
Identification of Candidate Genes in Breast Cancer Induced by Estrogen Plus Progestogens Using Bioinformatic Analysis


Abstract
1. Introduction
2. Results
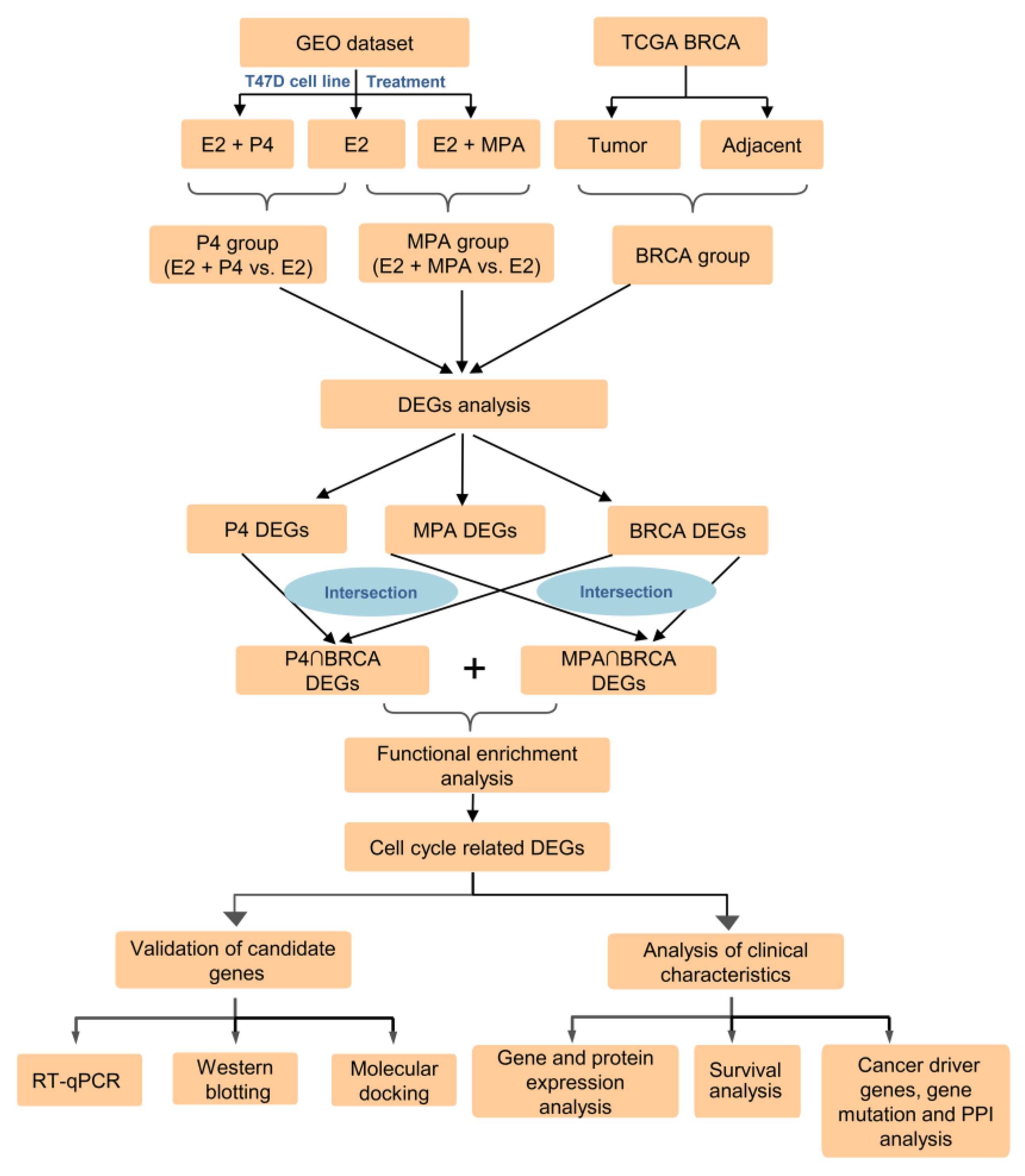
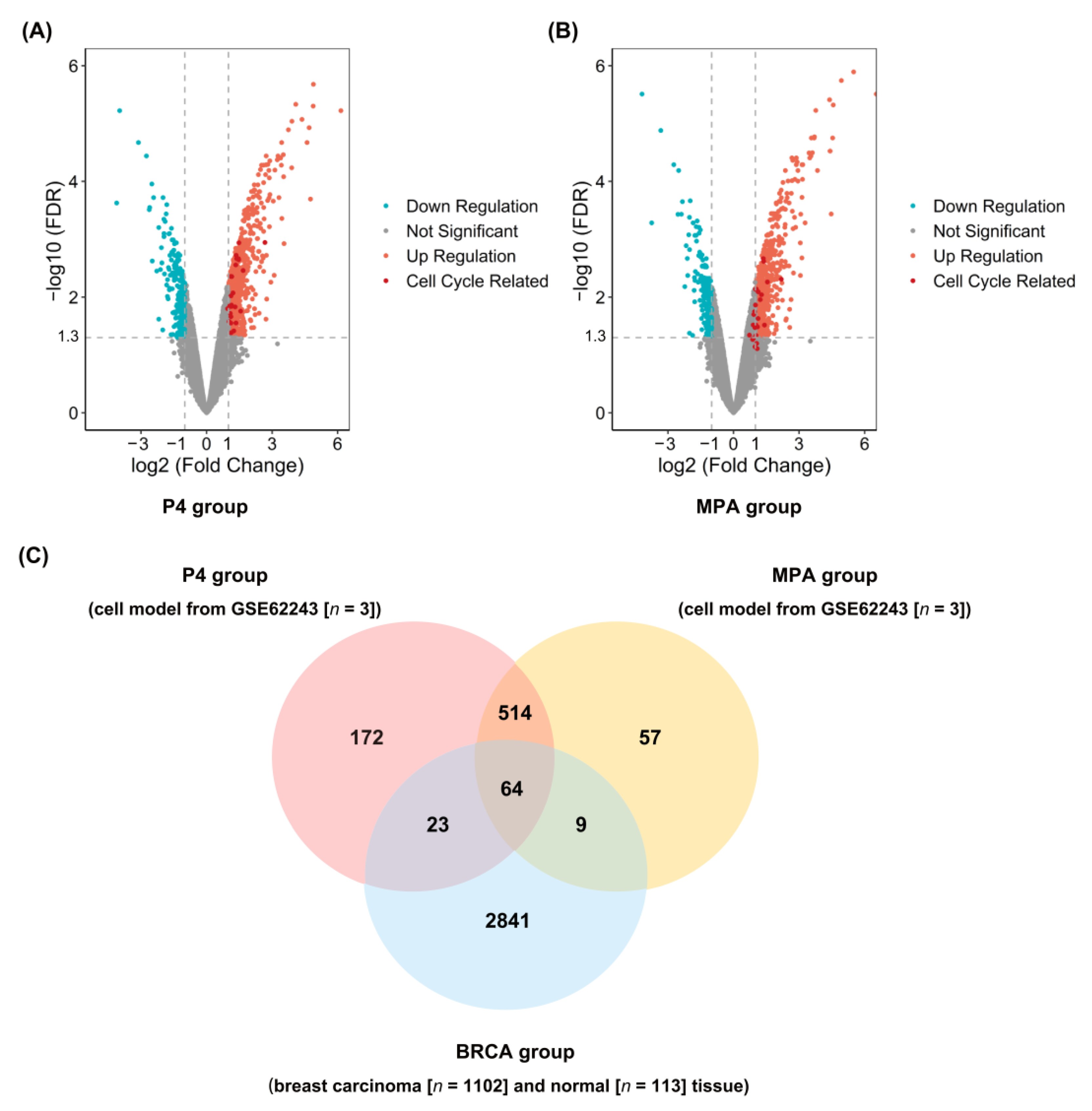
2.1. Identification of DEGs in P4, MPA, and BRCA Group
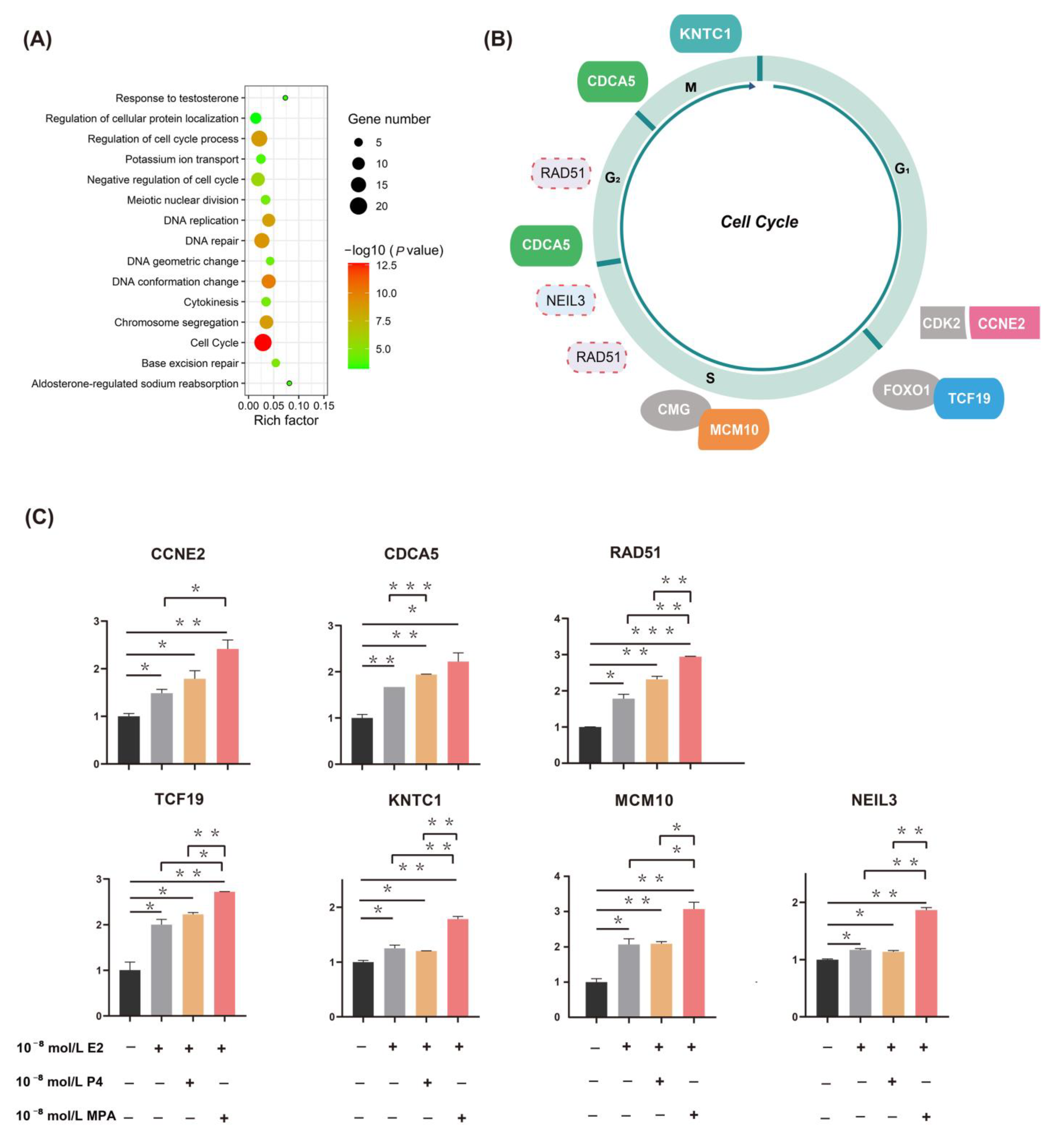
2.2. Functional Enrichment Analysis and qPCR Validation of DEGs
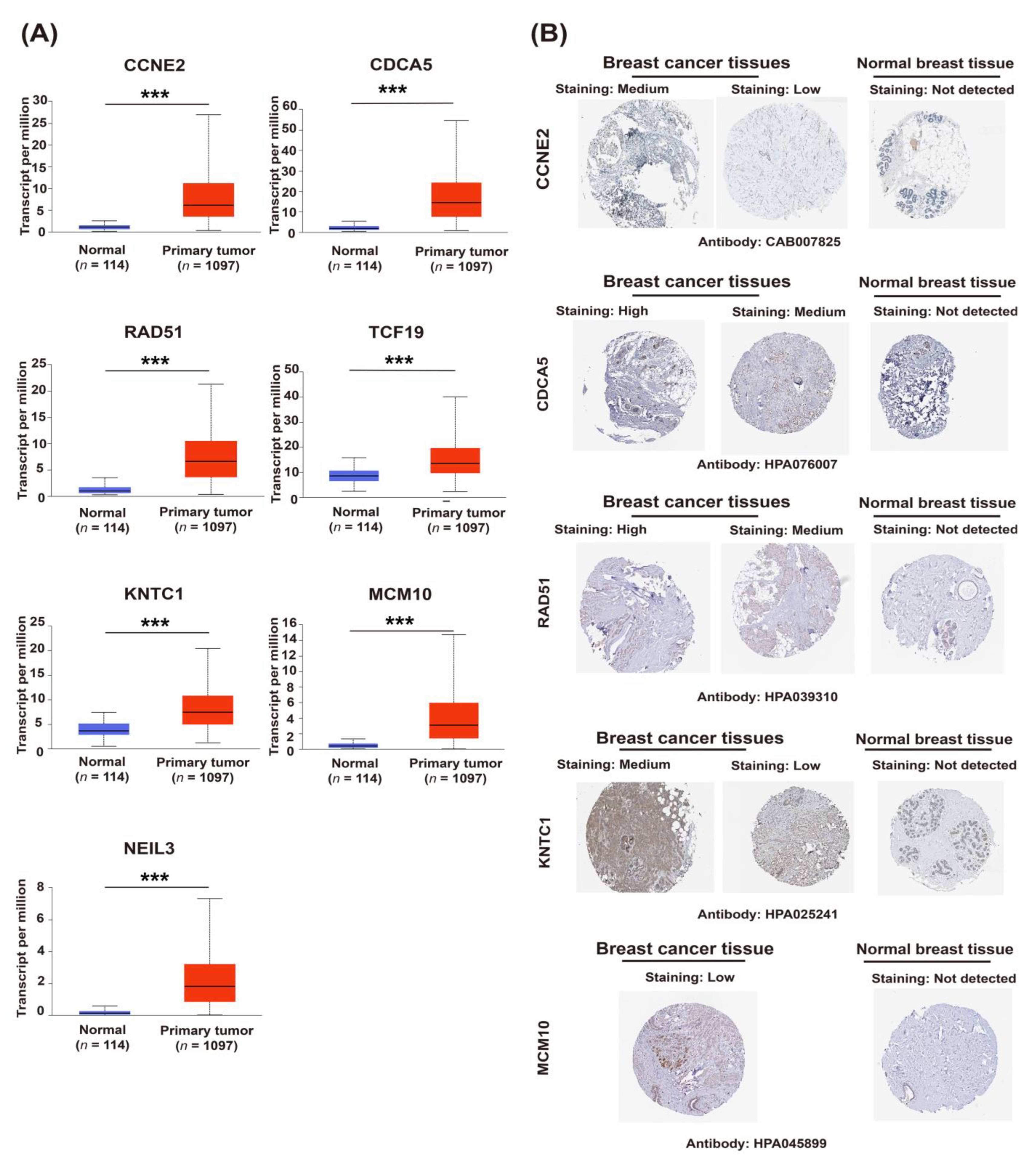
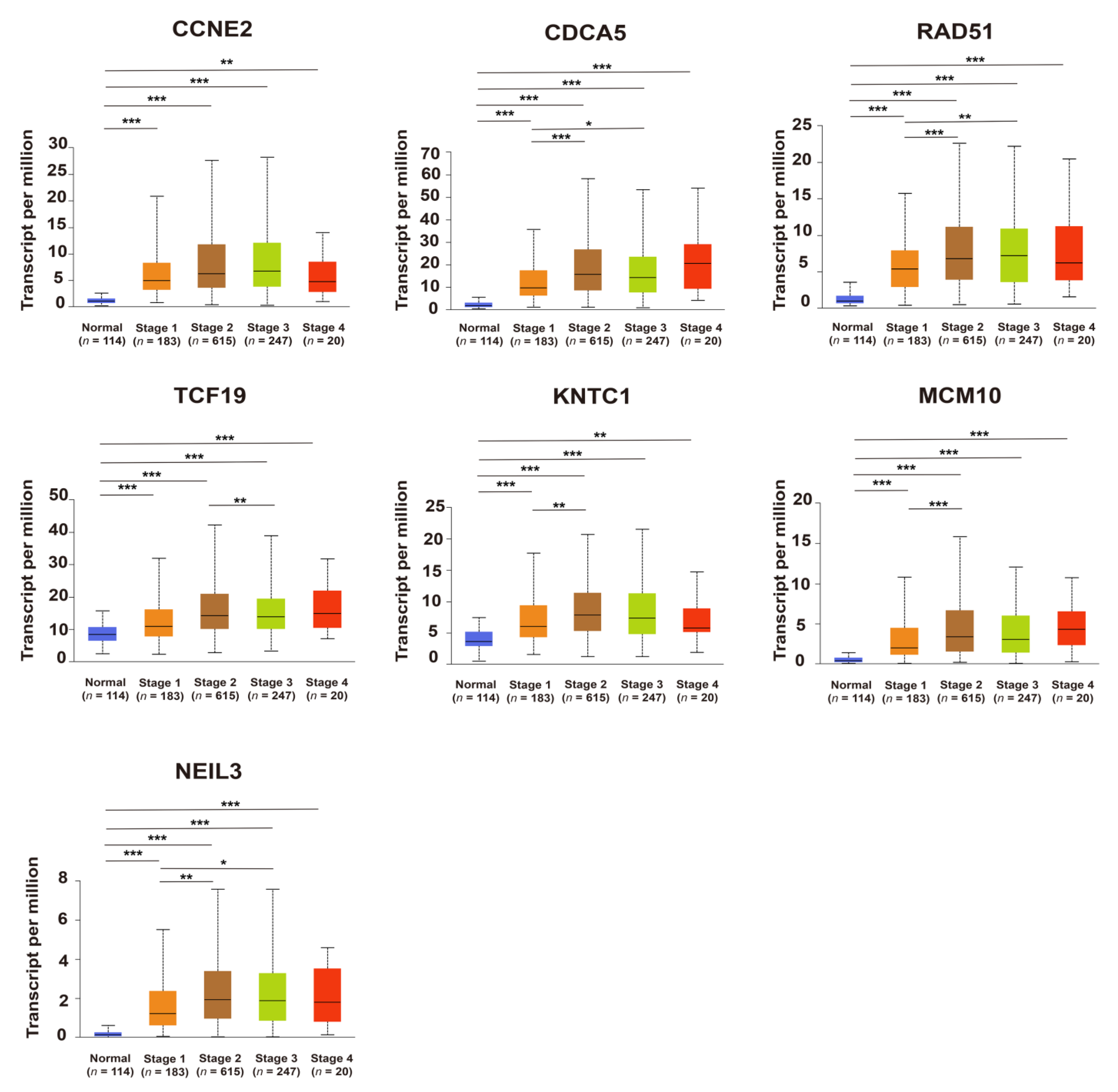
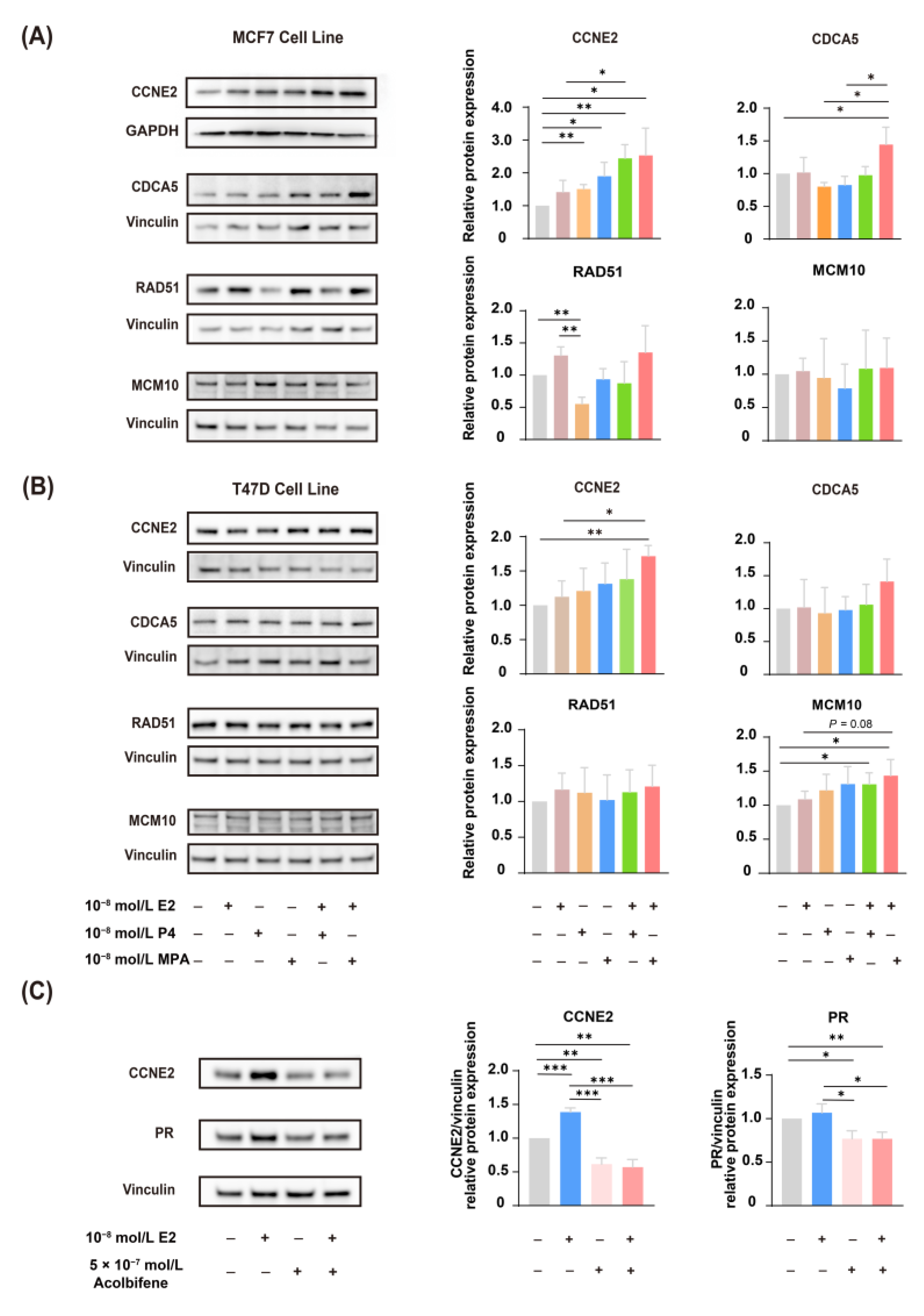
2.3. Upregulation of Candidate Genes Associated with Mammary Tumorigenesis and Progression
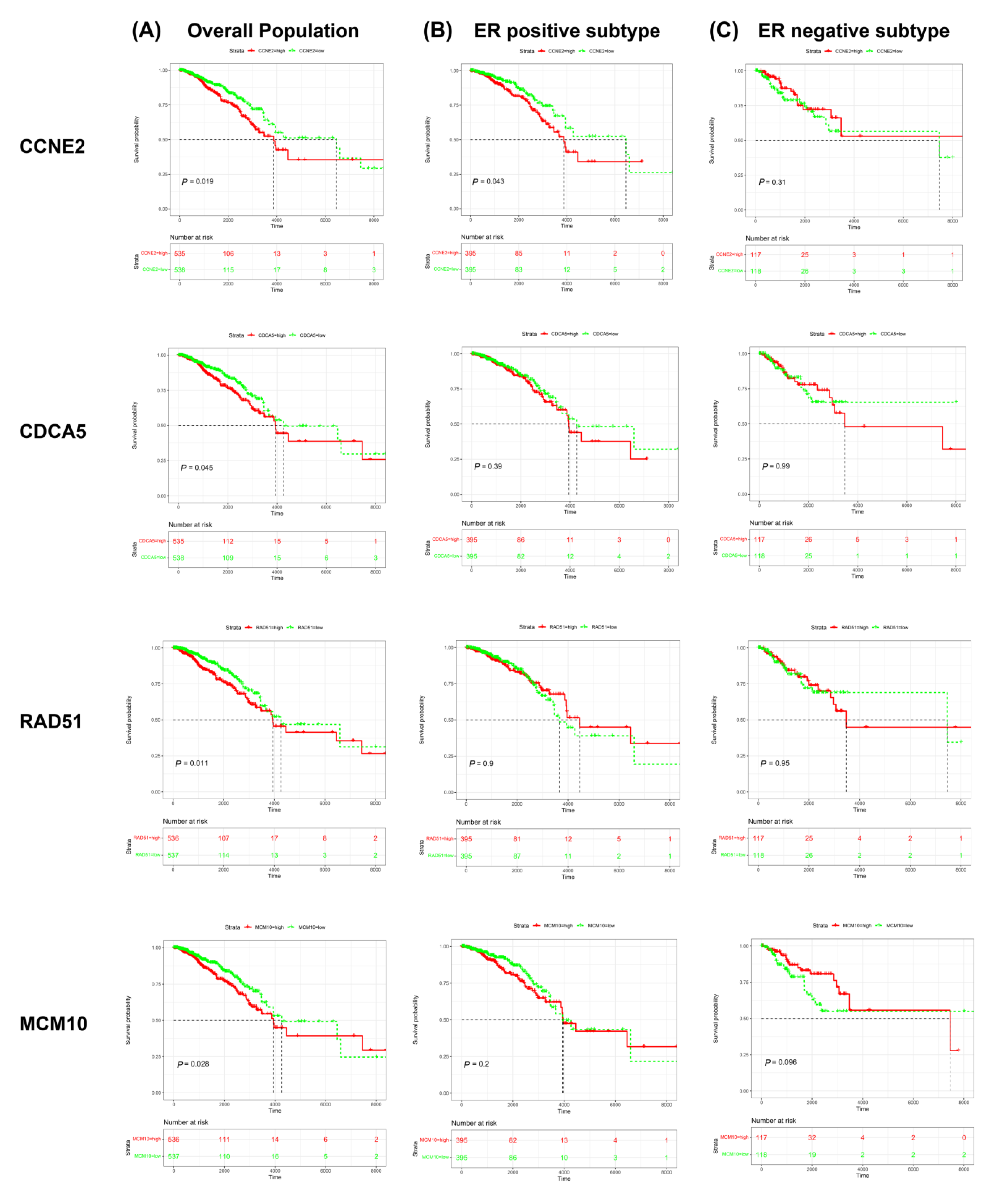
2.4. CCNE2 Expression Was Positively Correlated with Poor Outcomes in ER-Positive Breast Cancer Patients
2.5. CCNE2 Expression Was Correlated with ER
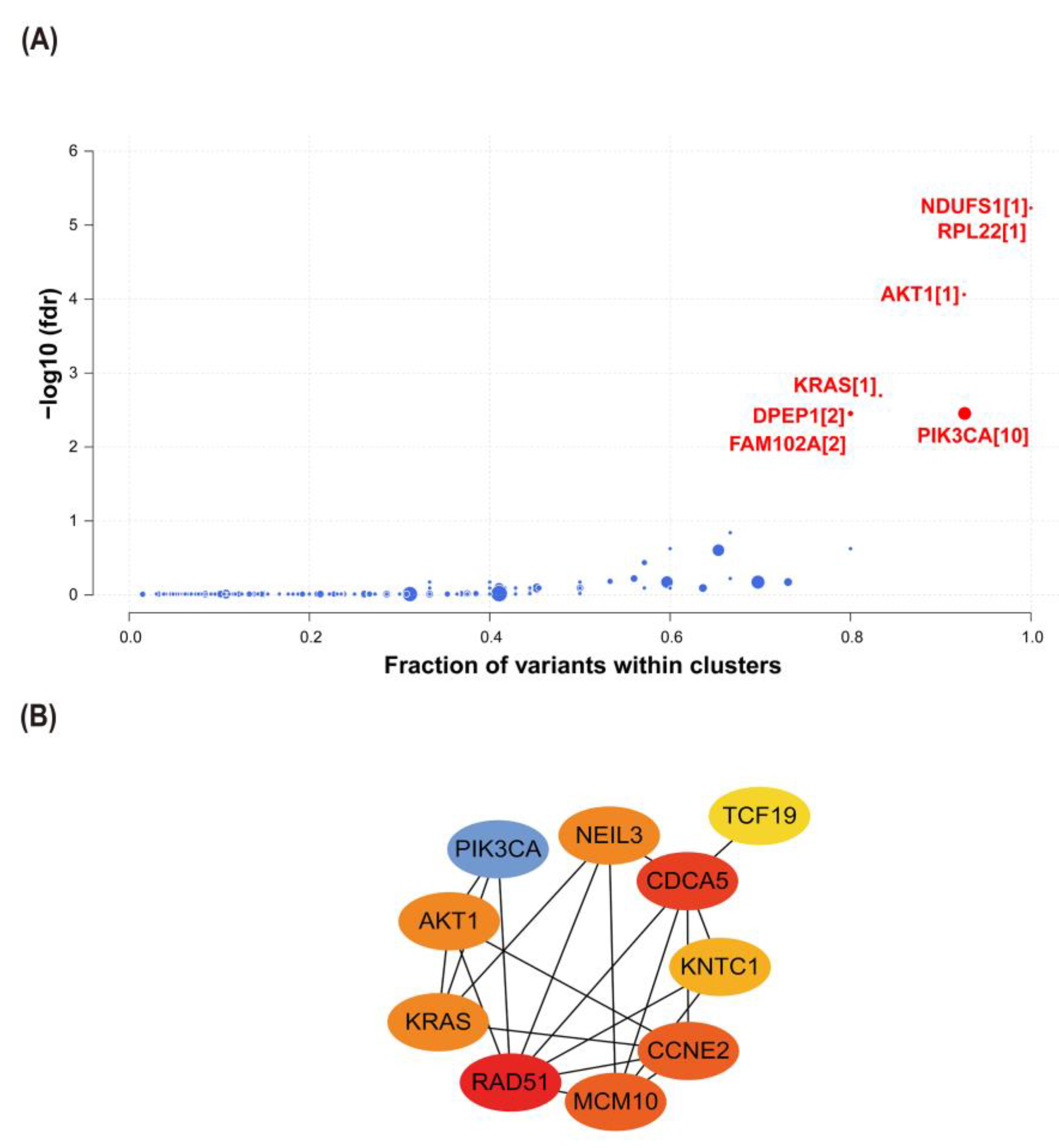
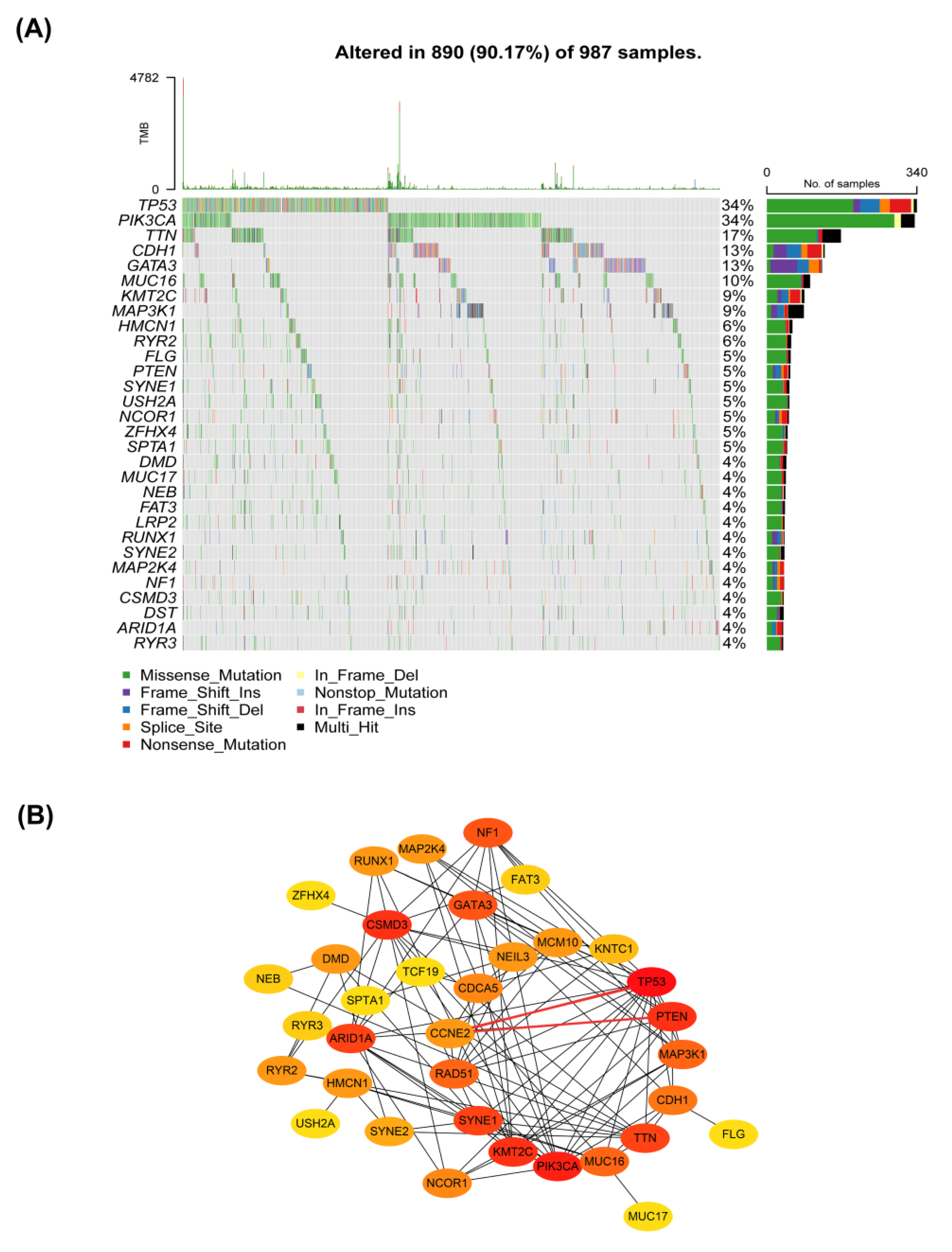
2.6. Interactions between CCNE2 and Breast Cancer Driver Genes
2.7. CCNE2 Related to Genomic Stability in Breast Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Data Sources and Data Preprocessing
4.2. Identification and Functional Enrichment Analysis of DEGs
4.3. Gene Expression, Protein Expression, and Survival Analysis of Candidate Genes
4.4. Prediction of Cancer Driver Genes
4.5. Gene Mutation Analysis
4.6. Protein–Protein Interaction (PPI) Network Analysis of Candidate Genes and Top Mutation Genes
4.7. Molecular Docking
4.8. Reagents
4.9. Cell Culture and Treatment
4.10. RT-qPCR
4.11. Western Blotting
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT1 | AKT serine/threonine kinase 1 |
| CCNE2 | Cyclin E2 |
| CDCA5 | Cell division cycle-associated 5 |
| DEGs | Differentially expressed genes |
| DPEP1 | Dipeptidase 1 |
| E2 | 17β-estradiol |
| ER | Estrogen receptor |
| EPT | Estrogen plus progestogens treatment |
| ET | Estrogen treatment |
| FAM102A | Family with sequence similarity 102 member A |
| FDR | False discovery rate |
| GEO | Gene Expression Omnibus |
| HPA | The Human Protein Atlas database |
| HR | Hazards ratio |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KNTC1 | Kinetochore-associated 1 |
| KRAS | KRAS proto-oncogene, GTPase |
| MCM10 | Minichromosome maintenance 10 replication initiation factor |
| MHT | Menopausal hormone therapy |
| MPA | Medroxyprogesterone acetate |
| MPA group | E2 + MPA vs. E2 |
| NEIL3 | Nei-like DNA glycosylase 3 |
| NDUFS1 | NADH:ubiquinone oxidoreductase core subunit S1 |
| P4 | Progesterone |
| P4 group | E2 + P4 vs. E2 |
| PPI | Protein–protein interaction |
| PR | Progesterone receptor |
| PTEN | Phosphatase and tensin homolog |
| RAD51 | RAD51 recombinase |
| RPL22 | Ribosomal protein L22 |
| RR | Relative risk |
| TCF19 | Transcription factor 19 |
| TCGA | The Cancer Genome Atlas |
| TCGA-BRCA | The Cancer Genome Atlas Breast Invasive Carcinoma database |
| TP53 | Tumor protein p53 |
| WHI | Women’s Health Initiative |
References
