
Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2

Abstract
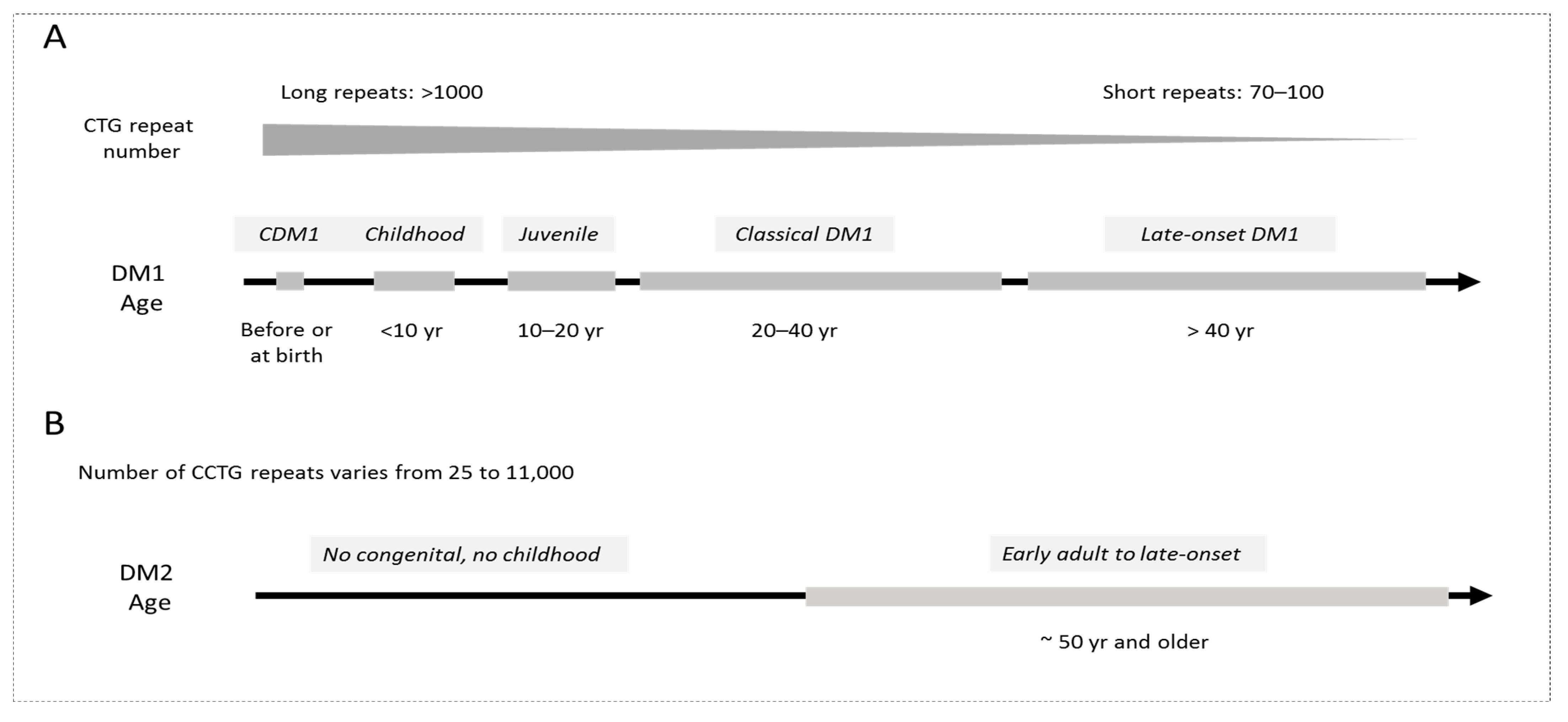
1. Introduction: From the Molecular Advances to Pre-Clinical and Clinical Studies in DM
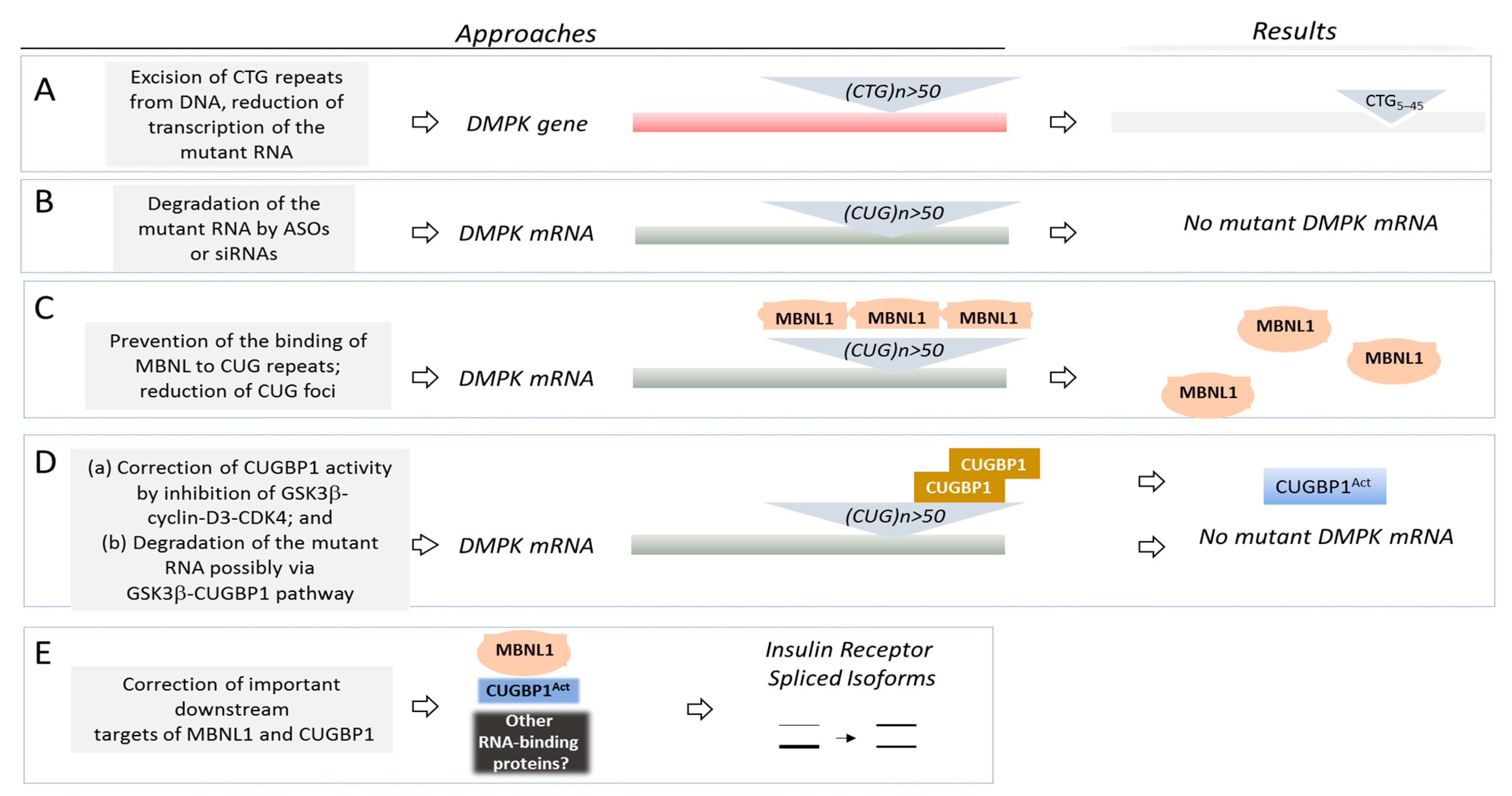
2. Therapeutic Targets in DM1 and DM2
2.1. Excision of the DM1 Mutation from the Genomic DNA
2.2. Degradation of the Mutant DMPK mRNA
2.3. Correction of Activities of RNA-Binding Proteins as the Therapeutic Approach for DM1 and CDM1
2.3.1. Tideglusib Treatments Corrected CNS and Muscle Defects in a Phase II Clinical Trial in Patients with CDM1
2.3.2. Small Molecules as Therapeutics Correcting MBNL1 and CUGBP1 in DM1
2.3.3. Downstream Targets of the Main RNA-Binding Proteins, Misregulated in DM1
3. Therapeutic Studies in DM2
4. Conclusions and Further Studies
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G., Jr.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy 2 is caused by CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [PubMed]
- Harper, P.S. Myotonic Dystrophy; WB Saunders: London, UK, 2001. [Google Scholar]
- Thornton, C.A. Myotonic Dystrophy. Neurol. Clin. 2014, 32, 705–719. [Google Scholar] [CrossRef]
- Day, J.W.; Ricker, K.; Jacobsen, J.F.; Rasmussen, L.J.; Dick, K.A.; Kress, W.; Schneider, C.; Koch, M.C.; Beilman, G.J.; Harrison, A.R.; et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology 2003, 60, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Meola, G. Myotonic dystrophy type 2: The 2020 update. Acta Myol. 2020, 39, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genom. 2010, 11, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1772. [Google Scholar] [PubMed]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying > 1,000 CTG repeats from the human DM1 locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef]
- Taneja, K.L.; McCurrach, M.; Schalling, M.; Housman, D.; Singer, R.H. Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell Biol. 1995, 128, 995–1002. [Google Scholar] [CrossRef]
- Timchenko, L. Correction of RNA-Binding Protein CUGBP1 and GSK3β Signaling as Therapeutic Approach for Congenital and Adult Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2019, 21, 94. [Google Scholar] [CrossRef]
