Myotonic Dystrophy: From Molecular Pathogenesis to Therapeutics

{kind=link}
Funding
Conflicts of Interest
References
- Tomé, S.; Gourdon, G. DM1 Phenotype Variability and Triplet Repeat Instability: Challenges in the Development of New Therapies. Int. J. Mol. Sci. 2020, 21, 457. [Google Scholar] [CrossRef]
- Cumming, S.A.; Jimenez-Moreno, C.; Okkersen, K.; Wenninger, S.; Daidj, F.; Hogarth, F.; Littleford, R.; Gorman, G.; Bassez, G.; Schoser, B.; et al. OPTIMISTIC Consortium. Genetic determinants of disease severity in the myotonic dystrophy type 1 OPTIMISTIC cohort. Neurology 2019, 93, e995–e1009. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.M.; Pearson, C.E. Disease-associated repeat instability and mismatch repair. DNA Repair 2016, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Peric, S.; Pesovic, J.; Savic-Pavicevic, D.; Rakocevic Stojanovic, V.; Meola, G. Molecular and Clinical Implications of Variant Repeats in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 23, 354. [Google Scholar] [CrossRef] [PubMed]
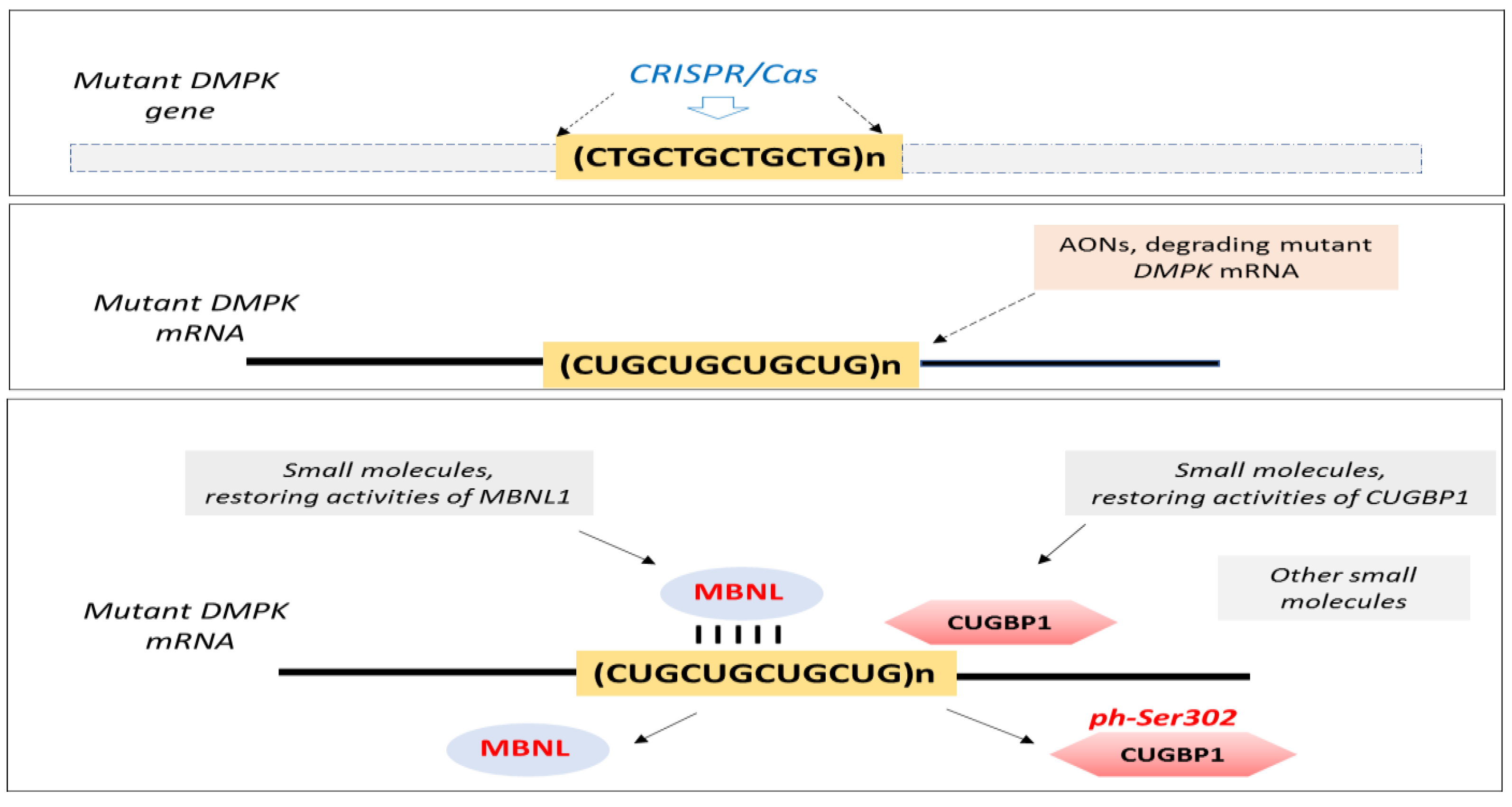
- Raaijmakers, R.H.L.; Ripken, L.; Ausems, C.R.M.; Wansink, D.G. CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities. Int. J. Mol. Sci. 2019, 20, 3689. [Google Scholar] [CrossRef] [PubMed]
- André, L.M.; van Cruchten, R.T.P.; Willemse, M.; Bezstarosti, K.; Demmers, J.A.A.; van Agtmaal, E.L.; Wansink, D.G.; Wieringa, B. Recovery in the Myogenic Program of Congenital Myotonic Dystrophy Myoblasts after Excision of the Expanded (CTG)n Repeat. Int. J. Mol. Sci. 2019, 20, 5685. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Jenquin, J.R.; Cleary, J.D.; Berglund, J.A. Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules. Int. J. Mol. Sci. 2019, 20, 4017. [Google Scholar] [CrossRef]
- García-Puga, M.; Saenz-Antoñanzas, A.; Matheu, A.; de Munain, A.L. Targeting Myotonic Dystrophy Type 1 with Metformin. Int. J. Mol. Sci. 2022, 23, 2901. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, L. Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2019, 21, 94. [Google Scholar] [CrossRef]
- Xing, X.; Kumari, A.; Brown, J.; Brook, J.D. Disrupting the Molecular Pathway in Myotonic Dystrophy. Int. J. Mol. Sci. 2021, 22, 13225. [Google Scholar] [CrossRef] [PubMed]
- Castel, A.L.; Overby, S.J.; Artero, R. MicroRNA-Based Therapeutic Perspectives in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 5600. [Google Scholar] [CrossRef]