- Zhang, G.Q.; Chen, J.L.; Luo, Y.; Mathur, M.B.; Anagnostis, P.; Nurmatov, U.; Talibov, M.; Zhang, J.; Hawrylowicz, C.M.; Lumsden, M.A.; et al. Menopausal hormone therapy and women’s health: An umbrella review. PLoS Med. 2021, 18, e1003731. [Google Scholar] [CrossRef] [PubMed]
- Baber, R.J.; Panay, N.; Fenton, A. 2016 IMS Recommendations on women’s midlife health and menopause hormone therapy. Climacteric 2016, 19, 109–150. [Google Scholar] [CrossRef] [PubMed]
- North American Menopause Society. The 2017 hormone therapy position statement of The North American Menopause Society. Menopause 2018, 25, 1362–1387. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Cho, M.K.; Cho, Y.J.; Chun, S.; Hong, S.H.; Hwang, K.R.; Jeon, G.H.; Joo, J.K.; Kim, S.K.; Lee, D.O.; et al. The 2020 Menopausal Hormone Therapy Guidelines. J. Menopausal. Med. 2020, 26, 69–98. [Google Scholar]
- Pinkerton, J.V. Hormone Therapy for Postmenopausal Women. N. Engl. J. Med. 2020, 382, 446–455. [Google Scholar] [CrossRef]
- Stuenkel, C.A.; Davis, S.R.; Gompel, A.; Lumsden, M.A.; Murad, M.H.; Pinkerton, J.V.; Santen, R.J. Treatment of Symptoms of the Menopause: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2015, 100, 3975–4011. [Google Scholar] [CrossRef]
- Santen, R.J.; Allred, D.C.; Ardoin, S.P.; Archer, D.F.; Boyd, N.; Braunstein, G.D.; Burger, H.G.; Colditz, G.A.; Davis, S.R.; Gambacciani, M.; et al. Postmenopausal hormone therapy: An Endocrine Society scientific statement. J. Clin. Endocrinol. Metab. 2010, 95 (Suppl. 1), s1–s66. [Google Scholar] [CrossRef]
- Lambrinoudaki, I. Menopausal hormone therapy and breast cancer risk: All progestogens are not the same. Case Rep. Womens Health 2021, 29, e00270. [Google Scholar] [CrossRef]
- Stevenson, J.C.; Farmer, R.D.T. HRT and breast cancer: A million women ride again. Climacteric 2020, 23, 226–228. [Google Scholar] [CrossRef]
- Langer, R.D.; Hodis, H.N.; Lobo, R.A.; Allison, M.A. Hormone replacement therapy—Where are we now? Climacteric 2021, 24, 3–10. [Google Scholar] [CrossRef]
- Flores, V.A.; Taylor, H.S. The Effect of Menopausal Hormone Therapies on Breast Cancer: Avoiding the Risk. Endocrinol. Metab. Clin. North Am. 2015, 44, 587–602. [Google Scholar] [CrossRef]
- Crawford, S.L.; Crandall, C.J.; Derby, C.A.; El Khoudary, S.R.; Waetjen, L.E.; Fischer, M.; Joffe, H. Menopausal hormone therapy trends before versus after 2002: Impact of the Women’s Health Initiative Study Results. Menopause 2018, 26, 588–597. [Google Scholar] [CrossRef]
- Steinkellner, A.R.; Denison, S.E.; Eldridge, S.L.; Lenzi, L.L.; Chen, W.; Bowlin, S.J. A decade of postmenopausal hormone therapy prescribing in the United States: Long-term effects of the Women’s Health Initiative. Menopause 2012, 19, 616–621. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar]
- Anderson, G.L.; Limacher, M.; Assaf, A.R.; Bassford, T.; Beresford, S.A.; Black, H.; Bonds, D.; Brunner, R.; Brzyski, R.; Caan, B.; et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004, 291, 1701–1712. [Google Scholar]
