.; Niso-Santano, M.; Alvarez-Erviti, L.; Pizarro-Estrella, E.; Gutiérrez-Martín, Y.; Alvarez-Barrientos, A.; Fuentes, J.M.; González-Polo, R.A.; et al. Mitochondrial Impairment Increases FL-PINK1 levels by Calcium-Dependent Gene Expression. Neuro. Biol. Dis. 2014, 62, 426–440. [Google Scholar] [CrossRef]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Shefa, U.; Jeong, N.Y.; Song, I.O.; Chung, H.-J.; Kim, D.; Jung, J.; Huh, Y. Mitophagy links oxidative stress conditions and neurodegenerative diseases. Neural Regen. Res. 2019, 14, 749–756. [Google Scholar] [PubMed]
- Triolo, M.; Hood, D.A. Mitochondrial Breakdown in Skeletal Muscle and the Emerging Role of the Lysosomes. Arch. Biochem. Biophys. 2019, 661, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhang, R.; Krigman, J.; McAdams, A.; Ozgen, S.; Sun, N. A Healthy Heart and a Healthy Brain: Looking at Mitophagy. Front. Cell Dev. Biol. 2020, 8, 294. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalón-García, I.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Talaverón-Rey, M.; Jackson, S.; Falcón-Moya, R.; Rodríguez-Moreno, A.; Sánchez-Alcázar, J.A. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef]
- Homayoun, H. Parkinson disease. Ann. Intern. Med. 2018, 169, ITC33–ITC48. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Onyango, I.G.; Khan, S.M.; Bennett, J.P. Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front. Biosci. 2017, 22, 854–872. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef]
- Do, C.B.; Tung, J.Y.; Dorfman, E.; Kiefer, A.K.; Drabant, E.M.; Francke, U.; Mountain, J.L.; Goldman, S.M.; Tanner, C.M.; Langston, J.W.; et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011, 7, e1002141. [Google Scholar] [CrossRef] [PubMed]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mitophagy in neurodegenerative diseases. Neurochem. Int. 2018, 117, 156–166. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H.V. PINK1-parkin-dependent mitophagy involves ubiquitiNation of mitofusins 1 and 2. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef]
- Zanon, A.; Kalvakuri, S.; Rakovic, A.; Foco, L.; Guida, M.; Schwienbacher, C.; Serafin, A.; Rudolph, F.; Trilck, M.; Grünewald, A.; et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum. Mol. Genet. 2017, 26, 2412–2425. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Yakhine-Diop, S.M.S.; Niso-Santano, M.; Rodríguez-Arribas, M.; Gómez-Sánchez, R.; Martínez-Chacón, G.; Uribe-Carretero, E.; Navarro-García, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M.; et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2466–2481. [Google Scholar] [CrossRef] [PubMed]
- Paisán-Ruiz, C.; Guevara, R.; Federoff, M.; Hanagasi, H.; Sina, F.; Elahi, E.; Schneider, S.A.; Schwingenschuh, P.; Bajaj, N.; Emre, M.; et al. Early-Onset L-Dopa-Responsive Parkinsonism with Pyramidal Signs due to ATP13A2, PLA2G6, FBXO7 and Spatacsin Mutations. Mov. Disord. 2010, 25, 1791–1800. [Google Scholar] [CrossRef]
- Lehman, N.L. The Ubiquitin Proteasome System in Neuropathology. Acta Neuropathol. 2009, 118, 329–347. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Xie, S.P.; Sathiyamoorthy, S.; Saw, W.T.; Sing, T.Y.; Ng, S.H.; Chua, H.P.H.; Tang, A.M.Y.; Shaffra, F.; Li, Z.; et al. F-box Protein 7 Mutations Promote Protein Aggregation in Mitochondria and Inhibit Mitophagy. Hum. Mol. Genet. 2015, 24, 6314–6330. [Google Scholar] [CrossRef]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.-W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef]
- Hao, L.-Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is Critical for Mitochondrial Function and Rescues PINK1 Loss of Function. Proc. Nat. Acad. Sci. USA 2010, 107, 9747–9752. Available online: https://www.pnas.org/content/107/21/9747 (accessed on 4 September 2020). [CrossRef] [PubMed]
- Mihoub, M.; Abdallah, J.; Richarme, G. Protein Repair from Glycation by Glyoxals by the DJ-1 Family Maillard Deglycases. In DJ-1/PARK7 Protein: Parkinson’s Disease, Cancer and Oxidative Stress-Induced Diseases; Ariga, H., Iguchi-Ariga, S.M.M., Eds.; Springer: Singapore, 2017; pp. 133–147. ISBN 978-981-10-6583-5. [Google Scholar]
- Moscovitz, O.; Ben-Nissan, G.; Fainer, I.; Pollack, D.; Mizrachi, L.; Sharon, M. The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat. Commun. 2015, 6, 6609. [Google Scholar] [CrossRef]
- Nichols, W.C.; Pankratz, N.; Hernandez, D.; Paisán-Ruíz, C.; Jain, S.; Halter, C.A.; Michaels, V.E.; Reed, T.; Rudolph, A.; Shults, C.W.; et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 2005, 365, 410–412. [Google Scholar] [CrossRef]
- Gilks, W.P.; Abou-Sleiman, P.M.; Gandhi, S.; Jain, S.; Singleton, A.; Lees, A.J.; Shaw, K.; Bhatia, K.P.; Bonifati, V.; Quinn, N.P.; et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 2005, 365, 415–416. [Google Scholar] [CrossRef]
- Duque, A.F.; Lopez, J.C.; Benitez, B.; Hernandez, H.; Yunis, J.J.; Fernandez, W.; Arboleda, H.; Arboleda, G. Analysis of the LRRK2 p.G2019S mutation in Colombian Parkinson’s Disease Patients. Colomb. Med. 2015, 46, 117–121. [Google Scholar] [CrossRef]
- Roosen, D.A.; Cookson, M.R. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol. Neurodegener 2016, 11, 73. [Google Scholar] [CrossRef]
- Su, Y.-C.; Guo, X.; Qi, X. Threonine 56 phosphorylation of Bcl-2 is required for LRRK2 G2019S-induced mitochondrial depolarization and autophagy. Biochim. Biophys. Acta 2015, 1852, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic parkinson’s disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, G.; Onofri, F.; Cirnaru, M.D.; Kaiser, C.J.O.; Jagtap, P.; Kastenmuller, A.; Pischedda, F.; Marte, A.; von Zweydorf, F.; Vogt, A.; et al. Leucine-Rich Repeat Kinase 2 Binds to Neuronal Vesicles through Protein Interactions Mediated by Its C-Terminal WD40 Domain. Mol. Cell. Biol. 2014, 34, 2147–2161. Available online: http://mcb.asm.org/cgi/doi/10.1128/MCB.00914-13 (accessed on 4 September 2020). [CrossRef]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-Rich Repeat Kinase 2 Regulates Autophagy through a Calcium-Dependent Pathway Involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef]
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107. [Google Scholar] [CrossRef]
- Esteves, A.R.; Cardoso, S.M. LRRK2 at the Crossroad Between Autophagy and Microtubule Trafficking: Insights into Parkinson’s Disease. Neuroscientist 2017, 23, 16–26. [Google Scholar] [CrossRef]
- Ganguly, U.; Chakrabarti, S.S.; Kaur, U.; Mukherjee, A.; Chakrabarti, S. Alpha-synuclein, Proteotoxicity and Parkinson’s Disease: Search for Neuroprotective Therapy. Curr. Neuropharmacol. 2018, 16, 1086–1097. [Google Scholar] [CrossRef]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J.; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Lehtonen, Š.; Sonninen, T.-M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.-L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Ashkenazi, A.; Jimenez-Sanchez, M.; Rubinsztein, D.C. The Parkinson’s disease-associated genes ATP13A2 and SYT11 regulate autophagy via a common pathway. Nat. Commun. 2016, 7, 11803. [Google Scholar] [CrossRef]
- Norris, K.L.; Hao, R.; Chen, L.-F.; Lai, C.-H.; Kapur, M.; Shaughnessy, P.J.; Chou, D.; Yan, J.; Taylor, J.P.; Engelender, S.; et al. Convergence of Parkin, PINK1, and α-Synuclein on Stress-Induced Mitochondrial Morphological Remodeling. J. Biol. Chem. 2015, 290, 13862–13874. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.H.; Alaimo, A.; Gorojod, R.M.; Porte Alcon, S.; Fuentes, F.; Coluccio Leskow, F.; Kotler, M.L. Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein. Mol. Cell. Neurosci. 2018, 88, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The Close Encounter Between Alpha-Synuclein and Mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef]