- Timchenko, L.T.; Timchenko, N.A.; Caskey, C.T.; Roberts, R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: Implications for myotonic dystrophy. Hum. Mol. Genet. 1996, 5, 115–121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Timchenko, L.T.; Miller, J.W.; Timchenko, N.A.; DeVore, D.R.; Datar, K.V.; Lin, L.; Roberts, R.; Caskey, C.T.; Swanson, M.S. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucl. Acids Res. 1996, 24, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Timchenko, L.T. Myotonic dystrophy: The role of RNA CUG triplet repeats. Am. J. Hum. Genet. 1999, 64, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.V.; Timchenko, L.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef]
- Miller, J.W.; Urbinati, C.R.; Teng-Umnuay, P.; Stenberg, M.G.; Byrne, B.J.; Thornton, C.A.; Swanson, M.S. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000, 19, 4439–4448. [Google Scholar] [CrossRef]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef]
- Michalowski, S.; Miller, J.W.; Urbinati, C.R.; Paliouras, M.; Swanson, M.S.; Griffith, J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucl. Acids Res. 1999, 27, 3534–3542. [Google Scholar] [CrossRef]
- Timchenko, N.A.; Cai, Z.-J.; Welm, A.L.; Reddy, S.; Ashizawa, T.; Timchenko, L.T. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J. Biol. Chem. 2001, 276, 7820–7826. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.-S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78. [Google Scholar] [CrossRef]
- Huichalaf, C.; Sakai, K.; Jin, B.; Jones, K.; Wang, G.-L.; Schoser, B.; Schneider-Gold, C.; Sarkar, P.; Pereira-Smith, O.M.; Timchenko, N.; et al. Expansion of CUG RNA repeats causes stress and inhibition of translation in Myotonic Dystrophy 1 (DM1) cells. FASEB J. 2010, 24, 3706–3719. [Google Scholar] [CrossRef]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3β mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 2012, 122, 4461–4472. [Google Scholar] [CrossRef]
- Wang, M.; Weng, W.-C.; Stock, L.; Lindquist, D.; Martinez, A.; Gourdon, G.; Timchenko, N.; Snape, M.; Timchenko, L. Correction of Glycogen Synthase Kinase 3β in Myotonic Dystrophy 1 Reduces the Mutant RNA and Improves Postnatal Survival of DMSXL Mice. Mol. Cell. Biol. 2019, 39, e00155-19. [Google Scholar] [CrossRef] [PubMed]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmüller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr. Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Pattamatta, A.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation in Neurological Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033019. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Cleary, J.D.; Liu, Y.; Bañez-Coronel, M.; Bubenik, J.L.; Ayhan, F.; Ashizawa, T.; Xia, G.; Clark, H.B.; Yachnis, A.T.; et al. RAN Translation Regulated by Muscleblind Proteins in Myotonic Dystrophy Type 2. Neuron 2017, 95, 1292–1305. [Google Scholar] [CrossRef] [PubMed]
