
Aquacobalamin Accelerates Orange II Destruction by Peroxymonosulfate via the Transient Formation of Secocorrinoid: A Mechanistic Study


Abstract
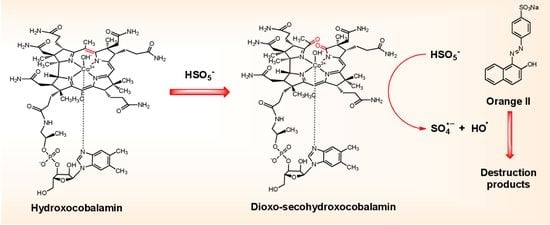
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Wang, J.; Wang, S. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants. Chem. Eng. J. 2018, 334, 1502–1517. [Google Scholar] [CrossRef]
- Zhang, B.-T.; Zhang, Y.; Teng, Y.; Fan, M. Sulfate Radical and Its Application in Decontamination Technologies. Crit. Rev. Environ. Sci. Technol. 2015, 45, 1756–1800. [Google Scholar] [CrossRef]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D. Degradation of Organic Contaminants in Water with Sulfate Radicals Generated by the Conjunction of Peroxymonosulfate with Cobalt, Environ. Sci. Technol. 2003, 37, 4790–4797. [Google Scholar] [CrossRef]
- Hu, P.; Long, M. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications. Appl. Catal. B Environ. 2016, 181, 103–117. [Google Scholar] [CrossRef]
- Shamir, D.; Meyerstein, D.; Katsaran, D.; Pochtarenko, L.; Yardeni, G.; Burg, A.; Albo, Y.; Kornweitz, H.; Zilbermann, I. Mechanisms of Reaction Between Co(II) Complexes and Peroxymonosulfate. Eur. J. Inorg. Chem. 2022, e202100646. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Qian, J.; Pan, B. Are Free Radicals the Primary Reactive Species in Co(II)-Mediated Activation of Peroxymonosulfate? New Evidence for the Role of the Co(II)–Peroxymonosulfate Complex. Environ. Sci. Technol. 2021, 55, 6397–6406. [Google Scholar] [CrossRef]
- Huang, Z.; Yao, Y.; Lu, J.; Chen, C.; Lu, W.; Huang, S.; Chen, W. The consortium of heterogeneous cobalt phthalocyanine catalyst and bicarbonate ion as a novel platform for contaminants elimination based on peroxymonosulfate activation. J. Hazard. Mater. 2016, 301, 214–221. [Google Scholar] [CrossRef]
- Wu, M.; Fu, K.; Deng, H.; Shi, J. Cobalt tetracarboxyl phthalocyanine-manganese octahedral molecular sieve (OMS-2) as a heterogeneous catalyst of peroxymonosulfate for degradation of diclofenac. Chemosphere 2019, 219, 756–765. [Google Scholar] [CrossRef]
- Giedyk, M.; Goliszewska, K.; Gryko, D. Vitamin B12 catalysed reactions. Chem. Soc. Rev. 2015, 44, 3391–3404. [Google Scholar] [CrossRef] [PubMed]
- Koide, T.; Ono, T.; Shimakoshi, H.; Hisaeda, Y. Functions of bioinspired pyrrole cobalt complexes–recently developed catalytic systems of vitamin B12 related complexes and porphycene complexes. Coord. Chem. Rev. 2022, 472, 214690. [Google Scholar] [CrossRef]
- Dereven’kov, I.A.; Salnikov, D.S.; Silaghi-Dumitrescu, R.; Makarov, S.V.; Koifman, O.I. Redox chemistry of cobalamin and its derivatives. Coord. Chem. Rev. 2016, 309, 68–83. [Google Scholar] [CrossRef]
- Cheng, J.; Shiota, Y.; Yamasaki, M.; Izukawa, K.; Tachi, Y.; Yoshizawa, K.; Shimakoshi, H. Mechanistic Study for the Reaction of B12 Complexes with m-Chloroperbenzoic Acid in Catalytic Alkane Oxidations. Inorg. Chem. 2022, 61, 9710–9724. [Google Scholar] [CrossRef]