- van der Zee, J.; Van Langenhove, T.; Kovacs, G.G.; Dillen, L.; Deschamps, W.; Engelborghs, S.; Matěj, R.; Vandenbulcke, M.; Sieben, A.; Dermaut, B.; et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014, 128, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s disease and the environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Ahfeldt, T.; Ordureau, A.; Bell, C.; Sarrafha, L.; Sun, C.; Piccinotti, S.; Grass, T.; Parfitt, G.M.; Paulo, J.A.; Yanagawa, F.; et al. Pathogenic pathways in early-onset autosomal recessive parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Rep. 2020, 14, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Krainc, D. The role of dopamine in the pathogenesis of GBA1-linked Parkinson’s disease. Neurobiol. Dis. 2019, 132, 104545. [Google Scholar] [CrossRef]
- Coskun, P.E.; Wyrembak, J.; Derbereva, O.; Melkonian, G.; Doran, E.; Lott, I.T.; Head, E.; Cotman, C.W.; Wallace, D.C. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. J. Alzheimers Dis. 2010, 20, S293–S310. [Google Scholar] [CrossRef] [PubMed]
- Spano, M.; Signorelli, M.; Vitaliani, R.; Aguglia, E.; Giometto, B. The possible involvement of mitochondrial dysfunctions in Lewy body dementia: A systematic review. Funct. Neurol. 2015, 30, 151–158. [Google Scholar] [CrossRef]
- Kragh, C.L.; Ubhi, K.; Wyss-Corey, T.; Masliah, E. Autophagy in dementias: Autophagy in dementias. Brain Pathol. 2012, 22, 99–109. [Google Scholar] [CrossRef]
- Kalaria, R.N. The pathology and pathophysiology of vascular dementia. Neuropharmacology 2018, 134, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- van der Zee, J.; Urwin, H.; Engelborghs, S.; Bruyland, M.; Vandenberghe, R.; Dermaut, B.; De Pooter, T.; Peeters, K.; Santens, P.; De Deyn, P.P.; et al. CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum. Mol. Genet. 2008, 17, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Sheehan, P.; Chen, S.; Yue, Z. Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Mol. Neurodegener. 2017, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Cook, C.; Liu, C.-C.; Kang, S.S.; Lin, W.-L.; DeTure, M.; Heckman, M.G.; Diehl, N.N.; Al-Shaikh, F.S.H.; et al. Mitophagy alterations in Alzheimer’s disease are associated with granulovacuolar degeneration and early tau pathology. Alzheimers Dement. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. On the central role of mitochondria dysfunction and oxidative stress in Alzheimer’s disease. Neurol. Sci. 2019, 40, 1527–1540. [Google Scholar] [CrossRef]
- Cai, Q.; Tammineni, P. Alterations in Mitochondrial Quality Control in Alzheimer’s Disease. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell. Neurosci. 2019, 98, 109–120. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. ELife 2017, 6, e21776. [Google Scholar] [CrossRef] [PubMed]
- Nuriel, T.; Peng, K.Y.; Ashok, A.; Dillman, A.A.; Figueroa, H.Y.; Apuzzo, J.; Ambat, J.; Levy, E.; Cookson, M.R.; Mathews, P.M.; et al. The Endosomal-Lysosomal Pathway Is Dysregulated by APOE4 Expression in vivo. Front. Neurosci. 2017, 11, 702. [Google Scholar] [CrossRef]
- Koenig, L.N.; McCue, L.M.; Grant, E.; Massoumzadeh, P.; Roe, C.M.; Xiong, C.; Moulder, K.L.; Wang, L.; Zazulia, A.R.; Kelly, P.; et al. Lack of Association between Acute Stroke, Post-Stroke Dementia, Race, and β-Amyloid Status. Neuroimage Clin. 2021, 29, 102553. [Google Scholar] [CrossRef]
- Molad, J.; Hallevi, H.; Korczyn, A.D.; Kliper, E.; Auriel, E.; Bornstein, N.M.; Ben Assayag, E. Vascular and Neurodegenerative Markers for the Prediction of Post-Stroke Cognitive Impairment: Results from the TABASCO Study. J. Alzheimers Dis. 2019, 70, 889–898. [Google Scholar] [CrossRef]
- Bordet, R.; Ihl, R.; Korczyn, A.D.; Lanza, G.; Jansa, J.; Hoerr, R.; Guekht, A. Towards the Concept of Disease-Modifier in Post-Stroke or Vascular Cognitive Impairment: A Consensus Report. BMC Med. 2017, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Lan, R.; Wu, J.-T.; Wu, T.; Ma, Y.-Z.; Wang, B.-Q.; Zheng, H.-Z.; Li, Y.-N.; Wang, Y.; Gu, C.-Q.; Zhang, Y. Mitophagy is Activated in Brain Damage Induced by Cerebral Ischemia and Reperfusion via the PINK1/Parkin/p62 Signalling Pathway. Brain Res. Bull. 2018, 142, 63–77. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, X. Role of Glucose Metabolism and ATP in Maintaining PINK1 Levels during Parkin-Mediated Mitochondrial Damage Responses. J. Biol. Chem. 2015, 290, 904–917. [Google Scholar] [CrossRef]
- Kumari, S.; Anderson, L.; Farmer, S.; Mehta, S.L.; Li, P.A. Hyperglycemia Alters Mitochondrial Fission and Fusion Proteins in Mice Subjected to Cerebral Ischemia and Reperfusion. Transl. Stroke Res. 2012, 3, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Shrestha, S.; Li, J.; Yu, X.; Chen, J.; Yan, F.; Ying, G.; Gu, C.; Wang, L.; Chen, G. Melatonin-Mediated Mitophagy Protects Against Early Brain Injury after Subarachnoid Hemorrhage through Inhibition of NLRP3 Inflammasome Activation. Sci. Rep. 2017, 7, 2417. [Google Scholar] [CrossRef]
- Zheng, G.; Wang, L.; Li, X.; Niu, X.; Xu, G.; Lv, P. Rapamycin alleviates cognitive impairment in murine vascular dementia: The enhancement of mitophagy by PI3K/AKT/mTOR axis. Tissue Cell 2021, 69, 101481. [Google Scholar] [CrossRef]
- Feng, J.; Chen, X.; Guan, B.; Li, C.; Qiu, J.; Shen, J. Inhibition of Peroxynitrite-Induced Mitophagy Activation Attenuates Cerebral Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 6369–6386. [Google Scholar] [CrossRef]
- Tang, Y.-C.; Tian, H.-X.; Yi, T.; Chen, H.-B. The Critical Roles of Mitophagy in Cerebral Ischemia. Protein Cell 2016, 7, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Dou, S.; Zhu, J.; Wang, H.; Xu, D.; Wang, C.; Cheng, B.; Bai, B. The Role of Mitophagy in Ischemic Stroke. Front. Neurol. 2020, 11. [Google Scholar]
- Wang, M.; Lee, H.; Elkin, K.; Bardhi, R.; Guan, L.; Chandra, A.; Geng, X.; Ding, Y. Detrimental and Beneficial Effect of Autophagy and a Potential Therapeutic Target after Ischemic Stroke. Evid. Based Complementary Altern. Med. 2020, 2020, e8372647. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Sureda, A.; Sanches-Silva, A.; Devi, K.P.; Ahmed, T.; Shahid, M.; Sobarzo-Sánchez, E.; Dacrema, M.; Daglia, M.; Braidy, N.; et al. Novel Therapeutic Strategies for Stroke: The Role of Autophagy. Crit. Rev. Clin. Lab. Sci. 2019, 56, 182–199. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun. 2017, 483, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Manfredi, G. Mitochondrial function, morphology, and axonal transport in amyotrophic lateral sclerosis. Antioxid Redox Signal 2009, 11, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denomiNators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Goldstein, L.H.; Abrahams, S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: Nature of impairment and implications for assessment. Lancet Neurol. 2013, 12, 368–380. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Nat. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Hadano, S.; Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Akatsuka, A.; Koike, M.; Aoki, M.; Uchiyama, Y.; Itoyama, Y.; Ikeda, J.-E. Loss of ALS2/Alsin exacerbates motor dysfunction in a SOD1H46R-expressing mouse ALS model by disturbing endolysosomal trafficking. PLoS ONE 2010, 5, e9805. [Google Scholar] [CrossRef] [PubMed]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in Neurodegeneration and Aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Pinho, B.R.; Soares, T.R.; de Proença, J.; Duchen, M.R.; Oliveira, J.M.A. Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol. Dis. 2016, 90, 51–57. [Google Scholar] [CrossRef]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Disatnik, M.-H.; Mochly-Rosen, D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol. Med. 2015, 7, 1307–1326. [Google Scholar] [CrossRef]