- Castel, A.L.; Overby, S.J.; Artero, R. MicroRNA-Based Therapeutic Perspectives in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 5600. [Google Scholar] [CrossRef]
- López-Morató, M.; Brook, J.D.; Wojciechowska, M. Small Molecules Which Improve Pathogenesis of Myotonic Dystrophy Type 1. Front. Neurol. 2018, 9, 349. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W.; Van Den Bosch, L. RNA toxicity in non-coding repeat expansion disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef]
- Mohan, A.; Goodwin, M.; Swanson, M.S. RNA–protein interactions in unstable microsatellite diseases. Brain Res. 2014, 1584, 3–14. [Google Scholar] [CrossRef][Green Version]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef]
- Glineburg, M.R.; Todd, P.K.; Charlet-Berguerand, N.; Sellier, C. Repeat-associated non-AUG (RAN) translation and other molecular mechanisms in Fragile X Tremor Ataxia Syndrome. Brain Res. 2018, 1693 (Pt A), 43–54. [Google Scholar] [CrossRef] [PubMed]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [PubMed]
- Daughters, R.S.; Tuttle, D.L.; Gao, W.; Ikeda, Y.; Moseley, M.L.; Ebner, T.J.; Swanson, M.S.; Ranum, L.P. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009, 5, e1000600. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Aleff, R.A.; Soragni, E.; Kalari, K.; Nie, J.; Tang, X.; Davila, J.; Kocher, J.P.; Patel, S.V.; Gottesfeld, J.M.; et al. RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J. Biol. Chem. 2015, 290, 5979–5990. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Izzo, M.; Battistini, J.; Provenzano, C.; Martelli, F.; Cardinali, B.; Falcone, G. Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing. Int. J. Mol. Sci. 2022, 23, 4622. [Google Scholar] [CrossRef]
- Liu, J.; Guo, Z.N.; Yan, X.L.; Yang, Y.; Huang, S. Brain Pathogenesis and Potential Therapeutic Strategies in Myotonic Dystrophy Type 1. Front. Aging Neurosci. 2021, 13, 755392. [Google Scholar] [CrossRef]
- van Agtmaal, E.L.; André, L.M.; Willemse, M.; Cumming, S.A.; Kessel, I.D.G.; van den Broek, W.J.A.A.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTG⋅CAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucl. Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef]
- Lo Scrudato, M.; Poulard, K.; Sourd, C.; Tomé, S.; Klein, A.F.; Corre, G.; Huguet, A.; Furling, D.; Gourdon, G.; Buj-Bello, A. Genome Editing of Expanded CTG Repeats within the Human DMPK Gene Reduces Nuclear RNA Foci in the Muscle of DM1 Mice. Mol. Ther. 2019, 27, 1372–1388. [Google Scholar] [CrossRef] [PubMed]
