
Aryl-Capped Lysine-Dehydroamino Acid Dipeptide Supergelators as Potential Drug Release Systems
 ,
,  ,
,  , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Gelation Study
2.3. Scanning Transmission Electron Microscopy (STEM) Studies
2.4. Circular Dichroism (CD) Spectroscopy
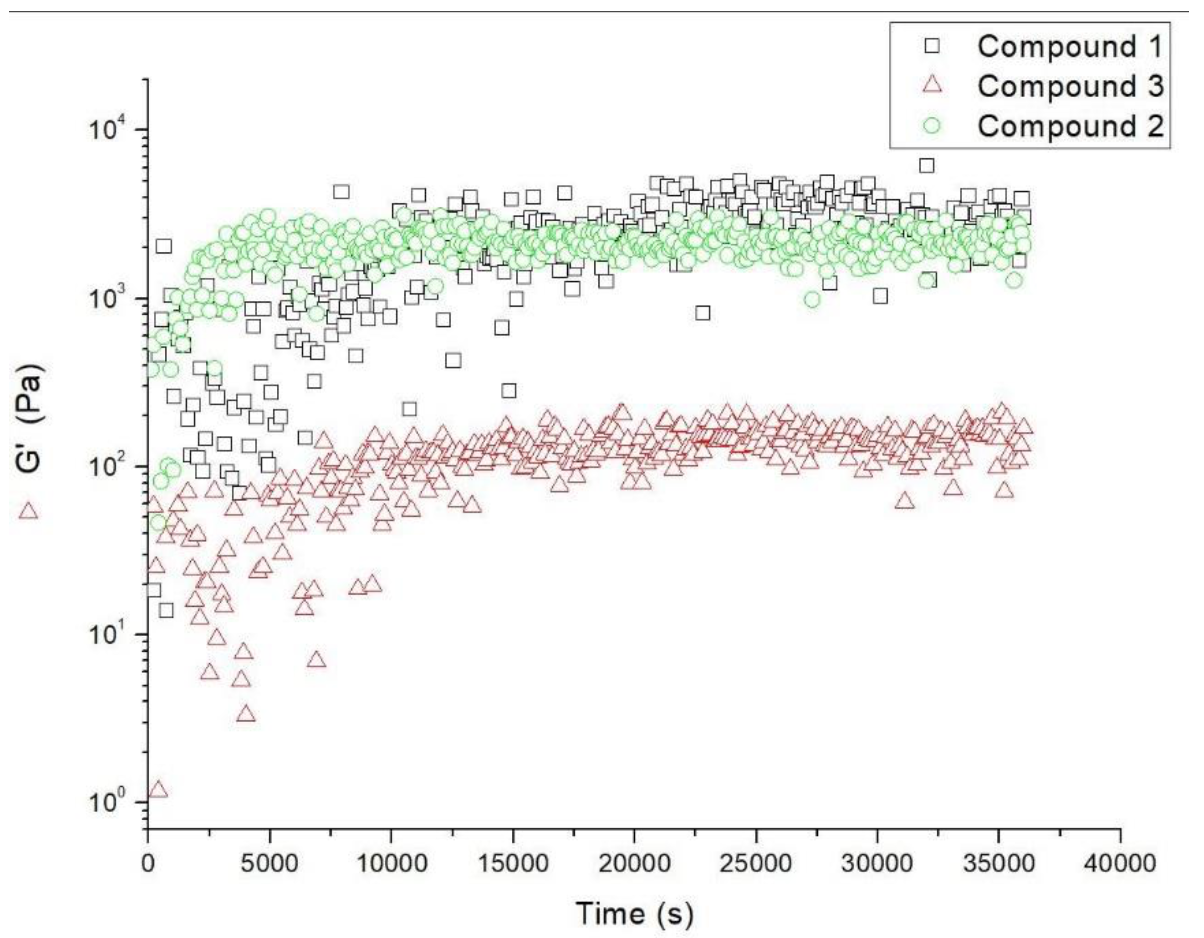
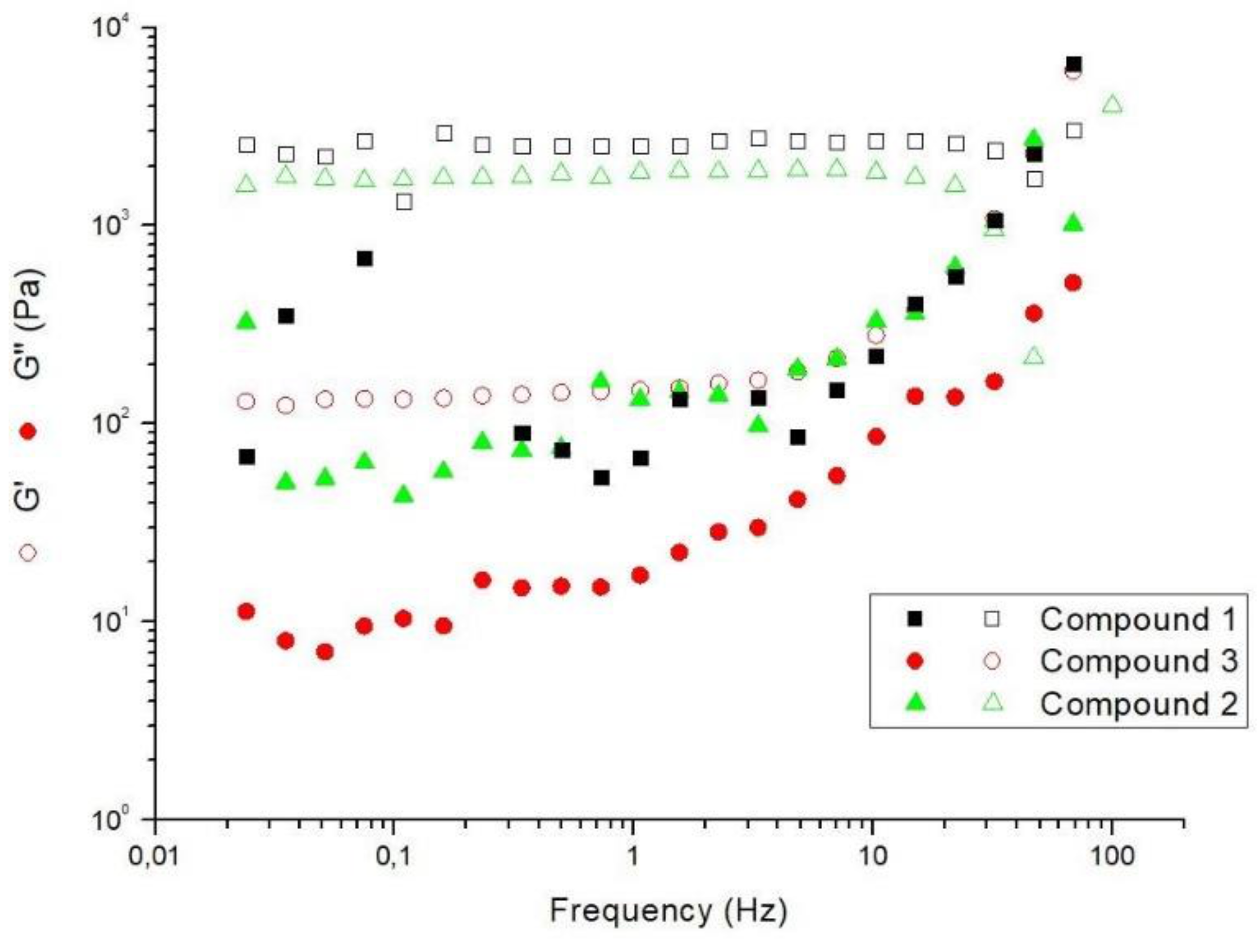
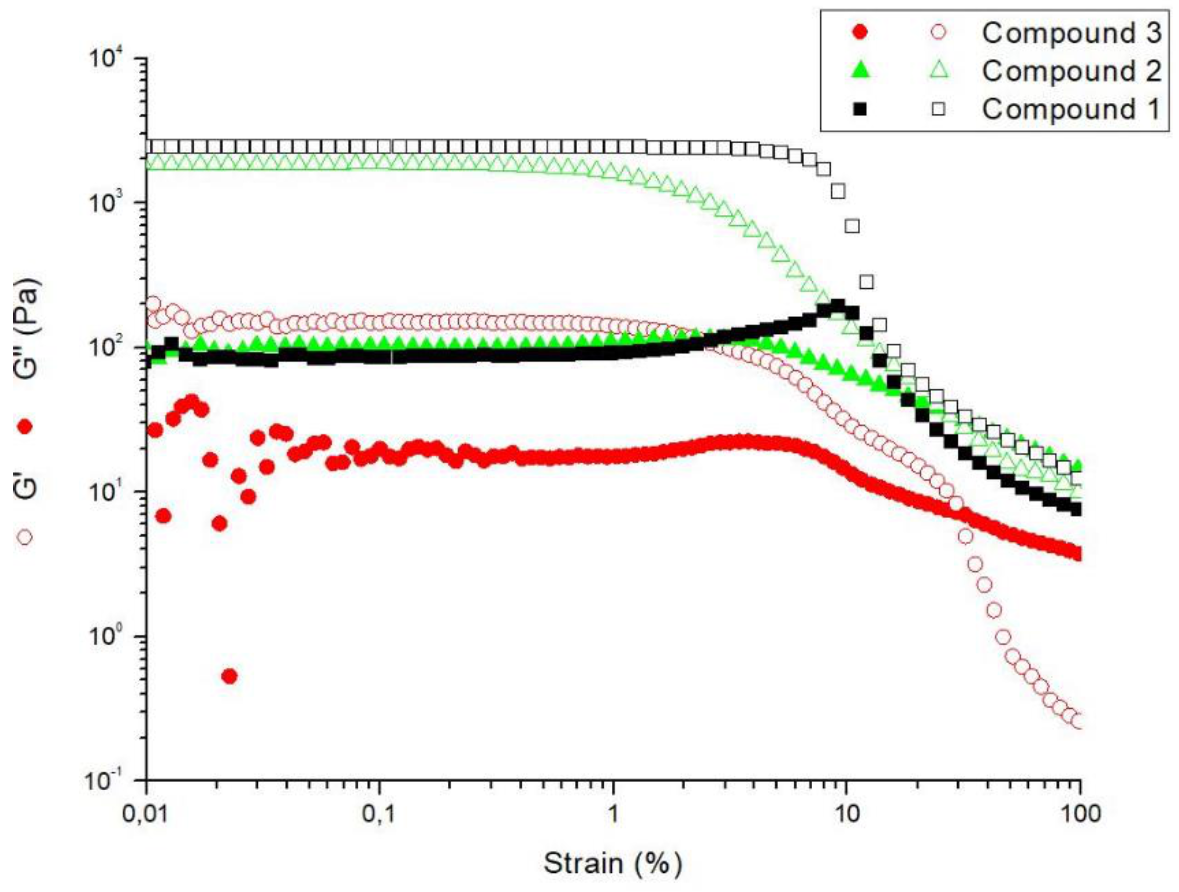
2.5. Rheological Studies
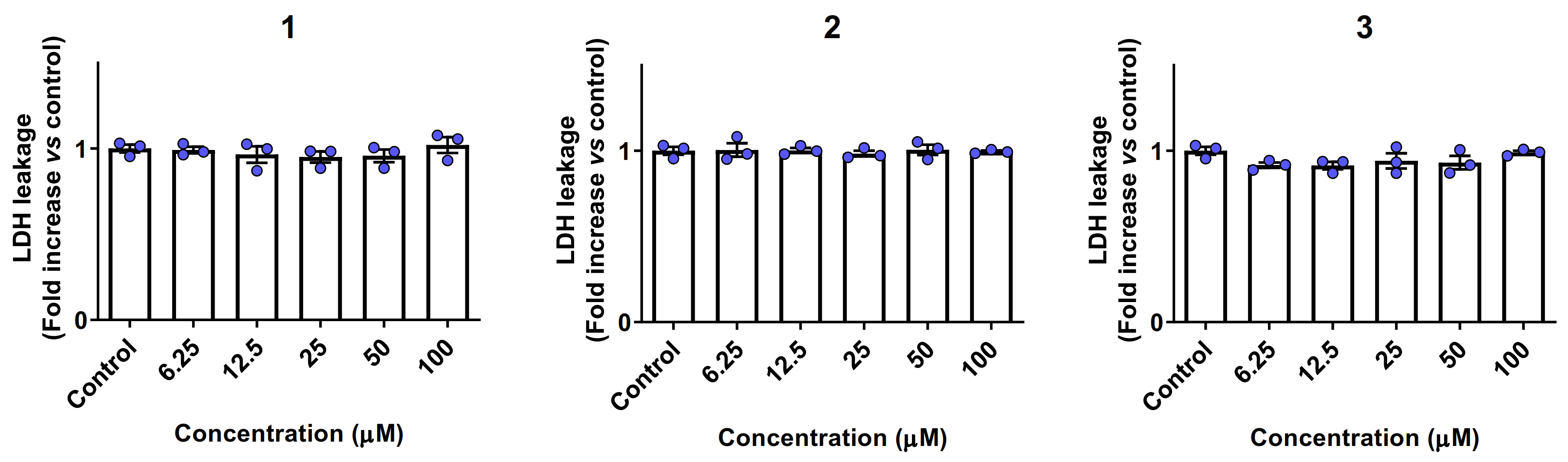
2.6. Biocompatibility and Cytotoxicity Studies
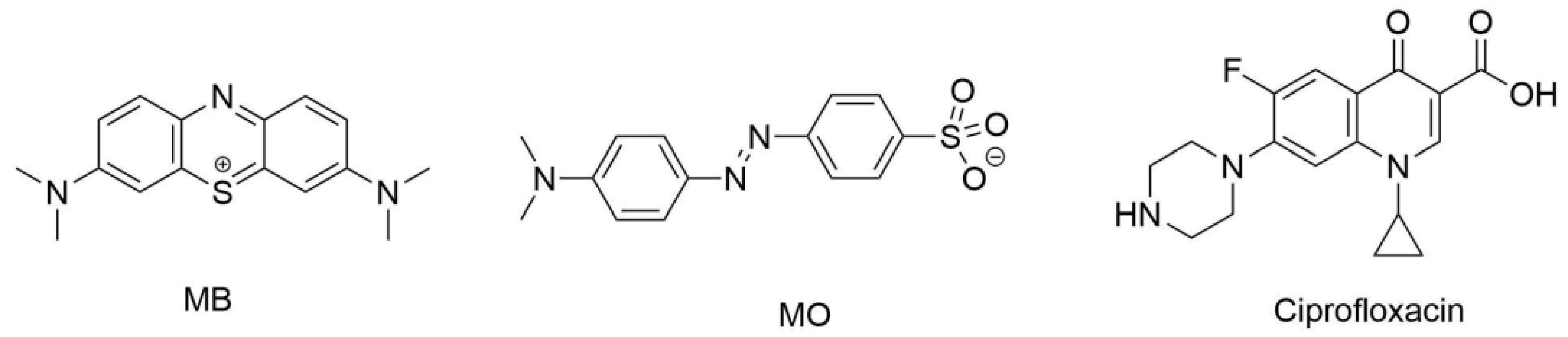
2.7. Drug Delivery Study
2.8. Molecular Modelling Studies
3. Conclusions
4. Materials and Methods
4.1. Synthesis
4.1.1. Synthesis of Naph-L-Lys(Naph)-Z-ΔPhe-OH (1)
4.1.2. Synthesis of Naph-L-Lys(Cbz)-Z-ΔPhe-OH (2)
4.1.3. Synthesis of Cbz-L-Lys(Cbz)-Z-ΔPhe-OH (3)
4.2. Hydrogel Preparation and Determination of Critical Gelation Concentration (CGC)
4.3. Scanning Transmission Electron Microscopy (STEM)
4.4. Circular Dichroísm Spectroscopy
4.5. Rheological Studies
4.6. Sustained Release Assays
4.7. Cell Culture
4.8. MTT Assay
4.9. LDH Leakage
4.10. Computer Modelling
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Philip, D.; Stoddart, J.F. Self-assembly in natural and unnatural systems. Angew. Chem. Int. Ed. 1996, 35, 1154–1196. [Google Scholar] [CrossRef]
- Mendes, A.C.; Baran, E.T.; Reis, R.L.; Azevedo, H.S. Self-assembly in nature: Using the principles of nature to create complex nanobiomaterials. WIREs Nanomed. Nanobiotechnol. 2015, 5, 582–612. [Google Scholar] [CrossRef] [PubMed]