- Bae, B.-I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.-J.; Snyder, S.H.; Sawa, A. Mutant huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Natl. Acad Sci. USA 2006, 103, 3405–3409. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.-J.; Rival, T.; Liévens, J.-C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; de Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.-J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 1–17. [Google Scholar] [CrossRef]
- Pecorelli, A.; Ferrara, F.; Messano, N.; Cordone, V.; Schiavone, M.L.; Cervellati, F.; Woodby, B.; Cervellati, C.; Hayek, J.; Valacchi, G. Alterations of Mitochondrial Bioenergetics, Dynamics, and Morphology Support the Theory of Oxidative Damage Involvement in Autism Spectrum Disorder. FASEB J. 2020, 34, 6521–6538. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Rapin, I. Epilepsy in autism. Lancet Neurol. 2002, 1, 352–358. [Google Scholar] [CrossRef]
- Tang, G.; Rios, P.G.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; DiMauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; De Vries, R.L.A.; Lebot, P.; Wikstrom, J.D.; Torgyekes, E.; Shirihai, O.S.; Przedborski, S.; Schon, E.A. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum. Mol. Genet. 2012, 21, 978–990. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Panoutsopoulos, A.; Riyadh, M.A.; Kaushik, G.; Halmai, J.; Levenson, R.; Zarbalis, K.S.; Giulivi, C. Beyond autophagy: A novel role for autism-linked Wdfy3 in brain mitophagy. Sci. Rep. 2018, 8, 11348. [Google Scholar] [CrossRef] [PubMed]
- Sirven, J.I. Epilepsy: A Spectrum Disorder. Cold Spring Harb. Perspect. Med. 2015, 5, a022848. [Google Scholar] [CrossRef] [PubMed]
- Besag, F.M. Epilepsy in PATIENTs with Autism: Links, Risks and Treatment Challenges. Neuropsychiatr. Dis. Treat. 2017, 14, 1–10. [Google Scholar] [CrossRef]
- Saneto, R.P. Epilepsy and Mitochondrial Dysfunction: A Single Center’s Experience. J. Inborn Errors Metab. Screen. 2017. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondrial Dysfunction and Oxidative Stress: A Contributing Link to Acquired Epilepsy? J. Bioenerg. Biomembr. 2010, 42, 449. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Eid, T.; Gruenbaum, S.E.; Dhaher, R.; Lee, T.-S.W.; Zhou, Y.; Danbolt, N.C. The glutamate-glutamine cycle in epilepsy. Adv. Neurobiol. 2016, 13, 351–400. [Google Scholar]
- Garrido-Maraver, J.; Paz, M.V.; Cordero, M.D.; Bautista-Lorite, J.; Oropesa-Ávila, M.; de la Mata, M.; Pavón, A.D.; de Lavera, I.; Alcocer-Gómez, E.; Galán, F.; et al. Critical role of AMP-activated protein kinase in the balance between mitophagy and mitochondrial biogenesis in MELAS disease. Biochim. Biophys. Acta 2015, 1852, 2535–2553. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-K.; Chen, S.-D.; Lin, K.-J.; Chuang, Y.-C. Seizure-induced oxidative stress in status epilepticus: Is antioxidant beneficial? Antioxid 2020, 9, 1029. [Google Scholar] [CrossRef]
- Ugarteburu, O.; Sánchez-Vilés, M.; Ramos, J.; Barcos-Rodríguez, T.; Garrabou, G.; García-Villoria, J.; Ribes, A.; Tort, F. physiopathological bases of the disease caused by hace1 mutations: Alterations in autophagy, mitophagy and oxidative stress response. JCM 2020, 9, 913. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Ravi, S.; Pradeep, B.E.; Subramanyam, K.N.; Choudhary, B.; Srinivasan, S.; Khanchandani, P. A novel loss-of-function mutation in HACE1 is linked to a genetic disorder in a patient from India. Hum. Genome Var. 2018, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, R.; Parry, D.A.; Nalbach, L.; Logan, C.V.; Strom, T.M.; Hartill, V.L.; Carr, I.M.; Korenke, G.C.; Uppal, S.; Ahmed, M.; et al. HACE1 deficiency causes an autosomal recessive neurodevelopmental syndrome. J. Med. Genet. 2015, 52, 797–803. [Google Scholar] [CrossRef]
- Akawi, N.; McRae, J.; Ansari, M.; Balasubramanian, M.; Blyth, M.; Brady, A.F.; Clayton, S.; Cole, T.; Deshpande, C.; Fitzgerald, T.W.; et al. Discovery of four recessive developmental disorders using probabilistic genotype and phenotype matching among 4125 families. Nat. Genet. 2015, 47, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Doccini, S.; Morani, F.; Nesti, C.; Pezzini, F.; Calza, G.; Soliymani, R.; Signore, G.; Rocchiccioli, S.; Kanninen, K.M.; Huuskonen, M.T.; et al. Proteomic and functional analyses in disease models reveal CLN5 protein involvement in mitochondrial dysfunction. Cell Death Discov. 2020, 6, 18. [Google Scholar] [CrossRef]
- Shi, R.; Zhu, S.; Li, V.; Gibson, S.B.; Xu, X.; Kong, J. BNIP3 interacting with lc3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. An emerging role for mitochondrial dynamics in schizophrenia. Schizophr. Res. 2017, 187, 26–32. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Owen, M.J.; Sawa, A.; Mortensen, P.B. Schizophrenia. Lancet 2016, 388, 86–97. [Google Scholar] [CrossRef]
- Kirkpatrick, B.; Xu, L.; Cascella, N.; Ozeki, Y.; Sawa, A.; Roberts, R.C. DISC1 Immunoreactivity at the Light and Ultrastructural Level in the Human Neocortex. J. Comp. Neurol. 2006, 497, 436–450. [Google Scholar] [CrossRef]
- Duff, B.J.; Macritchie, K.A.N.; Moorhead, T.W.J.; Lawrie, S.M.; Blackwood, D.H.R. Human brain imaging studies of disc1 in schizophrenia, bipolar disorder and depression: A systematic review. Schizophr. Res. 2013, 147, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, N.J.; Porteous, D.J. DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology 2012, 62, 1230–1241. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Duan, X.; Liu, C.Y.; Jang, M.-H.; Guo, J.U.; Pow-anpongkul, N.; Kang, E.; Song, H.; Ming, G. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 2009, 63, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.K.; James, R.; Christie, S.; Porteous, D.J. Disrupted in Schizophrenia 1 (DISC1): Subcellular targeting and induction of ring mitochondria. Mol. Cell. Neurosci. 2005, 30, 477–484. [Google Scholar] [CrossRef]
- Norkett, R.; Modi, S.; Kittler, J.T. Mitochondrial roles of the psychiatric disease risk factor DISC1. Schizophr. Res. 2017, 187, 47–54. [Google Scholar] [CrossRef]
- Ogawa, F.; Malavasi, E.L.V.; Crummie, D.K.; Eykelenboom, J.E.; Soares, D.C.; Mackie, S.; Porteous, D.J.; Millar, J.K. DISC1 complexes with TRAK1 and Miro1 to modulate anterograde axonal mitochondrial trafficking. Hum. Mol. Genet. 2014, 23, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-U.; Jeong, J.; Lee, H.; Mun, J.Y.; Kim, J.-H.; Lee, J.S.; Nguyen, M.D.; Han, S.S.; Suh, P.-G.; Park, S.K. Disrupted-in-schizophrenia 1 (DISC1) plays essential roles in mitochondria in collaboration with Mitofilin. Proc. Nat. Acad. Sci. USA 2010, 107, 17785–17790. [Google Scholar] [CrossRef]
- Piñero-Martos, E.; Ortega-Vila, B.; Pol-Fuster, J.; Cisneros-Barroso, E.; Ruiz-Guerra, L.; Medina-Dols, A.; Heine-Suñer, D.; Lladó, J.; Olmos, G.; Vives-Bauzà, C. Disrupted in schizophrenia 1 (DISC1) is a constituent of the mammalian mitochondrial contact site and cristae organizing system (MICOS) complex, and is essential for oxidative phosphorylation. Hum. Mol. Genet. 2016, 25, 4157–4169. [Google Scholar] [CrossRef] [PubMed]