- Chlebowski, R.T.; Anderson, G.L.; Aragaki, A.K.; Manson, J.E.; Stefanick, M.L.; Pan, K.; Barrington, W.; Kuller, L.H.; Simon, M.S.; Lane, D.; et al. Association of Menopausal Hormone Therapy with Breast Cancer Incidence and Mortality During Long-term Follow-up of the Women’s Health Initiative Randomized Clinical Trials. JAMA 2020, 324, 369–380. [Google Scholar] [CrossRef]
- Armeni, E.; Cano, A.; Rees, M.; Lambrinoudaki, I. Menopausal Hormone Therapy and breast cancer risk: Individualization is the key to safety. Maturitas 2020, 141, 85–86. [Google Scholar] [CrossRef]
- Fournier, A.; Berrino, F.; Clavel-Chapelon, F. Unequal risks for breast cancer associated with different hormone replacement therapies: Results from the E3N cohort study. Breast Cancer Res. Treat. 2008, 107, 103–111. [Google Scholar] [CrossRef]
- Purmonen, S.; Manninen, T.; Pennanen, P.; Ylikomi, T. Progestins regulate genes that can elicit both proliferative and antiproliferative effects in breast cancer cells. Oncol. Rep. 2008, 19, 1627–1634. [Google Scholar]
- Traboulsi, T.; El Ezzy, M.; Gleason, J.L.; Mader, S. Antiestrogens: Structure-activity relationships and use in breast cancer treatment. J. Mol. Endocrinol. 2017, 58, R15–R31. [Google Scholar] [CrossRef]
- Almutairi, M.S.; Al Suwayyid, A.N.; Aldarwesh, A.; Aboulwafa, O.M.; Attia, M.I. Antiestrogenic Activity and Possible Mode of Action of Certain New Nonsteroidal Coumarin-4-acetamides. Molecules 2020, 25, 1553. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, K.B.; McGuire, W.L. Estrogen control of progesterone receptor in human breast cancer. J. Biol. Chem. 1978, 253, 2223–2228. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Koseki, Y.; McGuire, W.L. Estrogen control of progesterone receptor in human breast cancer: Role of estradiol and antiestrogen. Endocrinology 1978, 103, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Kim, S. New and emerging factors in tumorigenesis: An overview. Cancer Manag. Res. 2015, 7, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Leylek, T.R.; Jeusset, L.M.; Lichtensztejn, Z.; McManus, K.J. Reduced Expression of Genes Regulating Cohesion Induces Chromosome Instability that May Promote Cancer and Impact Patient Outcomes. Sci. Rep. 2020, 10, 592. [Google Scholar] [CrossRef] [PubMed]
- Janic, A.; Valente, L.J.; Wakefield, M.J.; Di Stefano, L.; Milla, L.; Wilcox, S.; Yang, H.; Tai, L.; Vandenberg, C.J.; Kueh, A.J.; et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat. Med. 2018, 24, 947–953. [Google Scholar] [CrossRef]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Manson, J.E. Menopausal Hormone Therapy and Breast Cancer. Cancer J. 2022, 28, 169–175. [Google Scholar] [CrossRef]
- Murkes, D.; Conner, P.; Leifland, K.; Tani, E.; Beliard, A.; Lundström, E.; Söderqvist, G. Effects of percutaneous estradiol-oral progesterone versus oral conjugated equine estrogens-medroxyprogesterone acetate on breast cell proliferation and bcl-2 protein in healthy women. Fertil. Steril. 2011, 95, 1188–1191. [Google Scholar] [CrossRef]