- Dreiss, C.A. Hydrogel design strategies for drug delivery. Curr. Opin. Colloid Interface Sci. 2020, 48, 1–17. [Google Scholar] [CrossRef]
- Jayawarna, V.; Richardson, S.M.; Hirst, A.R.; Hodson, N.W.; Saiani, A.; Gough, J.E.; Ulijn, R.V. Introducing chemical functionality in Fmoc-peptide gels for cell culture. Acta Biomater. 2009, 5, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liu, M.; Zhang, Y.; Yin, J.; Pei, R. Nanocomposite hydrogels for tissue engineering applications. Nanoscale 2020, 12, 14976–14995. [Google Scholar] [CrossRef]
- Tavakoli, S.; Klar, A.S. Advanced hydrogels as wound dressings. Biomolecules 2020, 10, 1169. [Google Scholar] [CrossRef]
- Herrmann, A.; Haag, R.; Schedler, U. Hydrogels and their role in biosensing applications. Adv. Healthcare Mater. 2021, 10, 2100062. [Google Scholar] [CrossRef]
- Mancha Sánchez, E.; Gómez-Blanco, J.C.; López Nieto, E.; Casado, J.G.; Macías-García, A.; Díaz Díez, M.A.; Carrasco-Amador, J.P.; Torrejón Martín, D.; Sánchez-Margallo, F.M.; Pagador, J.B. Hydrogels for bioprinting: A systematic review of hydrogels synthesis, bioprinting parameters, and bioprinted structures behavior. Front. Bioeng. Biotechnol. 2020, 8, 776. [Google Scholar] [CrossRef]
- Kim, B.J.; Yang, D.; Xu, B. Emerging applications of supramolecular peptide assemblies. Trends Chem. 2020, 2, 71–83. [Google Scholar] [CrossRef]
- Adams, D.J. Dipeptide and tripeptide conjugates as low-molecular-weight hydrogelators. Macromol. Biosci. 2011, 11, 160–173. [Google Scholar] [CrossRef]