- Taya, S.; Shinoda, T.; Tsuboi, D.; Asaki, J.; Nagai, K.; Hikita, T.; Kuroda, S.; Kuroda, K.; Shimizu, M.; Hirotsune, S.; et al. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3ε complex through Kinesin-1. J. Neurosci. 2007, 27, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.; Orlovskaya, D.; Vikhreva, O.; Zimina, I.; Kolomeets, N.; Vostrikov, V.; Rachmanova, V. Electron microscopy of oligodendroglia in severe mental illness. Brain Res. Bull. 2001, 55, 597–610. [Google Scholar] [CrossRef]
- Uranova, N.A.; Vikhreva, O.V.; Rakhmanova, V.I.; Orlovskaya, D.D. Ultrastructural pathology of oligodendrocytes adjacent to microglia in prefrontal white matter in schizophrenia. NPJ Schizophrenia 2018, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vikhreva, O.V.; Rakhmanova, V.I.; Orlovskaya, D.D.; Uranova, N.A. Ultrastructural alterations of oligodendrocytes in prefrontal white matter in schizophrenia: A post-mortem morphometric study. Schizophr. Res. 2016, 177, 28–36. [Google Scholar] [CrossRef]
- Sachs, N.A.; Sawa, A.; Holmes, S.E.; Ross, C.A.; DeLisi, L.E.; Margolis, R.L. A frameshift mutation in Disrupted in Schizophrenia 1 in an American family with schizophrenia and schizoaffective disorder. Mol. Psychiatry 2005, 10, 758–764. [Google Scholar] [CrossRef]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Cataldo, A.M.; McPhie, D.L.; Lange, N.T.; Punzell, S.; Elmiligy, S.; Ye, N.Z.; Froimowitz, M.P.; Hassinger, L.C.; Menesale, E.B.; Sargent, L.W.; et al. Abnormalities in mitochondrial structure in cells from patients with bipolar disorder. Am. J. Pathol. 2010, 177, 575–585. [Google Scholar] [CrossRef]
- Sardon Puig, L.; Valera-Alberni, M.; Cantó, C.; Pillon, N.J. Circadian rhythms and mitochondria: Connecting the dots. Front. Genet. 2018, 9, 452. [Google Scholar] [CrossRef]
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713. [Google Scholar] [CrossRef]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta 2008, 1777, 860–866. [Google Scholar] [CrossRef]
- Scaini, G.; Fries, G.R.; Valvassori, S.S.; Zeni, C.P.; Zunta-Soares, G.; Berk, M.; Soares, J.C.; Quevedo, J. Perturbations in the apoptotic pathway and mitochondrial network dynamics in peripheral blood mononuclear cells from bipolar disorder patients. Transl. Psychiatry 2017, 7, e1111. [Google Scholar] [CrossRef] [PubMed]
- Kubli Dieter, A.; Gustafsson Åsa, B. Mitochondria and mitophagy. Circ. Res. 2012, 111, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Scaini, G.; Barichello, T.; Fries, G.R.; Kennon, E.A.; Andrews, T.; Nix, B.R.; Zunta-Soares, G.; Valvassori, S.S.; Soares, J.C.; Quevedo, J. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 inflammasome activation. Neuropsychopharmacology 2019, 44, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Denora, N.; Natile, G. An Updated View of Translocator Protein (TSPO). Int. J. Mol. Sci. 2017, 18, 2640. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Walder, K.; McGee, S.L.; Dean, O.M.; Tye, S.J.; Maes, M.; Berk, M. A model of the mitochondrial basis of bipolar disorder. Neurosci. Biobehav. Rev. 2017, 74, 1–20. [Google Scholar] [CrossRef]
- Lang, U.E.; Borgwardt, S. Molecular Mechanisms of Depression: Perspectives on New Treatment Strategies. CPB 2013, 31, 761–777. [Google Scholar] [CrossRef]
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Du, Y.; Diaz-Meco, M.-T.; Moscat, J.; Wooten, M.C.; Wooten, M.W. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim. Biophys. Acta 2013, 1833, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Kubota, M.; Miyauchi, T.; Noda, Y.; Mouri, A.; Nabeshima, T.; Kato, T. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol. Psychiatry 2006, 11, 577–593. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts inflammasome activation via elimiNation of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Zhao, T.; Du, Y.; Calderilla-Barbosa, L.; Yan, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. Behavioral effects of SQSTM1/p62 overexpression in mice: Support for a mitochondrial role in depression and anxiety. Behav. Brain Res. 2013, 248, 94–103. [Google Scholar] [CrossRef]
- Yki-Järvinen, H. Non-Alcoholic Fatty Liver Disease as a Cause and a Consequence of Metabolic Syndrome. Lancet Diabetes Endocrinol. 2014, 2, 901–910. [Google Scholar] [CrossRef]
- Mulder, P.; Morrison, M.C.; Verschuren, L.; Liang, W.; van Bockel, J.H.; Kooistra, T.; Wielinga, P.Y.; Kleemann, R. Reduction of Obesity-Associated White Adipose Tissue Inflammation by Rosiglitazone is Associated with Reduced Non-Alcoholic Fatty Liver Disease in LDLr-Deficient Mice. Sci. Rep. 2016, 6, 31542. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Makhlouf, H.R. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; French, S.W.; French, B.A.; Seitz, H.K.; Cohen, L.B.; Mueller, S.; Osna, N.A.; Kharbanda, K.K.; Seth, D.; Bautista, A.; et al. Alcoholic and non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2014, 97, 492–510. [Google Scholar] [CrossRef]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 2018, 28, 588–604.e5. [Google Scholar] [CrossRef]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ramshesh, V.K.; Rehman, H.; Liu, Q.; Theruvath, T.P.; Krishnasamy, Y.; Lemasters, J.J. Acute ethanol causes hepatic mitochondrial depolarization in mice: Role of ethanol metabolism. PLoS ONE 2014, 9, e91308. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ding, W.-X. A mechanistic review of mitophagy and its role in protection against alcoholic liver disease. Biomolecules 2015, 5, 2619–2642. [Google Scholar] [CrossRef]
- Williams, J.A.; Ni, H.-M.; Ding, Y.; Ding, W.-X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G324–G340. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Zhong, Z. Mitophagy in hepatocytes: Types, initiators and role in adaptive ethanol metabolism. Liver Res. 2018, 2, 125–132. [Google Scholar] [CrossRef]
- Szendroedi, J.; Phielix, E.; Roden, M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011, 8, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Rovira-Llopis, S.; Bañuls, C.; Diaz-Morales, N.; Hernandez-Mijares, A.; Rocha, M.; Victor, V.M. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017, 11, 637–645. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Walia, R.; Saikia, U.N.; Dhawan, V. Alterations in Mitochondrial Oxidative Stress and Mitophagy in Subjects with Prediabetes and Type 2 Diabetes Mellitus. Front. Endocrinol. 2017, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Czajka, A.; Ajaz, S.; Gnudi, L.; Parsade, C.K.; Jones, P.; Reid, F.; Malik, A.N. Altered mitochondrial function, mitochondrial dna and reduced metabolic flexibility in patients with diabetic nephropathy. EbioMedicine 2015, 2, 499–512. [Google Scholar] [CrossRef]
- Scheele, C.; Nielsen, A.R.; Walden, T.B.; Sewell, D.A.; Fischer, C.P.; Brogan, R.J.; Petrovic, N.; Larsson, O.; Tesch, P.A.; Wennmalm, K.; et al. Altered regulation of the PINK1 locus: A link between type 2 diabetes and neurodegeneration? FASEB J. 2007, 21, 3653–3665. [Google Scholar] [CrossRef] [PubMed]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef]
- Greene, N.P.; Lee, D.E.; Brown, J.L.; Rosa, M.E.; Brown, L.A.; Perry, R.A.; Henry, J.N.; Washington, T.A. Mitochondrial quality control, promoted by PGC-1α, is dysregulated by Western diet-induced obesity and partially restored by moderate physical activity in mice. Physiol. Rep. 2015, 3, e12470. [Google Scholar] [CrossRef]
- Heo, J.-W.; No, M.-H.; Park, D.-H.; Kang, J.-H.; Seo, D.Y.; Han, J.; Neufer, P.D.; Kwak, H.-B. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J. Physiol. Pharm. 2017, 21, 567–577. [Google Scholar] [CrossRef]