- Stanczyk, F.Z.; Hapgood, J.P.; Winer, S.; Mishell, D.R., Jr. Progestogens used in postmenopausal hormone therapy: Differences in their pharmacological properties, intracellular actions, and clinical effects. Endocr. Rev. 2013, 34, 171–208. [Google Scholar] [CrossRef]
- Payton, M.; Coats, S. Cyclin E2, the cycle continues. Int. J. Biochem. Cell Biol. 2002, 34, 315–320. [Google Scholar] [CrossRef]
- Caldon, C.E.; Musgrove, E.A. Distinct and redundant functions of cyclin E1 and cyclin E2 in development and cancer. Cell Div. 2010, 5, 2. [Google Scholar] [CrossRef]
- Sieuwerts, A.M.; Look, M.P.; Meijer-van Gelder, M.E.; Timmermans, M.; Trapman, A.M.; Garcia, R.R.; Arnold, M.; Goedheer, A.J.; de Weerd, V.; Portengen, H.; et al. Which cyclin E prevails as prognostic marker for breast cancer? Results from a retrospective study involving 635 lymph node-negative breast cancer patients. Clin. Cancer Res. 2006, 12, 3319–3328. [Google Scholar] [CrossRef]
- Chen, D.; Guo, W.; Qiu, Z.; Wang, Q.; Li, Y.; Liang, L.; Liu, L.; Huang, S.; Zhao, Y.; He, X. MicroRNA-30d-5p inhibits tumour cell proliferation and motility by directly targeting CCNE2 in non-small cell lung cancer. Cancer Lett. 2015, 362, 208–217. [Google Scholar] [CrossRef]
- Santen, R.J.; Yue, W. Cause or prevention of breast cancer with estrogens: Analysis from tumor biologic data, growth kinetic model and Women’s Health Initiative study. Climacteric 2019, 22, 3–12. [Google Scholar] [CrossRef]
- Santen, R.J.; Yue, W.; Heitjan, D.F. Modeling of the growth kinetics of occult breast tumors: Role in interpretation of studies of prevention and menopausal hormone therapy. Cancer Epidemiol. Biomarkers Prev. 2012, 21, 1038–1048. [Google Scholar] [CrossRef]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Feng, W.; Wang, C.; Liang, C.; Yang, H.; Chen, D.; Yu, X.; Zhao, W.; Geng, D.; Li, S.; Chen, Z.; et al. The Dysregulated Expression of KCNQ1OT1 and Its Interaction with Downstream Factors miR-145/CCNE2 in Breast Cancer Cells. Cell Physiol. Biochem. 2018, 49, 432–446. [Google Scholar] [CrossRef]
- Lee, C.; Fernandez, K.J.; Alexandrou, S.; Sergio, C.M.; Deng, N.; Rogers, S.; Burgess, A.; Caldon, C.E. Cyclin E2 Promotes Whole Genome Doubling in Breast Cancer. Cancers 2020, 12, 2268. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, L.; Dong, Y.; Tao, C.; Zhang, R.; Shao, H.; Shen, H. Effect of AKT1 (p. E17K) Hotspot Mutation on Malignant Tumorigenesis and Prognosis. Front. Cell Dev. Biol. 2020, 8, 573599. [Google Scholar] [CrossRef]
- Testa, J.R.; Tsichlis, P.N. AKT signaling in normal and malignant cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.L.; Wang, J. KRAS gene silencing inhibits the activation of PI3K-Akt-mTOR signaling pathway to regulate breast cancer cell epithelial-mesenchymal transition, proliferation and apoptosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3085–3096. [Google Scholar]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef]