- Adams, D.J.; Butler, M.F.; Frith, W.J.; Kirkland, M.; Mullen, L.; Sanderson, P. A new method for maintaining homogeneity during liquid–hydrogel transitions using low molecular weight hydrogelators. Soft Matter 2009, 5, 1856–1862. [Google Scholar] [CrossRef]
- Zhou, J.; Du, X.; Wang, J.; Yamagata, N.; Xu, B. Enzyme-instructed self-assembly of peptides containing phosphoserine to form supramolecular hydrogels as potential soft biomaterials. Front. Chem. Sci. Eng. 2017, 11, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Raeburn, J.; Pont, G.; Chen, L.; Cesbron, Y.; Lévyc, R.; Adams, D.J. Fmoc-diphenylalanine hydrogels: Understanding the variability in reported mechanical properties. Soft Matter 2012, 8, 1168–1174. [Google Scholar] [CrossRef]
- Haines, L.A.; Rajagopal, K.; Ozbas, B.; Salick, D.A.; Pochan, D.J.; Schneider, J.P. Light-activated hydrogel formation via the triggered folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2005, 127, 17025–17029. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Adams, D.J.; Cameron, P.J. Peptide based low molecular weight gelators. J. Mater. Chem. 2011, 21, 2024–2027. [Google Scholar] [CrossRef]
- Sangeetha, N.M.; Maitra, U. Supramolecular gels: Functions and uses. Chem. Soc. Rev. 2005, 34, 821–836. [Google Scholar] [CrossRef]
- Patterson, A.K.; Smith, D.K. Two-component supramolecular hydrogel for controlled drug release. Chem. Commun. 2020, 56, 11046–11049. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular hydrogelators and hydrogels: From soft matter to molecular biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Fichman, G.; Gazit, E. Self-assembly of short peptides to form hydrogels: Design of building blocks, physical properties and technological applications. Acta Biomater. 2014, 10, 1671–1682. [Google Scholar] [CrossRef]
- Wang, K.; Keasling, J.D.; Muller, S.J. Effects of the sequence and size of non-polar residues on self-assembly of amphiphilic peptides. Int. J. Biol. Macromol. 2005, 36, 232–240. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, H.; Liu., Y.; Mao., L.; Chen, W.; Zhu, Z.; Liu, W.; Zheng, W.; Zhao, Y.; Kong, D.; et al. A peptide-based nanofibrous hydrogel as a promising DNA nanovector for optimizing the efficacy of HIV vaccine. Nano Lett. 2014, 14, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Tang, Y.; Yamamoto, T.; Yao, Y.; Guterman, T.; Zilberzwige-Tal, S.; Adadi, N.; Ji, W.; Dvir, T.; Ramamoorthy, A.; et al. Unusual Two-Step Assembly of a Minimalistic Dipeptide-Based Functional Hypergelator. Adv. Mater. 2020, 32, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jervis, P.J.; Amorim, C.; Pereira, T.; Martins, J.A.; Ferreira, P.M.T. Dehydropeptide supramolecular hydrogels and nanostructures as potential peptidomimetic biomedical materials. Int. J. Mol. Sci. 2021, 22, 2528. [Google Scholar] [CrossRef] [PubMed]
- Veloso, S.R.; Jervis, P.J.; Silva, J.F.; Hilliou, L.; Moura, C.; Pereira, D.M.; Coutinho, P.J.G.; Martins, J.A.; Castanheira, E.M.S.; Ferreira, P.M. Supramolecular ultra-short carboxybenzyl-protected dehydropeptide-based hydrogels for drug delivery. Mater. Sci. Eng. C 2021, 122, 111869. [Google Scholar] [CrossRef] [PubMed]