- Ro, S.-H.; Jang, Y.; Bae, J.; Kim, I.M.; Schaecher, C.; Shomo, Z.D. Autophagy in adipocyte browning: Emerging drug target for intervention in obesity. Front. Physiol. 2019, 10, 22. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, A.; Oh, K.-J.; Lee, S.C.; Kim, W.K.; Bae, K.-H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. [Google Scholar] [CrossRef]
- Lu, X.; Altshuler-Keylin, S.; Wang, Q.; Chen, Y.; Henrique Sponton, C.; Ikeda, K.; Maretich, P.; Yoneshiro, T.; Kajimura, S. Mitophagy controls beige adipocyte maintenance through a Parkin-dependent and UCP1-independent mechanism. Sci. Signal. 2018, 11, eaap8526. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Zarrouk-Mahjoub, S. Mitochondrial vasculopathy. World J. Cardiol. 2016, 8, 333–339. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Sukhovershin, R.A.; Toledano Furman, N.E.; Tasciotti, E.; Trachtenberg, B.H. local inhibition of macrophage and smooth muscle cell proliferation to suppress plaque progression. Methodist Debakey Cardiovasc. J. 2016, 12, 141–145. [Google Scholar] [CrossRef]
- Wu, M.-Y.; Li, C.-J.; Hou, M.-F.; Chu, P.-Y. New Insights into the Role of inflammation in the pathogenesis of atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis: Update and Therapeutic Implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 2017, 95, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C.H.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef]
- Valle, I.; Álvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. PGC-1α Regulates the Mitochondrial Antioxidant Defense System in Vascular Endothelial Cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef]
- Münzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef]
- Finsterer Is atherosclerosis a mitochondrial disorder? Vasa 2007, 36, 229–240. [CrossRef]
- Orekhov, A.N.; Poznyak, A.V.; Sobenin, I.A.; Nikifirov, N.N.; Ivanova, E.A. Mitochondrion as a selective target for treatment of atherosclerosis: Role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr. Neuropharmacol. 2020, 18, 1064–1075. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2018, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Zhelankin, A.V.; Khasanova, Z.B.; Sinyov, V.V.; Medvedeva, L.V.; Sagaidak, M.O.; Makeev, V.J.; Kolmychkova, K.I.; Smirnova, A.S.; Sukhorukov, V.N.; et al. Heteroplasmic Variants of Mitochondrial DNA in Atherosclerotic Lesions of Human Aortic Intima. Biomolecules 2019, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Mitochondrial mutations are associated with atherosclerotic lesions in the human aorta. Clin. Dev. Immunol. 2012, 2012, 832464. [Google Scholar] [CrossRef]
- Liu, L.-P.; Cheng, K.; Ning, M.-A.; Li, H.-H.; Wang, H.-C.; Li, F.; Chen, S.-Y.; Qu, F.-L.; Guo, W.-Y. Association between peripheral blood cells mitochondrial DNA content and severity of coronary heart disease. Atherosclerosis 2017, 261, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Pek, N.M.Q.; Phua, Q.H.; Ho, B.X.; Pang, J.K.S.; Hor, J.-H.; An, O.; Yang, H.H.; Yu, Y.; Fan, Y.; Ng, S.-Y.; et al. Mitochondrial 3243A > G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. BioMed Res. Int. 2015, 2015, 825468. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Won, D.; Haidari, M. Leukocyte Recruitment to atherosclerotic lesions. Can. J. Cardiol. 2004, 20, 24B–28B. [Google Scholar] [PubMed]
- Pirillo, A.; Norata, G.D.; Catapano, A.L. LOX-1, OxLDL, and Atherosclerosis. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective autophagy in atherosclerosis: To die or to senesce? Oxid. Med. Cell Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef]
- Yang, Y.; Li, T.; Li, Z.; Liu, N.; Yan, Y.; Liu, B. Role of mitophagy in cardiovascular disease. Aging Dis. 2020, 11, 419–437. [Google Scholar] [CrossRef]
- Yu, E.P.K.; Reinhold, J.; Yu, H.; Starks, L.; Uryga, A.K.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.-F.; Logan, A.; et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arter. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef]
- He, L.; Zhou, Q.; Huang, Z.; Xu, J.; Zhou, H.; Lv, D.; Lu, L.; Huang, S.; Tang, M.; Zhong, J.; et al. PINK1/Parkin-mediated mitophagy promotes apelin-13-induced vascular smooth muscle cell proliferation by AMPKα and exacerbates atherosclerotic lesions. J. Cell. Physiol. 2019, 234, 8668–8682. [Google Scholar] [CrossRef]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821–28835. [Google Scholar] [CrossRef] [PubMed]
- Docherty, C.K.; Carswell, A.; Friel, E.; Mercer, J.R. Impaired mitochondrial respiration in human carotid plaque atherosclerosis: A potential role for Pink1 in vascular smooth muscle cell energetics. Atherosclerosis 2018, 268, 1–11. [Google Scholar] [CrossRef]
- Peng, X.; Chen, H.; Li, Y.; Huang, D.; Huang, B.; Sun, D. Effects of NIX-mediated mitophagy on ox-LDL-induced macrophage pyroptosis in atherosclerosis. Cell Biol. Int. 2020, 44, 1481–1490. [Google Scholar] [CrossRef]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The role of autophagy in the heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Ong, S.-B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef]
- Marín-García, J.; Akhmedov, A.T. Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 2016, 21, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef]
- Pei, H.; Yang, Y.; Zhao, H.; Li, X.; Yang, D.; Li, D.; Yang, Y. The Role of Mitochondrial Functional Proteins in ROS Production in Ischemic Heart Diseases. Oxid. Med. Cell Longev. 2016, 2016, 5470457. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Thomas, A. Mitophagy and mitochondrial quality control mechanisms in the heart. Curr. Pathobiol. Rep. 2017, 5, 161–169. [Google Scholar] [CrossRef]
- Shires, S.E.; Gustafsson, Å.B. Mitophagy and heart failure. J. Mol. Med. 2015, 93, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.-F.; Dohmen, P.M.; Choi, Y.-H.; Wahlers, T.; et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef]
- Kurmani, S.; Squire, I. Acute Heart Failure: Definition, Classification and Epidemiology. Curr. Heart Fail. Rep. 2017, 14, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, L.M.D.; Mellor, K.M.; Taylor, D.J.R.; Gottlieb, R.A. Myocardial autophagic energy stress responses—Macroautophagy, mitophagy, and glycophagy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1194–H1204. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Shires, S.; Gustafsson, Å.B. Regulating renewable energy: Connecting AMPKα2 to PINK1/parkin-mediated mitophagy in the heart. Circ. Res. 2018, 122, 649–651. [Google Scholar] [CrossRef]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. ampkα2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. USA 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Villarroya, F. SIRT3, a pivotal actor in mitochondrial functions: Metabolism, cell death and aging. Biochem. J. 2012, 444, 1–10. [Google Scholar] [CrossRef]
- Bochaton, T.; Crola-Da-Silva, C.; Pillot, B.; Villedieu, C.; Ferreras, L.; Alam, M.R.; Thibault, H.; Strina, M.; Gharib, A.; Ovize, M.; et al. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J. Mol. Cell. Cardiol. 2015, 84, 61–69. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Kageyama, Y.; Hoshijima, M.; Seo, K.; Bedja, D.; Sysa-Shah, P.; Andrabi, S.A.; Chen, W.; Höke, A.; Dawson, V.L.; Dawson, T.M.; et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014, 33, 2798–2813. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Wu, X.-D.; Liu, X.-H.; Li, P.-F. Mitophagy imbalance in cardiomyocyte ischaemia/reperfusion injury. Acta Physiol. 2019, 225, e13228. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Madungwe, N.B.; da Cruz Junho, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharm. 2017, 174, 4329–4344. [Google Scholar] [CrossRef]