- Győrffy, B.; Bottai, G.; Lehmann-Che, J.; Kéri, G.; Orfi, L.; Iwamoto, T.; Desmedt, C.; Bianchini, G.; Turner, N.C.; de Thè, H.; et al. TP53 mutation-correlated genes predict the risk of tumor relapse and identify MPS1 as a potential therapeutic kinase in TP53-mutated breast cancers. Mol. Oncol. 2014, 8, 508–519. [Google Scholar] [CrossRef]
- Thu, Y.M.; Bielinsky, A.-K. Enigmatic roles of Mcm10 in DNA replication. Trends Biochem. Sci. 2013, 38, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Izumi, M.; Yatagai, F.; Hanaoka, F. Cell cycle-dependent proteolysis and phosphorylation of human Mcm10. J. Biol. Chem. 2001, 276, 48526–48531. [Google Scholar] [CrossRef]
- Im, J.S.; Ki, S.H.; Farina, A.; Jung, D.S.; Hurwitz, J.; Lee, J.K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 15628–15632. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Bang, S.W.; Jeon, Y.; Kang, S.; Hwang, D.S. Knockdown of human MCM10 exhibits delayed and incomplete chromosome replication. Biochem. Biophys. Res. Commun. 2008, 365, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Miotto, B.; Chibi, M.; Xie, P.; Koundrioukoff, S.; Moolman-Smook, H.; Pugh, D.; Debatisse, M.; He, F.; Zhang, L.; Defossez, P.A. The RBBP6/ZBTB38/MCM10 axis regulates DNA replication and common fragile site stability. Cell Rep. 2014, 7, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Mahadevappa, R.; Neves, H.; Yuen, S.M.; Jameel, M.; Bai, Y.; Yuen, H.F.; Zhang, S.D.; Zhu, Y.; Lin, Y.; Kwok, H.F. DNA Replication Licensing Protein MCM10 Promotes Tumor Progression and Is a Novel Prognostic Biomarker and Potential Therapeutic Target in Breast Cancer. Cancers 2018, 10, 282. [Google Scholar] [CrossRef]
- Zhang, N.; Pati, D. Sororin is a master regulator of sister chromatid cohesion and separation. Cell Cycle 2012, 11, 2073–2083. [Google Scholar] [CrossRef]
- Schmitz, J.; Watrin, E.; Lénárt, P.; Mechtler, K.; Peters, J.M. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Curr. Biol. 2007, 17, 630–636. [Google Scholar] [CrossRef]
- Shen, A.; Liu, L.; Chen, H.; Qi, F.; Huang, Y.; Lin, J.; Sferra, T.J.; Sankararaman, S.; Wei, L.; Chu, J.; et al. Cell division cycle associated 5 promotes colorectal cancer progression by activating the ERK signaling pathway. Oncogenesis 2019, 8, 19. [Google Scholar] [CrossRef]
- Phan, N.N.; Wang, C.Y.; Li, K.L.; Chen, C.F.; Chiao, C.C.; Yu, H.G.; Huang, P.L.; Lin, Y.C. Distinct expression of CDCA3, CDCA5, and CDCA8 leads to shorter relapse free survival in breast cancer patient. Oncotarget 2018, 9, 6977–6992. [Google Scholar] [CrossRef]
- Shen, Z.; Yu, X.; Zheng, Y.; Lai, X.; Li, J.; Hong, Y.; Zhang, H.; Chen, C.; Su, Z.; Guo, R. CDCA5 regulates proliferation in hepatocellular carcinoma and has potential as a negative prognostic marker. Onco. Targets Ther. 2018, 11, 891–901. [Google Scholar] [CrossRef]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46. [Google Scholar] [CrossRef]
- Yamamoto, A.; Taki, T.; Yagi, H.; Habu, T.; Yoshida, K.; Yoshimura, Y.; Yamamoto, K.; Matsushiro, A.; Nishimune, Y.; Morita, T. Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol. Gen. Genet. 1996, 251, 1–12. [Google Scholar]