- Amorim, C.; Veloso, S.R.S.; Castanheira, E.M.S.; Hilliou, L.; Pereira, R.B.; Pereira, D.M.; Martins, J.A.; Jervis, P.J.; Ferreira, P.M.T. Bolaamphiphilic bis-dehydropeptide hydrogels as potential drug release systems. Gels 2021, 7, 52. [Google Scholar] [CrossRef]
- Veloso, S.R.S.; Martins, J.A.; Hilliou, L.; Amorim, C.O.; Amaral, V.S.; Almeida, B.G.; Jervis, P.J.; Moreira, R.; Pereira, D.M.; Coutinho, P.J.G.; et al. Dehydropeptide-based plasmonic magnetogels: A supramolecular composite nanosystem for multimodal cancer therapy. J. Mater. Chem. B 2020, 8, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Jervis, P.J.; Hilliou, L.; Pereira, R.B.; Pereira, D.M.; Martins, J.A.; Ferreira, P.M.T. Evaluation of a model photo-caged dehydropeptide as a stimuli-responsive supramolecular hydrogel. Nanomaterials 2021, 11, 704. [Google Scholar] [CrossRef]
- Vilaça, H.; Pereira, G.; Castro, T.G.; Hermenegildo, B.F.; Shi, J.; Faria, T.Q.; Micaêlo, N.; Brito, R.M.M.; Xu, B.; Castanheira, E.M.S.; et al. New self-assembled supramolecular hydrogels based on dehydropeptides. J. Mater. Chem. B 2015, 3, 6355–6367. [Google Scholar] [CrossRef]
- Carvalho, A.; Gallo, J.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Hilliou, L.; Ferreira, P.M.T.; Bañobre-López, M.; Martins, J.A. Magnetic dehydrodipeptide-based self-assembled hydrogels for theragnostic applications. Nanomaterials 2019, 9, 54126. [Google Scholar] [CrossRef]