- Iliodromitis, E.K.; Lazou, A.; Kremastinos, D.T. Ischemic preconditioning: Protection against myocardial necrosis and apoptosis. Vasc Health Risk Manag. 2007, 3, 629–637. [Google Scholar] [PubMed]
- Zhang, W.; Siraj, S.; Zhang, R.; Chen, Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 2017, 13, 1080–1081. [Google Scholar] [CrossRef]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef]
- Lu, W.; Sun, J.; Yoon, J.S.; Zhang, Y.; Zheng, L.; Murphy, E.; Mattson, M.P.; Lenardo, M.J. Mitochondrial Protein pgam5 regulates mitophagic protection against cell necroptosis. PLoS ONE 2016, 11, e0147792. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Pennanen, C.; Parra, V.; López-Crisosto, C.; Morales, P.E.; Del Campo, A.; Gutierrez, T.; Rivera-Mejías, P.; Kuzmicic, J.; Chiong, M.; Zorzano, A.; et al. Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2+-calcineurin signaling pathway. J. Cell. Sci. 2014, 127, 2659–2671. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Trincado, C.; García-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Bauer, E.P.; Klövekorn, W.-P.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, A.S.; Brunskill, E.W.; Marreez, Y.; Benner, B.J.; Regula, K.M.; Kirschenbaum, L.A.; Dorn, G.W. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J. Biol. Chem. 2006, 281, 1442–1448. [Google Scholar] [CrossRef]
- Dorn, G.W. Mitochondrial pruning by Nix and BNip3: An essential function for cardiac-expressed death factors. J. Cardiovasc Transl. Res. 2010, 3, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.-Y.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian J. Ophthalmol. 2012, 60, 428–431. [Google Scholar] [PubMed]
- Kern, T.S.; Barber, A.J. Retinal ganglion cells in diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group; Lachin, J.M.; Genuth, S.; Cleary, P.; Davis, M.D.; Nathan, D.M. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N. Engl. J. Med. 2000, 342, 381–389. [Google Scholar] [PubMed]
- Diabetes Control and Complications Trial Research Group; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [PubMed]
- Bixler, G.V.; Vanguilder, H.D.; Brucklacher, R.M.; Kimball, S.R.; Bronson, S.K.; Freeman, W.M. Chronic insulin treatment of diabetes does not fully normalize alterations in the retinal transcriptome. BMC Med. Genom. 2011, 4, 40. [Google Scholar] [CrossRef]
- Chew, E.Y.; Davis, M.D.; Danis, R.P.; Lovato, J.F.; Perdue, L.H.; Greven, C.; Genuth, S.; Goff, D.C.; Leiter, L.A.; Ismail-Beigi, F.; et al. The effects of medical management on the progression of diabetic retinopathy in persons with type 2 diabetes: The Action to Control Cardiovascular Risk in Diabetes (ACCORD) Eye Study. Ophthalmology 2014, 121, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Madsen–Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial dna damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef]
- García-Quintans, N.; Prieto, I.; Sánchez-Ramos, C.; Luque, A.; Arza, E.; Olmos, Y.; Monsalve, M. Regulation of endothelial dynamics by PGC-1α relies on ROS control of VEGF-A signaling. Free Radic. Biol. Med. 2016, 93, 41–51. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Therapeutic targets for altering mitochondrial dysfunction associated with diabetic retinopathy. Expert Opin. Targets 2018, 22, 233–245. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Kowluru, A.; Mishra, M.; Kumar, B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog. Retin. Eye Res. 2015, 48, 40–61. [Google Scholar] [CrossRef]
- Mishra, M.; Kowluru, R.A. DNA Methylation-a potential source of mitochondria dna base mismatch in the development of diabetic retinopathy. Mol. Neurobiol. 2019, 56, 88–101. [Google Scholar] [CrossRef]
- dos Santos, J.M.; Tewari, S.; Goldberg, A.F.X.; Kowluru, R.A. Mitochondria biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849–1860. [Google Scholar] [CrossRef]
- Kanwar, M.; Chan, P.-S.; Kern, T.S.; Kowluru, R.A. Oxidative damage in the retinal mitochondria of diabetic mice: Possible protection by superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3805–3811. [Google Scholar] [CrossRef]
- Altomare, E.; Grattagliano, I.; Vendemaile, G.; Micelli-Ferrari, T.; Signorile, A.; Cardia, L. Oxidative protein damage in human diabetic eye: Evidence of a retinal participation. Eur. J. Clin. Investig. 1997, 27, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Xu, X.; Bi, H.; Zhu, Q.; Wu, J.; Xia, X.; Ren, Q.; Ho, P.C.P. Expression modification of uncoupling proteins and MnSOD in retinal endothelial cells and pericytes induced by high glucose: The role of reactive oxygen species in diabetic retinopathy. Exp. Eye Res. 2006, 83, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Abrahan, C.E.; Insua, M.F.; Politi, L.E.; German, O.L.; Rotstein, N.P. Oxidative stress promotes proliferation and dedifferentiation of retina glial cells in vitro. J. Neurosci. Res. 2009, 87, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xie, W.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells 2019, 8, 213. [Google Scholar] [CrossRef]
- Masser, D.R.; Otalora, L.; Clark, N.W.; Kinter, M.T.; Elliott, M.H.; Freeman, W.M. Functional changes in the neural retina occur in the absence of mitochondrial dysfunction in a rodent model of diabetic retinopathy. J. Neurochem. 2017, 143, 595–608. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Abbas, S.N. Diabetes-induced mitochondrial dysfunction in the retina. Invest. Ophthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef]
- Bek, T. Mitochondrial dysfunction and diabetic retinopathy. Mitochondrion 2017, 36, 4–6. [Google Scholar] [CrossRef]
- Rosa, M.D.; Distefano, G.; Gagliano, C.; Rusciano, D.; Malaguarnera, L. Autophagy in diabetic retinopathy. Curr. Neuropharmacol. 2016, 14, 810–825. [Google Scholar] [CrossRef]
- Wang, R.; Lu, L.; Guo, Y.; Lin, F.; Chen, H.; Chen, W.; Chen, M. Effect of Glucagon-like Peptide-1 on high-glucose-induced oxidative stress and cell apoptosis in human endothelial cells and its underlying mechanism. J. Cardiovasc. Pharm. 2015, 66, 135–140. [Google Scholar] [CrossRef]
- Zhang, Y.; Xi, X.; Mei, Y.; Zhao, X.; Zhou, L.; Ma, M.; Liu, S.; Zha, X.; Yang, Y. High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/PINK1/Parkin signal pathway. Biomed. Pharm. 2019, 111, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Masaki, S.; Matsuo, Y.; Chen, Z.; Tian, H.; Yodoi, J. Thioredoxin/Txnip: Redoxisome, as a redox switch for the pathogenesis of diseases. Front. Immunol. 2014, 4, 514. [Google Scholar] [CrossRef]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis. 2010, 1, e65. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.P. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Xiao, R.-P. Role of mitofusin 2 in cardiovascular oxidative injury. J. Mol. Med. 2010, 88, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Kowluru, R.A. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8739–8746. [Google Scholar] [CrossRef]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis 2019, 1865, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Schwartz, M. NIX-ing mitochondria: From development to pathology. EMBO J. 2017, 36, 1650–1652. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Martínez, L.; Sierra-Filardi, E.; McGreal, R.S.; Salazar-Roa, M.; Mariño, G.; Seco, E.; Durand, S.; Enot, D.; Graña, O.; Malumbres, M.; et al. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017, 36, 1688–1706. [Google Scholar] [CrossRef]