- Richardson, C. RAD51, genomic stability, and tumorigenesis. Cancer Lett. 2005, 218, 127–139. [Google Scholar] [CrossRef]
- Richardson, C.; Stark, J.M.; Ommundsen, M.; Jasin, M. Rad51 overexpression promotes alternative double-strand break repair pathways and genome instability. Oncogene 2004, 23, 546–553. [Google Scholar] [CrossRef]
- Son, M.Y.; Hasty, P. Homologous recombination defects and how they affect replication fork maintenance. AIMS Genet. 2018, 5, 192–211. [Google Scholar] [CrossRef]
- Maacke, H.; Opitz, S.; Jost, K.; Hamdorf, W.; Henning, W.; Krüger, S.; Feller, A.C.; Lopens, A.; Diedrich, K.; Schwinger, E.; et al. Over-expression of wild-type Rad51 correlates with histological grading of invasive ductal breast cancer. Int. J. Cancer 2000, 88, 907–913. [Google Scholar] [CrossRef]
- Gazy, I.; Zeevi, D.A.; Renbaum, P.; Zeligson, S.; Eini, L.; Bashari, D.; Smith, Y.; Lahad, A.; Goldberg, M.; Ginsberg, D.; et al. TODRA, a lncRNA at the RAD51 Locus, Is Oppositely Regulated to RAD51, and Enhances RAD51-Dependent DSB (Double Strand Break) Repair. PLoS ONE 2015, 10, e0134120. [Google Scholar] [CrossRef]
- Sen, S.; Sanyal, S.; Srivastava, D.K.; Dasgupta, D.; Roy, S.; Das, C. Transcription factor 19 interacts with histone 3 lysine 4 trimethylation and controls gluconeogenesis via the nucleosome-remodeling-deacetylase complex. J. Biol. Chem. 2017, 292, 20362–20378. [Google Scholar] [CrossRef]
- Mondal, P.; Sen, S.; Klein, B.J.; Tiwary, N.; Gadad, S.S.; Kutateladze, T.G.; Roy, S.; Das, C. TCF19 Promotes Cell Proliferation through Binding to the Histone H3K4me3 Mark. Biochemistry 2020, 59, 389–399. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Chen, G.; Deng, C.; Tang, J.M.; Xie, L.; Zhou, H.Y.; Ye, X.; Zhang, D.K.; Shi, R.Q.; Tian, D.; et al. TCF19 contributes to cell proliferation of non-small cell lung cancer by inhibiting FOXO1. Cell Biol. Int. 2019, 43, 1416–1424. [Google Scholar] [CrossRef]
- Ji, P.; Chang, J.; Wei, X.; Song, X.; Yuan, H.; Gong, L.; Li, Y.; Ding, D.; Zhang, E.; Yan, C.; et al. Genetic variants associated with expression of TCF19 contribute to the risk of head and neck cancer in Chinese population. J. Med. Genet. 2021, 59, 335–345. [Google Scholar] [CrossRef]
- Alshabi, A.M.; Shaikh, I.A.; Vastrad, C. Exploring the Molecular Mechanism of the Drug-Treated Breast Cancer Based on Gene Expression Microarray. Biomolecules 2019, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.K.; Jablonski, S.A.; Starr, D.A.; Goldberg, M.L.; Yen, T.J. Human Zw10 and ROD are mitotic checkpoint proteins that bind to kinetochores. Nat. Cell Biol. 2000, 2, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Karess, R. Rod-Zw10-Zwilch: A key player in the spindle checkpoint. Trends Cell Biol. 2005, 15, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.; Woldhuis, R.R.; Boudewijn, I.M.; van den Berg, A.; Kluiver, J.; Kok, K.; Terpstra, M.M.; Guryev, V.; de Vries, M.; Vermeulen, C.J.; et al. Age-related gene and miRNA expression changes in airways of healthy individuals. Sci. Rep. 2019, 9, 3765. [Google Scholar] [CrossRef]