- Lehene, M.; Plesa, D.; Ionescu-Zinca, S.; Iancu, S.D.; Leopold, N.; Makarov, S.V.; Brânzanic, A.M.V.; Silaghi-Dumitrescu, R. Adduct of Aquacobalamin with Hydrogen Peroxide. Inorg. Chem. 2021, 60, 12681–12684. [Google Scholar] [CrossRef]
- Salnikov, D.S.; Makarov, S.V.; Koifman, O.I. The radical versus ionic mechanisms of reduced cobalamin inactivation by tert-butyl hydroperoxide and hydrogen peroxide in aqueous solution. New J. Chem. 2021, 45, 535–543. [Google Scholar] [CrossRef]
- Dereven’kov, I.A.; Makarov, S.V.; Makarova, A.S. Mechanism of aquacobalamin decomposition in aqueous aerobic solutions containing glucose oxidase and glucose. React. Kinet. Mech. Catal. 2021, 133, 73–84. [Google Scholar] [CrossRef]
- Shahadat, H.M.; Younus, H.A.; Ahmad, N.; Shiguo, Z.; Zhuiykov, S.; Verpoort, F. Macrocyclic cyanocobalamin (vitamin B12) as a homogeneous electrocatalyst for water oxidation under neutral conditions. Chem. Commun. 2020, 56, 1968–1971. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Liao, R.-Z. Mechanism of water oxidation catalyzed by vitamin B12: Redox non-innocent nature of corrin ligand and crucial role of phosphate. Chin. Chem. Lett. 2022, 33, 358–361. [Google Scholar] [CrossRef]
- Hou, J.; Lin, J.; Fu, H.; Wan, Y.; Qu, X.; Xu, Z.; Zheng, S. Vitamin B12 derived CoCNx composite confined in SBA-15 as highly effective catalyst to activate peroxymonosulfate for naproxen degradation. Chem. Eng. J. 2020, 389, 124344. [Google Scholar] [CrossRef]
- Zhiyong, Y.; Bensimon, M.; Laub, D.; Kiwi-Minsker, L.; Jardim, W.; Mielczarski, E.; Mielczarski, J.; Kiwi, J. Accelerated photodegradation (minute range) of the commercial azo-dye Orange II mediated by Co3O4/Raschig rings in the presence of oxone. J. Mol. Catal. A: Chem. 2007, 272, 11–19. [Google Scholar] [CrossRef]
- Raja, P.; Bensimon, M.; Klehm, U.; Albers, P.; Laub, D.; Kiwi-Minsker, L.; Renken, A.; Kiwi, J. Highly dispersed PTFE/Co3O4 flexible films as photocatalyst showing fast kinetic performance for the discoloration of azo-dyes under solar irradiation. J. Photochem. Photobiol. A Chem. 2007, 187, 332–338. [Google Scholar] [CrossRef]
- Shi, P.; Su, R.; Wan, F.; Zhu, M.; Li, D.; Xu, S. Co3O4 nanocrystals on graphene oxide as a synergistic catalyst for degradation of Orange II in water by advanced oxidation technology based on sulfate radicals. Appl. Catal. B: Environ. 2012, 123–124, 265–272. [Google Scholar] [CrossRef]
- Shi, P.; Su, R.; Zhu, S.; Zhu, M.; Li, D.; Xu, S. Supported cobalt oxide on graphene oxide: Highly efficient catalysts for the removal of Orange II from water. J. Hazard. Mater. 2012, 229–230, 331–339. [Google Scholar] [CrossRef]
- Kräutler, B. The Photooxygenation of Heptamethyl Coα, Coβ-Dicyanocobyrinate. Helv. Chim. Acta. 1982, 65, 1941–1948. [Google Scholar] [CrossRef]
- Chemaly, S.M.; Brown, K.L.; Fernandes, M.A.; Munro, O.Q.; Grimmer, C.; Marques, H.M. Probing the Nature of the CoIII Ion in Corrins: The Structural and Electronic Properties of Dicyano- and Aquacyanocobyrinic Acid Heptamethyl Ester and a Stable Yellow Dicyano- and Aquacyanocobyrinic Acid Heptamethyl Ester. Inorg. Chem. 2011, 50, 8700–8718. [Google Scholar] [CrossRef]