- Raaijmakers, R.H.L.; Ripken, L.; Ausems, C.R.M.; Wansink, D.G. CRISPR/Cas Applications in Myotonic Dystrophy: Expanding Opportunities. Int. J. Mol. Sci. 2019, 20, 3689. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.F. Shortening of trinucleotide repeats using specific endonucleases: A possible approach to a gene therapy? Trends Genet. 2015, 31, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Mulders, S.A.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef]
- Thornton, C.A.; Wang, E.; Carrell, E.M. Myotonic dystrophy: Approach to therapy. Curr. Opin. Genet. Dev. 2017, 44, 135–140. [Google Scholar] [CrossRef]
- Yadava, R.S.; Yu, Q.; Mandal, M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Systemic therapy in an RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3′UTR RNA. Hum. Mol. Genet. 2020, 29, 1440–1453. [Google Scholar] [CrossRef]
- Ionis Reports Setback on DMPKRx Program for Myotonic Dystrophy. Available online: https://strongly.mda.org/ionis-reports-setback-dmpkrx-program-myotonic-dystrophy/ (accessed on 29 July 2022).
- Klein, A.F.; Varela, M.A.; Arandel, L.; Hollan, A.; Naouar, N.; Arzumanov, A.; Seoane, D.; Revillod, L.; Bassez, G.; Ferry, A.; et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Investig. 2019, 129, 4739–4744. [Google Scholar] [CrossRef]
- Hu, N.; Antoury, L.; Baran, T.M.; Mitra, S.; Bennett, C.F.; Rigo, F.; Foster, T.H.; Wheeler, T.M. Non-invasive monitoring of alternative splicing outcomes to identify candidate therapies for myotonic dystrophy type 1. Nat. Commun. 2018, 9, 5227–5241. [Google Scholar] [CrossRef]
- Ait Benichou, S.; Jauvi, D.; De-Serres-Berard, T.; Bennett, F.; Rigo, F.; Gourdon, G.; Boutjdir, M.; Chahine, M.; Puymirat, J. Enhanced Delivery of Ligand-Conjugated Antisense Oligonucleotides (C16-HA-ASO) Targeting DMPK Transcripts for the Treatment of Myotonic Dystrophy Type 1. Hum. Gene Ther. 2022, 33, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Benichou, A.S.; Jauvin, D.; De Serres-Bérard, T.; Pierre, M.; Ling, K.K.; Bennett, C.F.; Rigo, F.; Gourdon, G.; Chahine, M.; Puymirat, J. Antisense oligonucleotides as a potential treatment for brain deficits observed in myotonic dystrophy type 1. Gene Ther. 2022. [CrossRef] [PubMed]
- Dugal-Tessier, J.; Thirumalairajan, S.; Jain, N. Antibody-Oligonucleotide Conjugates: A Twist to Antibody-Drug Conjugates. J. Clin. Med. 2021, 10, 838. [Google Scholar] [CrossRef] [PubMed]
- Study of AOC 1001 in Adult Myotonic Dystrophy Type 1 (DM1) Patients (MARINA). Available online: https://clinicaltrials.gov/ct2/show/NCT05027269 (accessed on 29 July 2022).