- Zhou, J.; Chan, J.; Lambelé, M.; Yusufzai, T.; Stumpff, J.; Opresko, P.L.; Thali, M.; Wallace, S.S. NEIL3 Repairs Telomere Damage during S Phase to Secure Chromosome Segregation at Mitosis. Cell Rep. 2017, 20, 2044–2056. [Google Scholar] [CrossRef]
- Tran, O.T.; Tadesse, S.; Chu, C.; Kidane, D. Overexpression of NEIL3 associated with altered genome and poor survival in selected types of human cancer. Tumour Biol. 2020, 42, 1010428320918404. [Google Scholar] [CrossRef]
- Wood, C.E.; Register, T.C.; Cline, J.M. Transcriptional profiles of progestogen effects in the postmenopausal breast. Breast Cancer Res. Treat. 2009, 114, 233–242. [Google Scholar] [CrossRef]
- Atwood, C.S.; Ekstein, S.F. Human versus non-human sex steroid use in hormone replacement therapies part 1: Preclinical data. Mol. Cell Endocrinol. 2019, 480, 12–35. [Google Scholar] [CrossRef]
- Sitruk-Ware, R. Pharmacological profile of progestins. Maturitas 2004, 47, 277–283. [Google Scholar] [CrossRef]
- Cabeza, M.; Heuze, Y.; Sánchez, A.; Garrido, M.; Bratoeff, E. Recent advances in structure of progestins and their binding to progesterone receptors. J. Enzyme Inhib. Med. Chem. 2015, 30, 152–159. [Google Scholar] [CrossRef]
- King, R.J.; Whitehead, M.I. Assessment of the potency of orally administered progestins in women. Fertil. Steril. 1986, 46, 1062–1066. [Google Scholar] [CrossRef]
- Ward, A.V.; Matthews, S.B.; Fettig, L.M.; Riley, D.; Finlay-Schultz, J.; Paul, K.V.; Jackman, M.; Kabos, P.; MacLean, P.S.; Sartorius, C.A. Estrogens and Progestins Cooperatively Shift Breast Cancer Cell Metabolism. Cancers 2022, 14, 1776. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Sartorius, C.A. Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: A hypothesis. J. Clin. Endocrinol. Metab. 2008, 93, 3295–3298. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Sartorius, C.A. 90 YEARS OF PROGESTERONE: Progesterone and progesterone receptors in breast cancer: Past, present, future. J. Mol. Endocrinol. 2020, 65, T49–T63. [Google Scholar] [CrossRef]
- Need, E.F.; Selth, L.A.; Trotta, A.P.; Leach, D.A.; Giorgio, L.; O’Loughlin, M.A.; Smith, E.; Gill, P.G.; Ingman, W.V.; Graham, J.D.; et al. The unique transcriptional response produced by concurrent estrogen and progesterone treatment in breast cancer cells results in upregulation of growth factor pathways and switching from a Luminal A to a Basal-like subtype. BMC Cancer 2015, 15, 791. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Pontén, F.; Schwenk, J.M.; Asplund, A.; Edqvist, P.H. The Human Protein Atlas as a proteomic resource for biomarker discovery. J. Intern. Med. 2011, 270, 428–446. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Arnedo-Pac, C.; Mularoni, L.; Muiños, F.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUSTL: A sequence-based clustering method to identify cancer drivers. Bioinformatics 2019, 35, 4788–4790. [Google Scholar] [CrossRef]