- Pugina, R.A.; Denisova, E.A.; Ivlev, P.A.; Salnikov, D.S.; Makarov, S.V. Synthesis of vitamin B12 derivatives with sodium hydroxymethanesulfinate. J. Porphyr. Phthalocyanines 2018, 22, 1092–1098. [Google Scholar] [CrossRef]
- Xia, L.; Cregan, A.G.; Berben, L.A.; Brasch, N.E. Studies on the Formation of Glutathionylcobalamin: Any Free Intracellular Aquacobalamin Is Likely to Be Rapidly and Irreversibly Converted to Glutathionylcobalamin. Inorg. Chem. 2004, 43, 6848–6857. [Google Scholar] [CrossRef]
- Wagner, F. Reactions of the cyano and alkyl cobalamins. Proc. R. Soc. A 1965, 288, 344–347. [Google Scholar] [CrossRef]
- Kwon, B.G.; Ryu, S.; Yoon, J. Determination of hydroxyl radical rate constants in a continuous flow system using competition kinetics. J. Ind. Eng. Chem. 2009, 15, 809–812. [Google Scholar] [CrossRef]
- Thomas, J.K. Rates of Reaction of the Hydroxyl Radical. Trans. Faraday Soc. 1965, 61, 702–707. [Google Scholar] [CrossRef]
- Clifton, C.L.; Huie, R.E. Rate constants for hydrogen abstraction reactions of the sulfate radical, SO4−. Alcohols. Int. J. Chem. Kin. 1989, 21, 677–687. [Google Scholar] [CrossRef]
- Xiao, G.; Xu, T.; Faheem, M.; Xi, Y.; Zhou, T.; Moryani, H.T.; Bao, J.; Du, J. Evolution of Singlet Oxygen by Activating Peroxydisulfate and Peroxymonosulfate: A Review. Int. J. Environ. Res. Public Health 2021, 18, 3344. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.F.; Upton, M.W. Studies on singlet oxygen in aqueous solution. Part 3. The decomposition of peroxy-acids. J. Chem. Soc. Dalton Trans. 1985, 1151–1153. [Google Scholar] [CrossRef]
- Matheson, I.B.C.; Lee, J. Chemical reaction rates of amino acids with singlet oxygen. Photochem. Photobiol. 1979, 29, 879–881. [Google Scholar] [CrossRef]
- Charbouillot, T.; Brigante, M.; Mailhot, G.; Maddigapu, P.R.; Minero, C.; Vione, D. Performance and selectivity of the terephthalic acid probe for radical •OH as a function of temperature, pH and composition of atmospherically relevant aqueous media. J. Photochem. Photobiol. A Chem. 2011, 222, 70–76. [Google Scholar] [CrossRef]
- Dereven’kov, I.A.; Makarov, S.V.; Brânzanic, A.M.V.; Silaghi-Dumitrescu, R.; Molodtsov, P.A.; Pokrovskaya, E.A. Formation of hydroxyl radical in aqueous solutions containing selenite and glutathione. Polyhedron 2021, 198, 115072. [Google Scholar] [CrossRef]
- Neta, P.; Madhavan, V.; Zemel, H.; Fessenden, R.W. Rate constants and mechanism of the reaction of sulfate radical anion with aromatic compounds. J. Am. Chem. Soc. 1977, 99, 163–164. [Google Scholar] [CrossRef]
- Matthews, R.W.; Sangster, D.F. Measurement by Benzoate Radiolytic Decarboxylation of Relative Rate Constants for Hydroxyl Radical Reactions. J. Phys. Chem. 1965, 69, 1938–1946. [Google Scholar] [CrossRef]
- Hoigné, J.; Bader, H. The role of hydroxyl radical reactions in ozonation processes in aqueous solutions. Water Res. 1976, 10, 377–386. [Google Scholar] [CrossRef]