- Magaña, J.J.; Cisneros, B. Perspectives on gene therapy in myotonic dystrophy type 1. J. Neurosci. Res. 2011, 89, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Safety and Efficacy of Tideglusib in Congenital Myotonic Dystrophy (REACH CDM X). Available online: https://clinicaltrials.gov/ct2/show/NCT05004129 (accessed on 29 July 2022).
- Reddy, K.; Jenquin, J.R.; Cleary, J.D.; Berglund, J.A. Mitigating RNA Toxicity in Myotonic Dystrophy using Small Molecules. Int. J. Mol. Sci. 2019, 20, 4017. [Google Scholar] [CrossRef]
- Pascual-Gilabert, M.; Lopez-Castel, A.; Artero, R. Myotonic dystrophy type 1 drug development: A pipeline toward the market. Drug Discov. Today 2021, 26, 1765–1772. [Google Scholar] [CrossRef]
- Costales, M.G.; Childs-Disney, J.L.; Haniff, H.S.; Disney, M.D. How We Think about Targeting RNA with Small Molecules. J. Med. Chem. 2020, 63, 8880–8900. [Google Scholar] [CrossRef]
- Ondono, R.; Lirio, Á.; Elvira, C.; Álvarez-Marimon, E.; Provenzano, C.; Cardinali, B.; Pérez-Alonso, M.; Perálvarez-Marín, A.; Borrell, J.I.; Falcone, G.; et al. Design of novel small molecule base-pair recognizers of toxic CUG RNA transcripts characteristics of DM1. Comput. Struct. Biotechnol. J. 2020, 19, 51–61. [Google Scholar] [CrossRef]
- Xing, X.; Kumari, A.; Brown, J.; Brook, J.D. Disrupting the Molecular Pathway in Myotonic Dystrophy. Int. J. Mol. Sci. 2021, 22, 13225. [Google Scholar] [CrossRef]
- Warf, M.B.; Nakamori, M.; Matthys, C.M.; Thornton, C.A.; Berglund, J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Nat. Acad. Sci. USA 2009, 106, 18551–18556. [Google Scholar] [CrossRef]
- Nakamori, M.; Katarzyna Taylor, K.; Mochizuki, H.; Sobczak, K.; Takahashi, M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Annals of Clin. Transl. Neurol. 2016, 3, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Jenquin, J.R.; Yang, H.; Huigens, R.W., 3rd; Nakamori, M.; Berglund, J.A. Combination Treatment of Erythromycin and Furamidine Provides Additive and Synergistic Rescue of Mis-Splicing in Myotonic Dystrophy Type 1 Models. ACS Pharmacol. Transl. Sci. 2019, 2, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Bargiela, A.; Sabater-Arcis, M.; Jorge Espinosa-Espinosa, J.; Miren Zulaica, M.; Munain, A.L.M.; Artero, R. Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. USA 2019, 116, 25203–25213. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.; Jenquin, J.R.; McConnell, O.L.; Cleary, J.D.; Richardson, J.I.; Pinto, B.S.; Haerle, M.C.; Delgado, E.; Planco, L.; Nakamori, M.; et al. A CTG repeat-selective chemical screen identifies microtubule inhibitors as selective modulators of toxic CUG RNA levels. Proc. Natl. Acad. Sci. USA 2019, 116, 20991–21000. [Google Scholar] [CrossRef] [PubMed]
- Angelbello, A.J.; Benhamou, R.I.; Rzuczek, S.G.; Choudhary, S.; Tang, Z.; Chen, J.L.; Roy, M.; Wang, K.W.; Yildirim, I.; Jun, A.S.; et al. A Small Molecule that Binds an RNA Repeat Expansion Stimulates Its Decay via the Exosome Complex. Cell Chem. Biol. 2021, 28, 34–45. [Google Scholar] [CrossRef]
- Angelbello, A.J.; Rzuczek, S.G.; Mckee, K.K.; Chen, J.L.; Olafson, H.; Cameron, M.D.; Moss, W.N.; Wang, E.T.; Disney, M.D. Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 7799–7804. [Google Scholar] [CrossRef]
- Zhang, F.; Bodycombe, N.E.; Haskell, K.M.; Sun, Y.L.; Wang, E.T.; Morris, C.A.; Jones, L.H.; Wood, L.D.; Pletcher, M.T. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 2017, 26, 3056–3068. [Google Scholar] [CrossRef]
- Lee, J.; Bai, Y.; Chembazhi, U.V.; Peng, S.; Yum, K.; Luu, L.M.; Hagler, L.D.; Serrano, J.F.; Chan, H.Y.E.; Kalsotra, A.; et al. Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2019, 116, 8709–8714. [Google Scholar] [CrossRef]
- Chakraborty, M.; Sellier, C.; Ney, M.; Pascal, V.; Charlet-Berguerand, N.; Artero, R.; Llamusi, B. Daunorubicin reduces MBNL1 sequestration caused by CUG-repeat expansion and rescues cardiac dysfunctions in a Drosophila model of myotonic dystrophy. Disease Models Mechanisms 2018, 11, dmm032557. [Google Scholar] [CrossRef]
- Available online: Myotonic-Dystrophy-Drug-Development-Pipeline-as-of-March-8th-2021-v2.pdf (accessed on 29 July 2022).