- Rashid, M.; Alasiri, A.; Al Balwi, M.A.; Alkhaldi, A.; Alsuhaibani, A.; Alsultan, A.; Alharbi, T.; Alomair, L.; Almuzzaini, B. Identification of CSF3R Mutations in B-Lineage Acute Lymphoblastic Leukemia Using Comprehensive Cancer Panel and Next-Generation Sequencing. Genes 2021, 12, 1326. [Google Scholar] [CrossRef]
- Korbolina, E.E.; Bryzgalov, L.O.; Ustrokhanova, D.Z.; Postovalov, S.N.; Poverin, D.V.; Damarov, I.S.; Merkulova, T.I. A Panel of rSNPs Demonstrating Allelic Asymmetry in Both ChIP-seq and RNA-seq Data and the Search for Their Phenotypic Outcomes through Analysis of DEGs. Int. J. Mol. Sci. 2021, 22, 7240. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Helfmann, S.; Neumann, P.; Tittmann, K.; Moser, T.; Ficner, R.; Reisinger, E. The crystal structure of the C2A domain of otoferlin reveals an unconventional top loop region. J. Mol. Biol. 2011, 406, 479–490. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System; version 1.8; Schrödinger, Inc.: New York, NY, USA, 2015.
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Tan, C.C.; Wu, X.R.; He, R.; Zhang, X.; Wang, Q.S.; Li, X.Q.; Zhang, R.; Feng, Y.M. FOXF2 deficiency accelerates the visceral metastasis of basal-like breast cancer by unrestrictedly increasing TGF-β and miR-182-5p. Cell Death Differ. 2020, 27, 2973–2987. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Primer Sequence |
|---|---|
| 18S | F: 5′-AGGAATTCCCAGTAAGTGCG-3′ R: 5′-GCCTCACTAAACCATCCAA-3′ |
| CCNE2 | F: 5′-TCAAGACGAAGTAGCCGTTTAC-3′ R: 5′-TGACATCCTGGGTAGTTTTCCTC-3’ |
| CDCA5 | F: 5′-AGAAAGTCAGGCGTTCCTACAG-3′ R: 5′-GGGAGATTCCAGGGAGAGTCAT-3′ |
| RAD51 | F: 5′-TCTCTGGCAGTGATGTCCTGGA-3′ R: 5′-TAAAGGGCGGTGGCACTGTCTA-3′ |
| TCF19 | F: 5′-AGGCTGGAATTGAGTGATGGAGAC-3′ R: 5′-GTCCTGAGGCTTGACTCGTACTTGTT-3′ |
| KNTC1 | F: 5′-ATAGTCAACCCAGAGTGGGCTGT-3′ R: 5′-TTTCACGTTTTTCGTCCTGCG-3′ |
| MCM10 | F: 5′-GAAGAAGGTTACGCCACAGAG-3′ R: 5′-TTTACAGGTTCCCAGGTCAAG-3′ |
| NEIL3 | F: 5′-GGTCTCCACCCAGCTGTTAAAG-3′ R: 5’-CACGTATCATTTTCATGAGGTGATG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, Y.; Huang, H.; Shi, J.; Jin, H. Identification of Candidate Genes in Breast Cancer Induced by Estrogen Plus Progestogens Using Bioinformatic Analysis. Int. J. Mol. Sci. 2022, 23, 11892. https://doi.org/10.3390/ijms231911892
Deng Y, Huang H, Shi J, Jin H. Identification of Candidate Genes in Breast Cancer Induced by Estrogen Plus Progestogens Using Bioinformatic Analysis. International Journal of Molecular Sciences. 2022; 23(19):11892. https://doi.org/10.3390/ijms231911892
Chicago/Turabian StyleDeng, Yu, He Huang, Jiangcheng Shi, and Hongyan Jin. 2022. "Identification of Candidate Genes in Breast Cancer Induced by Estrogen Plus Progestogens Using Bioinformatic Analysis" International Journal of Molecular Sciences 23, no. 19: 11892. https://doi.org/10.3390/ijms231911892
APA StyleDeng, Y., Huang, H., Shi, J., & Jin, H. (2022). Identification of Candidate Genes in Breast Cancer Induced by Estrogen Plus Progestogens Using Bioinformatic Analysis. International Journal of Molecular Sciences, 23(19), 11892. https://doi.org/10.3390/ijms231911892