- Carpino, L.A.; Han, G.Y. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972, 37, 3404–3409. [Google Scholar] [CrossRef]
- Truong, W.T.; Su, Y.; Gloria, D.; Braet, F.; Thordarson, P. Dissolution and degradation of Fmoc-diphenylalanine self-assembled gels results in necrosis at high concentrations in vitro. Biomater. Sci. 2015, 3, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Zhang, C.; Dai, Y.; Li, X.; Pan, Y.; Huang, W.; Qian, H.; Ge, L. Injectable self-assembled peptide hydrogels for glucose-mediated insulin delivery. Biomater. Sci. 2018, 6, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.L.; Toriki, E.S.; Tucker, N.D.J.; Nilsson, B.L. Electrostatic interactions regulate the release of small molecules from supramolecular hydrogels. J. Mater. Chem. B 2020, 8, 6366–6377. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef] [PubMed]
- PyMOL. The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field Versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843. [Google Scholar] [CrossRef]
- Alper, H.E.; Levy, R.M. Computer simulations of the dielectric properties of water: Studies of the simple point charge and transferrable intermolecular potential models. J. Chem. Phys. 1989, 91, 1242–1251. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting crystal structures: The Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003, 90, 4. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Critical Gelation Concentration (CGC) | pH | cLogP 1 | |
|---|---|---|---|---|
| wt% | mM | |||
| 1 | 0.05 | 0.80 | 4.8 | 6.16 |