- Ketley, A.; Chen, C.Z.; Li, X.; Arya, S.; Robinson, T.E.; Granados-Riveron, J.; Udosen, I.; Morris, G.E.; Holt, I.; Furling, D.; et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum. Mol. Genet. 2014, 23, 1551–1562. [Google Scholar] [CrossRef]
- Ketley, A.; Wojciechowska, M.; Ghidelli-Disse, S.; Bamborough, P.; Ghosh, T.K.; Morato, M.L.; Sedehizadeh, S.; Malik, N.A.; Tang, Z.; Powalowska, P.; et al. CDK12 inhibition reduces abnormalities in cells from patients with myotonic dystrophy and in a mouse model. Sci. Transl. Med. 2020, 12, eaaz2415. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Laustriat, D.; Gide, J.; Barrault, L.; Chautard, E.; Benoit, C.; Auboeuf, D.; Boland, A.; Battail, C.; Artiguenave, F.; Deleuze, J.-F.; et al. In Vitro and In Vivo Modulation of Alternative Splicing by the Biguanide Metformin. Mol. Ther. Nucl. Acids. 2015, 4, e262. [Google Scholar] [CrossRef]
- García-Puga, M.; Saenz-Antoñanzas, A.; Matheu, A.; de Munain, A.L. Targeting Myotonic Dystrophy Type 1 with Metformin. Int. J. Mol. Sci. 2022, 23, 2901. [Google Scholar] [CrossRef]
- Bassez, G.; Hogrel, E.A.J.-Y.; Arrouasse, R.; Baghdoyan, S.; Bhugaloo, H.; Gourlay-Chu, M.-L.; Le Corvoisier, P.; Peschanski, M. Improved mobility with metformin in patients with myotonic dystrophy type 1: A randomized controlled trial. Brain 2018, 141, 2855–2865. [Google Scholar] [CrossRef] [PubMed]
- García-Puga, M.; Saenz-Antoñanzas, A.; Fernández-Torrón, R.; Munain, A.L.; Matheu, A. Myotonic Dystrophy type 1 cells display impaired metabolism and mitochondrial dysfunction that are reversed by metformin. Aging 2020, 12, 6260–6275. [Google Scholar] [CrossRef] [PubMed]
- Brockhoff, M.; Rion, N.; Chojnowska, K.; Wiktorowicz, T.; Eickhorst, C.; Erne, B.; Frank, S.; Angelini, C.; Furling, D.; Rüegg, M.A.; et al. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J. Clin. Investig. 2017, 127, 549–563. [Google Scholar] [CrossRef]
- Ravel-Chapuis, A.; Al-Rewashdy, A.; Bélange, G.; Jasmin, B.J. Pharmacological and physiological activation of AMPK improves the spliceopathy in DM1 mouse muscle. Hum. Mol. Genet. 2018, 27, 3361–3376. [Google Scholar] [CrossRef]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef]
- Sznajder, Ł.J.; Thomas, J.D.; Carrell, E.M.; Reid, T.; McFarland, K.N.; Cleary, J.D.; Oliveira, R.; Nutter, C.A.; Bhatt, K.; Sobczak, K.; et al. Intron retention induced by microsatellite expansions as a disease biomarker. Proc. Natl. Acad. Sci. USA 2018, 115, 4234–4239. [Google Scholar] [CrossRef]
- Assessment of Myotonic Dystrophy Type 2 Research and Drug Development with Recommendations for Investment. Available online: https://www.myotonic.org/sites/default/files/pages/files/Review-of-DM2-FNL-2019-09-24 (accessed on 29 July 2022).