- Mahadevan, M.S.; Yadava, R.S.; Mandal, M. Cardiac Pathology in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2021, 22, 11874. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A. Myotonic Dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef]
- Benichou, A.S.; Jauvin, D.; De Serres-Bérard, T.; Pierre, M.; Ling, K.K.; Bennett, C.F.; Rigo, F.; Gourdon, G.; Chahine, M.; Puymirat, J. Antisense oligonucleotides as a potential treatment for brain deficits observed in myotonic dystrophy type 1. Gene Ther. 2022. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Yadava, R.S.; Yu, Q.; Mandal, M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Systemic therapy in an RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3’UTR RNA. Hum. Mol. Genet. 2020, 29, 1440–1453. [Google Scholar] [CrossRef] [PubMed]
- Sznajder, Ł.J.; Swanson, M.S. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 3365. [Google Scholar] [CrossRef]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmüller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Potier, B.; Lallemant, L.; Parrot, S.; Huguet-Lachon, A.; Gourdon, G.; Dutar, P.; Gomes-Pereira, M. DM1 Transgenic Mice Exhibit Abnormal Neurotransmitter Homeostasis and Synaptic Plasticity in Association with RNA Foci and Mis-Splicing in the Hippocampus. Int. J. Mol. Sci. 2022, 23, 592. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Espinosa, J.; González-Barriga, A.; López-Castel, A.; Artero, R. Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies. Int. J. Mol. Sci. 2022, 23, 1441. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timchenko, L. Myotonic Dystrophy: From Molecular Pathogenesis to Therapeutics. Int. J. Mol. Sci. 2022, 23, 11954. https://doi.org/10.3390/ijms231911954
Timchenko L. Myotonic Dystrophy: From Molecular Pathogenesis to Therapeutics. International Journal of Molecular Sciences. 2022; 23(19):11954. https://doi.org/10.3390/ijms231911954
Chicago/Turabian StyleTimchenko, Lubov. 2022. "Myotonic Dystrophy: From Molecular Pathogenesis to Therapeutics" International Journal of Molecular Sciences 23, no. 19: 11954. https://doi.org/10.3390/ijms231911954
APA StyleTimchenko, L. (2022). Myotonic Dystrophy: From Molecular Pathogenesis to Therapeutics. International Journal of Molecular Sciences, 23(19), 11954. https://doi.org/10.3390/ijms231911954