| 2 | 0.07 | 1.18 | 5.1 | 5.39 |
| 3 | 0.20 | 3.57 | 5.0 | 6.42 |
| Hydrogel | G’ (Pa) | G″ (Pa) |
|---|---|---|
| 1 | 3.63 × 103 | 221 |
| 2 | 1.83 × 103 | 145 |
| 3 | 1.84 × 102 | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, C.B.P.; Pereira, R.B.; Pereira, D.M.; Hilliou, L.; Castro, T.G.; Martins, J.A.; Jervis, P.J.; Ferreira, P.M.T. Aryl-Capped Lysine-Dehydroamino Acid Dipeptide Supergelators as Potential Drug Release Systems. Int. J. Mol. Sci. 2022, 23, 11811. https://doi.org/10.3390/ijms231911811
Oliveira CBP, Pereira RB, Pereira DM, Hilliou L, Castro TG, Martins JA, Jervis PJ, Ferreira PMT. Aryl-Capped Lysine-Dehydroamino Acid Dipeptide Supergelators as Potential Drug Release Systems. International Journal of Molecular Sciences. 2022; 23(19):11811. https://doi.org/10.3390/ijms231911811
Chicago/Turabian StyleOliveira, Carlos B. P., Renato B. Pereira, David M. Pereira, Loic Hilliou, Tarsila G. Castro, José A. Martins, Peter J. Jervis, and Paula M. T. Ferreira. 2022. "Aryl-Capped Lysine-Dehydroamino Acid Dipeptide Supergelators as Potential Drug Release Systems" International Journal of Molecular Sciences 23, no. 19: 11811. https://doi.org/10.3390/ijms231911811
APA StyleOliveira, C. B. P., Pereira, R. B., Pereira, D. M., Hilliou, L., Castro, T. G., Martins, J. A., Jervis, P. J., & Ferreira, P. M. T. (2022). Aryl-Capped Lysine-Dehydroamino Acid Dipeptide Supergelators as Potential Drug Release Systems. International Journal of Molecular Sciences, 23(19), 11811. https://doi.org/10.3390/ijms231911811