- Kim, E.Y.; Barefield, D.Y.; Vo, A.H.; Gacita, A.M.; Schuster, E.J.; Wyatt, E.J.; Davis, J.L.; Dong, B.; Sun, C.; Page, P.; et al. Distinct pathological signatures in human cellular models of myotonic dystrophy subtypes. JCI Insight 2019, 4, e122686. [Google Scholar] [CrossRef] [PubMed]
- Armas, P.; Coux, G.; Weiner, A.M.J.; Calcaterra, N.B. What’s new about CNBP? Divergent functions and activities for a conserved nucleic acid binding protein. Biochim. Biophys. Acta 2021, 1865, 129996. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Wei, C.; Schoser, B.; Meola, G.; Timchenko, N.; Timchenko, L. Reduction of toxic RNAs in myotonic dystrophies type 1 and type 2 by the RNA helicase p68/DDX5. Proc. Natl. Acad. Sci. USA 2015, 112, 8041–8804. [Google Scholar] [CrossRef] [PubMed]
- Jenquin, J.R.; O’Brien, A.P.; Poukalov, K.; Lu, Y.; Frias, J.A.; Shorrock, H.K.; Richardson, J.I.; Mazdiyasni, H.; Yang, H.; Huigens III, R.W.; et al. Molecular characterization of myotonic dystrophy fibroblast cell lines for use in small molecule screening. iScience 2022, 25, 104198. [Google Scholar] [CrossRef]
- Deng, J.; Guan, X.-X.; Zhu, Y.-B.; Deng, H.-T.; Li, G.-X.; Guo, Y.-C.; Jin, P.; Duan, R.-H.; Huang, W. Reducing the Excess Activin Signaling Rescues Muscle Degeneration in Myotonic Dystrophy Type 2 Drosophila Model. J. Pers. Med. 2022, 12, 385. [Google Scholar] [CrossRef]
- Wagner-Griffin, S.; Abe, M.; Benhamou, R.I.; Angelbello, A.J.; Vishnu, K.; Chen, J.L.; Childs-Disney, J.L.; Disney, M.D. A Druglike Small Molecule that Targets r(CCUG) Repeats in Myotonic Dystrophy Type 2 Facilitates Degradation by RNA Quality Control Pathways. J. Med. Chem. 2021, 64, 8474–8485. [Google Scholar] [CrossRef]
- Benhamou, R.I.; Angelbello, A.J.; Wang, E.T.; Disney, M.D. A Toxic RNA Catalyzes the Cellular Synthesis of Its Own Inhibitor, Shunting It to Endogenous Decay Pathways. Cell. Chem. Biol. 2020, 27, 223–231. [Google Scholar] [CrossRef]
- Childs-Disney, J.L.; Yildirim, I.; Park, H.; Lohman, J.R.; Guan, L.; Tran, T.; Sarkar, P.; Schatz, G.C.; Disney, M.D. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem Biol. 2014, 9, 538–550. [Google Scholar] [CrossRef]
- Wong, C.H.; Fu, Y.; Ramisetty, S.R.; Baranger, A.M.; Zimmerman, S.C. Selective inhibition of MBNL1-CCUG interaction by small molecules toward potential therapeutic agents for myotonic dystrophy type 2 (DM2). Nucl. Acids Res. 2011, 39, 8881–8890. [Google Scholar] [CrossRef][Green Version]
- Pellier, C.; Cerro-Herreros, E.; Blatter, M.; Freyermuth, F.; Gaucherot, A.; Ruffenach, F.; Sarkar, P.; Puymirat, J.; Udd, B.; Day, J.W.; et al. rbFOX1/MBNL1 competition for CCUG RNA repeats binding contributes to myotonic dystrophy type 1/type 2 differences. Nat. Commun. 2018, 9, 2009. [Google Scholar] [CrossRef]
- Huichalaf, C.; Schoser, B.; Schneider-Gold, C.; Jin, B.; Sarkar, P.; Timchenko, L. Reduction of the rate of protein translation in patients with myotonic dystrophy 2. J. Neurosci. 2009, 29, 9042–9049. [Google Scholar] [CrossRef] [PubMed]
- Raheem, O.; Olufemi, S.E.; Bachinski, L.L.; Vihola, A.; Sirito, M.; Holmlund-Hmpf, J.; Haapasalo, H.; Li, Y.P.; Udd, B.; Krahe, R. Mutant (CCTG)n expansion causes abnormal expression of zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2. Am. J. Pathol. 2010, 177, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, R.; Hamel, F.; Beaulieu, D.; Patry, L.; Haineault, C.; Tarnopolsky, M.; Schoser, B.; Puymirat, J. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. 2009, 36, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Botta, A.; Caldarola, S.; Vallo, L.; Bonifazi, E.; Fruci, D.; Gullotta, F.; Massa, R.; Novelli, G.; Loreni, F. Effect of the [CCTG]n repeat expansion on ZNF9 expression in myotonic dystrophy type II (DM2). Biochim. Biophys. Acta 2006, 1762, 329–334. [Google Scholar]