- Duan, P.; Liu, X.; Liu, B.; Akram, M.; Li, Y.; Pan, J.; Yue, Q.; Gao, B.; Xu, X. Effect of phosphate on peroxymonosulfate activation: Accelerating generation of sulfate radical and underlying mechanism. Appl. Catal. B Environ. 2021, 298, 120532. [Google Scholar] [CrossRef]
- Weeks, J.T.; Rabani, J.J. The Pulse Radiolysis of Deaerated Aqueous Carbonate Solutions. I. Transient Optical Spectrum and Mechanism. II. pK for OH Radicals. J. Phys. Chem. 1966, 70, 2100–2106. [Google Scholar] [CrossRef]
- Dogliotti, L.; Hayon, E. Flash photolysis of per[oxydi]sulfate ions in aqueous solutions. The sulfate and ozonide radical anions. J. Phys. Chem. 1967, 71, 2511–2516. [Google Scholar] [CrossRef]
- Perry, C.B.; Fernandes, M.A.; Brown, K.L.; Zou, X.; Valente, E.J.; Marques, H.M. Probing the Nature of the CoIII Ion in Cobalamins − Spectroscopic and Structural Investigations of the Reactions of Aquacobalamin (Vitamin B12a) with Ambident Nucleophiles. Eur. J. Inorg. Chem. 2003, 2095–2107. [Google Scholar] [CrossRef]
- Nowakowska, M.; Chemaly, S.M.; Rousseau, A.L.; Govender, P.P.; Varadwaj, P.R.; Varadwaj, A.; Yamashita, K.; Marques, H.M. Probing the nature of the Co(III) ion in corrins: The reactions of aquacyano-5-seco-cobyrinic acid heptamethyl ester with anionic ligands. Inorg. Chim. Acta 2019, 484, 402–413. [Google Scholar] [CrossRef]
- Salnikov, D.S.; Dereven’kov, I.A.; Artyushina, E.N.; Makarov, S.V. Interaction of cyanocobalamin with sulfur-containing reducing agents in aqueous solutions. Russ. J. Phys. Chem. A 2013, 87, 44–48. [Google Scholar] [CrossRef]
- Barker, H.A.; Smyth, R.D.; Weissbach, H.; Toohey, J.I.; Ladd, J.N.; Volcani, B.E. Isolation and Properties of Crystalline Cobamide Coenzymes Containing Benzimidazole or 5,6-Dimethylbenzimidazole. J. Biol. Chem. 1960, 235, 480–488. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dereven’kov, I.A.; Sakharova, E.S.; Osokin, V.S.; Makarov, S.V. Aquacobalamin Accelerates Orange II Destruction by Peroxymonosulfate via the Transient Formation of Secocorrinoid: A Mechanistic Study. Int. J. Mol. Sci. 2022, 23, 11907. https://doi.org/10.3390/ijms231911907
Dereven’kov IA, Sakharova ES, Osokin VS, Makarov SV. Aquacobalamin Accelerates Orange II Destruction by Peroxymonosulfate via the Transient Formation of Secocorrinoid: A Mechanistic Study. International Journal of Molecular Sciences. 2022; 23(19):11907. https://doi.org/10.3390/ijms231911907
Chicago/Turabian StyleDereven’kov, Ilia A., Ekaterina S. Sakharova, Vladimir S. Osokin, and Sergei V. Makarov. 2022. "Aquacobalamin Accelerates Orange II Destruction by Peroxymonosulfate via the Transient Formation of Secocorrinoid: A Mechanistic Study" International Journal of Molecular Sciences 23, no. 19: 11907. https://doi.org/10.3390/ijms231911907
APA StyleDereven’kov, I. A., Sakharova, E. S., Osokin, V. S., & Makarov, S. V. (2022). Aquacobalamin Accelerates Orange II Destruction by Peroxymonosulfate via the Transient Formation of Secocorrinoid: A Mechanistic Study. International Journal of Molecular Sciences, 23(19), 11907. https://doi.org/10.3390/ijms231911907