- Sin, J.; Andres, A.M.; Taylor, D.J.R.; Weston, T.; Hiraumi, Y.; Stotland, A.; Kim, B.J.; Huang, C.; Doran, K.S.; Gottlieb, R.A. Mitophagy is Required for Mitochondrial Biogenesis and Myogenic Differentiation of C2C12 Myoblasts. Autophagy 2016, 12, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite Cells in Ageing: Use it or Lose it. Open Biol. 2020, 10, 200048. [Google Scholar] [CrossRef]
- de Souza, G.T.; Zanette, R.d.S.S.; do Amaral, D.L.A.S.; da Guia, F.C.; Maranduba, C.P.; de Souza, C.M.; Guimarães, E.d.S.G.; Rettore, J.V.P.; Rabelo, N.C.; do Carmo, A.M.R.; et al. Satellite Cells: Regenerative Mechanisms and Applicability in Muscular Dystrophy. Stem Cells Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Muñoz-Cánoves, P.; Martinez-Vicente, M. Dysfunctional Autophagy is a Driver of Muscle Stem Cell Functional Decline with Aging. Autophagy 2016, 12, 612–613. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D.; Kang, C.; Zhang, T. The role of Mitochondria in Redox Signaling of Muscle Homeostasis. J. Sport Health Sci. 2020, 9, 386–393. [Google Scholar] [CrossRef]
- Romanello, V.; Sandri, M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front. Physiol. 2016, 6, 422. [Google Scholar] [CrossRef]
- Picard, M.; White, K.; Turnbull, D.M. Mitochondrial Morphology, Topology, and Membrane Interactions in Skeletal Muscle: A Quantitative Three-Dimensional Electron Microscopy Study. J. Appl. Physiol. 2013, 114, 161–171. [Google Scholar] [CrossRef]
- Casuso, R.A.; Huertas, J.R. The Emerging Role of Skeletal Muscle Mitochondrial Dynamics in Exercise and Ageing. Ageing Res. Rev. 2020, 58, 101025. [Google Scholar] [CrossRef]
- Erlich, A.T.; Hood, D.A. Mitophagy Regulation in Skeletal Muscle: Effect of Endurance Exercise and Age. J. Sci. Sport Exerc. 2019, 1, 228–236. [Google Scholar] [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-khat, D.; Hood, D.A. Autophagy and Mitophagy Flux in Young and Aged Skeletal Muscle Following Chronic Contractile Activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- Balan, E.; Schwalm, C.; Naslain, D.; Nielens, H.; Francaux, M.; Deldicque, L. Regular Endurance Exercise Promotes Fission, Mitophagy, and Oxidative Phosphorylation in Human Skeletal Muscle Independently of Age. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Liu, H.-W.; Chang, Y.-C.; Chan, Y.-C.; Hu, S.-H.; Liu, M.-Y.; Chang, S.-J. Dysregulations of mitochondrial quality control and autophagic flux at an early age lead to progression of sarcopenia in SAMP8 mice. Biogerontology 2020, 21, 367–380. [Google Scholar] [CrossRef]
- Kim, Y.; Triolo, M.; Erlich, A.T.; Hood, D.A. Regulation of Autophagic And Mitophagic Flux during Chronic Contractile Activity-Induced Muscle Adaptations. Pflug. Arch. Eur. J. Physiol. 2019, 471, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Deval, C.; Calonne, J.; Coudy-Gandilhon, C.; Vazeille, E.; Bechet, D.; Polge, C.; Taillandier, D.; Attaix, D.; Combaret, L. Mitophagy and Mitochondria Biogenesis Are Differentially Induced in Rat Skeletal Muscles during Immobilization and/or Remobilization. Int. J. Mol. Sci. 2020, 21, 3691. [Google Scholar] [CrossRef] [PubMed]
- Leermakers, P.A.; Kneppers, A.E.M.; Schols, A.M.W.J.; Kelders, M.C.J.M.; de Theije, C.C.; Verdijk, L.B.; van Loon, L.J.C.; Langen, R.C.J.; Gosker, H.R. Skeletal Muscle Unloading Results in Increased Mitophagy and Decreased Mitochondrial Biogenesis Regulation. Muscle Nerve 2019, 60, 769–778. [Google Scholar] [CrossRef]
- Oost, L.J.; Kustermann, M.; Armani, A.; Blaauw, B.; Romanello, V. Fibroblast Growth Factor 21 Controls Mitophagy and Muscle Mass. J. Cachexia Sarcopenia Muscle 2019, 10, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Alshudukhi, A.A.; Zhu, J.; Huang, D.; Jama, A.; Smith, J.D.; Wang, Q.J.; Esser, K.A.; Ren, H. Lipin-1 Regulates Bnip3–Mediated Mitophagy in Glycolytic Muscle. FASEB J. 2018, 32, 6796–6807. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria Mediate Cell Membrane Repair and Contribute to Duchenne Muscular Dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; Engel, W.K.; Nogalska, A. Sporadic inclusion-body myositis: A degenerative Muscle Disease Associated with Aging, Impaired Muscle Protein Homeostasis and abnormal Mitophagy. Biochim. Biophys. Acta (Bba) Mol. Basis Dis. 2015, 1852, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Rygiel, K.A.; Miller, J.; Grady, J.P.; Rocha, M.C.; Taylor, R.W.; Turnbull, D.M. Mitochondrial and Inflammatory Changes in Sporadic Inclusion Body Myositis. Neuropathol. Appl. Neurobiol. 2015, 41, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Sebori, R.; Kuno, A.; Hosoda, R.; Hayashi, T.; Horio, Y. Resveratrol Decreases Oxidative Stress by Restoring Mitophagy and Improves the Pathophysiology of Dystrophin-Deficient Mdx Mice. Oxid. Med. Cell. Longev. 2018, 2018, 9179270. [Google Scholar] [CrossRef] [PubMed]
- Heilman, J.; Andreux, P.; Tran, N.; Rinsch, C.; Blanco-Bose, W. Safety Assessment of Urolithin A, a Metabolite Produced by the Human Gut Microbiota Upon Dietary Intake of Plant Derived Ellagitannins and Ellagic Acid. Food Chem. Toxicol. 2017, 108, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The Mitophagy Activator Urolithin A is Safe and Induces a Molecular Signature of Improved Mitochondrial and Cellular Health in Humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Ma, X.; McKeen, T.; Zhang, J.; Ding, W.-X. Role and Mechanisms of Mitophagy in Liver Diseases. Cells 2020, 9, 837. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-Activated Protein Kinase (AMPK) Action in Skeletal Muscle via Direct Phosphorylation of PGC-1α. Proc. Nat. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.; Anand, R.; Altinoluk-Hambüchen, S.; Ezzahoini, H.; Stefanski, A.; Iram, A.; Bergmann, L.; Urbach, J.; Böhler, P.; Hänsel, J.; et al. SIRT4 Interacts with OPA1 and Regulates Mitochondrial Quality Control and Mitophagy. Aging (Albany NY) 2017, 9, 2163–2189. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent Regulation of Hypoxic Induction of the Cell Death Factors BNIP3 and NIX in Human Tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Fei, P.; Wang, W.; Kim, S.; Wang, S.; Burns, T.F.; Sax, J.K.; Buzzai, M.; Dicker, D.T.; McKenna, W.G.; Bernhard, E.J.; et al. Bnip3L is Induced by p53 under Hypoxia, and Its Knockdown Promotes Tumor Growth. Cancer Cell 2004, 6, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Kim, M.R.; Lee, H.J.; Lee, H.M.; Kim, G.C.; Lee, Y.S.; Nam, H.Y.; Lee, M.; Jang, H.J.; Lee, K.E.; et al. p53/BNIP3-Dependent Mitophagy Limits Glycolytic Shift in Radioresistant Cancer. Oncogene 2019, 38, 3729–3742. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; de las Heras, M.M.G.; Gabandé-Rodríguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A Key Player in the Inflammatory Response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef]
- Sun, L.; Shen, R.; Agnihotri, S.K.; Chen, Y.; Huang, Z.; Büeler, H. Lack of PINK1 Alters Glia Innate Immune Responses and Enhances Inflammation-Induced, Nitric Oxide-Mediated Neuron Death. Sci. Rep. 2018, 8, 383. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-Inflammatory Effect of IL-10 Mediated by Metabolic Reprogramming of Macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Tan, S.; Yu, C.Y.; Sim, Z.W.; Low, Z.S.; Lee, B.; See, F.; Min, N.; Gautam, A.; Chu, J.J.H.; Ng, K.W.; et al. Pomegranate Activates TFEB to Promote Autophagy-Lysosomal Fitness and Mitophagy. Sci. Rep. 2019, 9, 727. [Google Scholar] [CrossRef]