- Margolis, J.M.; Schoser, B.G.; Moseley, M.L.; Day, J.W.; Ranum, L.P. DM2 intronic expansions: Evidence for CCUG accumulation without flanking sequence or effects on ZNF9 mRNA processing or protein expression. Hum. Mol. Genet. 2006, 15, 1808–1815. [Google Scholar] [CrossRef] [PubMed]
- Massa, R.; Panico, M.B.; Caldarola, S.; Fusco, F.R.; Sabatelli, P.; Terracciano, C.; Botta, A.; Novelli, G.; Bernardi, G.; Loreni, F. The myotonic dystrophy type 2 (DM2) gene product zinc finger protein 9 (ZNF9) is associated with sarcomeres and normally localized in DM2 patients’ muscles. Neuropathol. Appl. Neurobiol. 2010, 36, 275–284. [Google Scholar] [CrossRef]
- Coni, S.; Falconio, F.A.; Marzullo, M.; Munafò, M.; Zuliani, B.; Mosti, F.; Fatica, A.; Ianniello, Z.; Bordone, R.; Macone, A.; et al. Translational control of polyamine metabolism by CNBP is required for Drosophila locomotor function. Elife 2021, 10, e69269. [Google Scholar] [CrossRef]
- Benhalevy, D.; Gupta, S.K.; Danan, C.H.; Ghosal, S.; Sun, H.-W.; Kazemier, H.G.; Paeschke, K.; Hafner, M.; Juranek, S.A. The human CCHC-type Zinc Finger Nucleic Acid Binding Protein binds G-rich elements in target mRNA coding sequences and promotes translation. Cell Rep. 2017, 18, 2979–2990. [Google Scholar] [CrossRef]
- Chen, W.; Wang, Y.; Abe, Y.; Cheney, L.; Udd, B.; Li, Y.-P. Haploinsufficiency for Znf9 in Znf9 +/− mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J. Mol. Biol. 2007, 368, 8–17. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; SchneiderGold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of cellular nucleic acid binding protein encoded by a myotonic dystrophy type 2 gene causes muscle atrophy. Mol. Cell. Biol. 2018, 38, e00649–e00717. [Google Scholar]
- Chen, Y.; Sharma, S.; Assis, P.A.; Jiang, Z.; Elling, R.; Olive, A.J.; Hang, S.; Bernier, J.; Huh, J.R.; Sassetti, C.M.; et al. CNBP controls IL-12 gene transcription and Th1 immunity. J. Exp. Med. 2018, 215, 3136–3150. [Google Scholar] [CrossRef] [PubMed]
- Mahyera, A.S.; Schneider, T.; Halliger-Keller, B.; Schrooten, K.; Hörner, E.M.; Rost, S.; Kress, W. Distribution and Structure of DM2 Repeat Tract Alleles in the German Population. Front. Neurol. 2018, 9, 463. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeted Mechanism of DM1 | Company | Phase I | Phase II | Outcome of Phase II | Phase III |
|---|---|---|---|---|---|
| Correction of RNA-binding protein CUGBP1 and degradation of the mutant RNA by small molecule GSK3 inhibitor tideglusib | AMO Pharma | Drug safety is known | Phase 2 completed | Reduction of CNS and muscle defects | Active |
| Correction of splicing of Insulin Receptor and other splicing events by metformin | Tor Vergata | Drug safety is known | Phase 2a completed | Mobility and gait improvement | Active |
| Correction of MBNL1 activity, reduction of CUG foci, reduction of myotonia by erythromycin | Osaka University Hospital | Drug safety is known | Active | ||
| Degradation of the mutant DMPK mRNA by AON | Ionis | Phase ½ completed | Phase ½ completed | Poor penetration into skeletal muscle | |
| Degradation of the mutant DMPK mRNA by AOC | Avidis | Phase ½ in progress |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timchenko, L. Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2. Int. J. Mol. Sci. 2022, 23, 10491. https://doi.org/10.3390/ijms231810491
Timchenko L. Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2. International Journal of Molecular Sciences. 2022; 23(18):10491. https://doi.org/10.3390/ijms231810491
Chicago/Turabian StyleTimchenko, Lubov. 2022. "Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2" International Journal of Molecular Sciences 23, no. 18: 10491. https://doi.org/10.3390/ijms231810491
APA StyleTimchenko, L. (2022). Development of Therapeutic Approaches for Myotonic Dystrophies Type 1 and Type 2. International Journal of Molecular Sciences, 23(18), 10491. https://doi.org/10.3390/ijms231810491