- Wang, W.; Wang, M.; Ruan, Y.; Tan, J.; Wang, H.; Yang, T.; Li, J.; Zhou, Q. Ginkgolic Acids Impair Mitochondrial Function by Decreasing Mitochondrial Biogenesis and Promoting FUNDC1-Dependent Mitophagy. J. Agric. Food Chem. 2019, 67, 10097–10106. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Frison, M.; Alvarez, M.S.; Bertrand, H.; Wells, G.; Campanella, M. Reversible Keap1 Inhibitors Are Preferential Pharmacological Tools to Modulate Cellular Mitophagy. Sci. Rep. 2017, 7, 10303. [Google Scholar] [CrossRef]
- Takahashi, D.; Arimoto, H. Targeting Selective Autophagy by AUTAC Degraders. Autophagy 2020, 16, 765–766. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Moriyama, J.; Nakamura, T.; Miki, E.; Takahashi, E.; Sato, A.; Akaike, T.; Itto-Nakama, K.; Arimoto, H. AUTACs: Cargo-Specific Degraders Using Selective Autophagy. Mol. Cell 2019, 76, 797–810.e10. [Google Scholar] [CrossRef]
- Gurusamy, N.; Lekli, I.; Mukherjee, S.; Ray, D.; Ahsan, M.K.; Gherghiceanu, M.; Popescu, L.M.; Das, D.K. Cardioprotection by resveratrol: A novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc. Res. 2010, 86, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Chen, G.; Deng, Y.; Tang, H.; Ling, L.; Zhou, X.; Song, X.; Yang, P.; Liu, Y.; Li, Z.; et al. Polydatin post-treatment alleviates myocardial ischaemia/reperfusion injury by promoting autophagic flux. Clin. Sci. 2016, 130, 1641–1653. [Google Scholar] [CrossRef]
- Ma, S.; Chen, J.; Feng, J.; Zhang, R.; Fan, M.; Han, D.; Li, X.; Li, C.; Ren, J.; Wang, Y.; et al. Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid. Med. Cell Longev. 2018, 2018, 9286458. [Google Scholar] [CrossRef]
- Díaz-Casado, M.E.; Lima, E.; García, J.A.; Doerrier, C.; Aranda, P.; Sayed, R.K.; Guerra-Librero, A.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Melatonin rescues zebrafish embryos from the parkinsonian phenotype restoring the parkin/PINK1/DJ-1/MUL1 network. J. Pineal. Res. 2016, 61, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Dhawan, V. Metformin promotes mitophagy in mononuclear cells: A potential in vitro model for unraveling metformin’s mechanism of action. Ann. N. Y. Acad. Sci. 2020, 1463, 23–36. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, M. Metformin rescues Parkin protein expression and mitophagy in high glucose-challenged human renal epithelial cells by inhibiting NF-κB via PP2A activation. Life Sci. 2020, 246, 117382. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, W.K.; Lee, Y.-H.; Kang, E.S.; Cha, B.-S.; Lee, B.-W. Metformin restores parkin-mediated mitophagy, suppressed by Cytosolic p53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Y.; Zhang, Y.; Liu, J.; Yao, Z.; Zhang, C. Protective effects of metformin against osteoarthritis through upregulation of SIRT3-mediated PINK1/Parkin-dependent mitophagy in primary chondrocytes. Biosci. Trends 2019, 12, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I.; et al. Ursolic and oleanolic acids induce mitophagy in A549 human lung cancer cells. Molecules 2019, 24, 3444. [Google Scholar] [CrossRef]
- Barini, E.; Miccoli, A.; Tinarelli, F.; Mulholland, K.; Kadri, H.; Khanim, F.; Stojanovski, L.; Read, K.D.; Burness, K.; Blow, J.J.; et al. The anthelmintic drug niclosamide and its analogues activate the Parkinson’s disease associated protein kinase PINK1. Chembiochem 2018, 19, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Wu, M.; Zhang, L.; Fu, W.; Yang, J.; Zhang, B.; Zheng, Z.; Zhang, H.; Lao, Y.; Xu, H. Gerontoxanthone I and macluraxanthone induce mitophagy and attenuate ischemia/reperfusion injury. Front. Pharm. 2020, 11, 452. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Q.; Lou, Y.; Xu, J.; Feng, Z.; Chen, Y.; Tang, Q.; Zheng, G.; Zhang, Z.; Wu, Y.; et al. Salidroside attenuates neuroinflammation and improves functional recovery after spinal cord injury through microglia polarization regulation. J. Cell Mol. Med. 2018, 22, 1148–1166. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, T.; Chen, J.; Shao, Z.; Wang, K.; Yan, Y.; Wu, C.; Lin, J.; Wang, H.; Gao, W.; et al. Parkin-mediated mitophagy as a potential therapeutic target for intervertebral disc degeneration. Cell Death Dis. 2018, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef]
- Nisoli, E.; Clementi, E.; Paolucci, C.; Cozzi, V.; Tonello, C.; Sciorati, C.; Bracale, R.; Valerio, A.; Francolini, M.; Moncada, S.; et al. Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 2003, 299, 896–899. [Google Scholar] [CrossRef]
- Borniquel, S.; Valle, I.; Cadenas, S.; Lamas, S.; Monsalve, M. Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1alpha. FASEB J. 2006, 20, 1889–1891. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-Y.; Kang, M.-J.; Kim, K.-H.; Han, P.-L.; Kim, H.-S.; Ha, J.-Y.; Son, J.H. Nitric oxide induction of Parkin translocation in PTEN-induced putative kinase 1 (PINK1) deficiency: Functional role of neuronal nitric oxide synthase during mitophagy. J. Biol. Chem. 2015, 290, 10325–10335. [Google Scholar] [CrossRef]
- Kim, K.-H.; Song, K.; Yoon, S.-H.; Shehzad, O.; Kim, Y.-S.; Son, J.H. Rescue of PINK1 protein null-specific mitochondrial complex IV deficits by ginsenoside Re activation of nitric oxide signaling. J. Biol. Chem. 2012, 287, 44109–44120. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Kluge, A.F.; Lagu, B.R.; Maiti, P.; Jaleel, M.; Webb, M.; Malhotra, J.; Mallat, A.; Srinivas, P.A.; Thompson, J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018, 28, 2655–2659. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Corsetti, G. Natural Compounds and Autophagy: Allies Against Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 555409. [Google Scholar] [CrossRef] [PubMed]
- Varghese, N.; Werner, S.; Grimm, A.; Eckert, A. Dietary Mitophagy Enhancer: A Strategy for Healthy Brain Aging? Antioxidants 2020, 9, 932. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, S.; Liu, J.; Wu, X.; Zhou, S.; Dai, K.; Kou, Y. Mitophagy Contributes to the Pathogenesis of Inflammatory Diseases. Inflammation 2018, 41, 1590–1600. [Google Scholar] [CrossRef]
- Yuk, J.-M.; Silwal, P.; Jo, E.-K. Inflammasome and mitophagy connection in health and disease. Int. J. Mol. Sci. 2020, 21, 4714. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Patergnani, S.; Bonora, M.; Pedriali, G.; Tarocco, A.; Bouhamida, E.; Marchi, S.; Ancora, G.; Anania, G.; Wieckowski, M.R.; et al. Mitophagy in cardiovascular diseases. J. Clin. Med. 2020, 9, 892. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Tian, H.; Chen, P.; Wang, D.; Ren, J.; Zhang, Y. Physical exercise and selective autophagy: Benefit and risk on cardiovascular health. Cells 2019, 8, 1436. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, H.; Wu, Y.; Zhu, Y.; Zhang, J.; Yang, G.; Yan, Q.; Li, J.; Li, T.; Liu, L. Mitochondrial-Derived Vesicles Protect Cardiomyocytes Against Hypoxic Damage. Front. Cell Dev. Biol. 2020, 8, 214. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. https://doi.org/10.3390/ijms22083903
Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, Monsalve M. Mitophagy in Human Diseases. International Journal of Molecular Sciences. 2021; 22(8):3903. https://doi.org/10.3390/ijms22083903
Chicago/Turabian StyleDoblado, Laura, Claudia Lueck, Claudia Rey, Alejandro K. Samhan-Arias, Ignacio Prieto, Alessandra Stacchiotti, and Maria Monsalve. 2021. "Mitophagy in Human Diseases" International Journal of Molecular Sciences 22, no. 8: 3903. https://doi.org/10.3390/ijms22083903
APA StyleDoblado, L., Lueck, C., Rey, C., Samhan-Arias, A. K., Prieto, I., Stacchiotti, A., & Monsalve, M. (2021). Mitophagy in Human Diseases. International Journal of Molecular Sciences, 22(8), 3903. https://doi.org/10.3390/ijms22083903