
Hepatocyte-Specific Deficiency of DAX-1 Protects Mice from Acetaminophen-Induced Hepatotoxicity by Activating NRF2 Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DAX-1 Deficiency Ameliorates APAP-Induced Hepatocellular Damage in Primary Hepatocytes
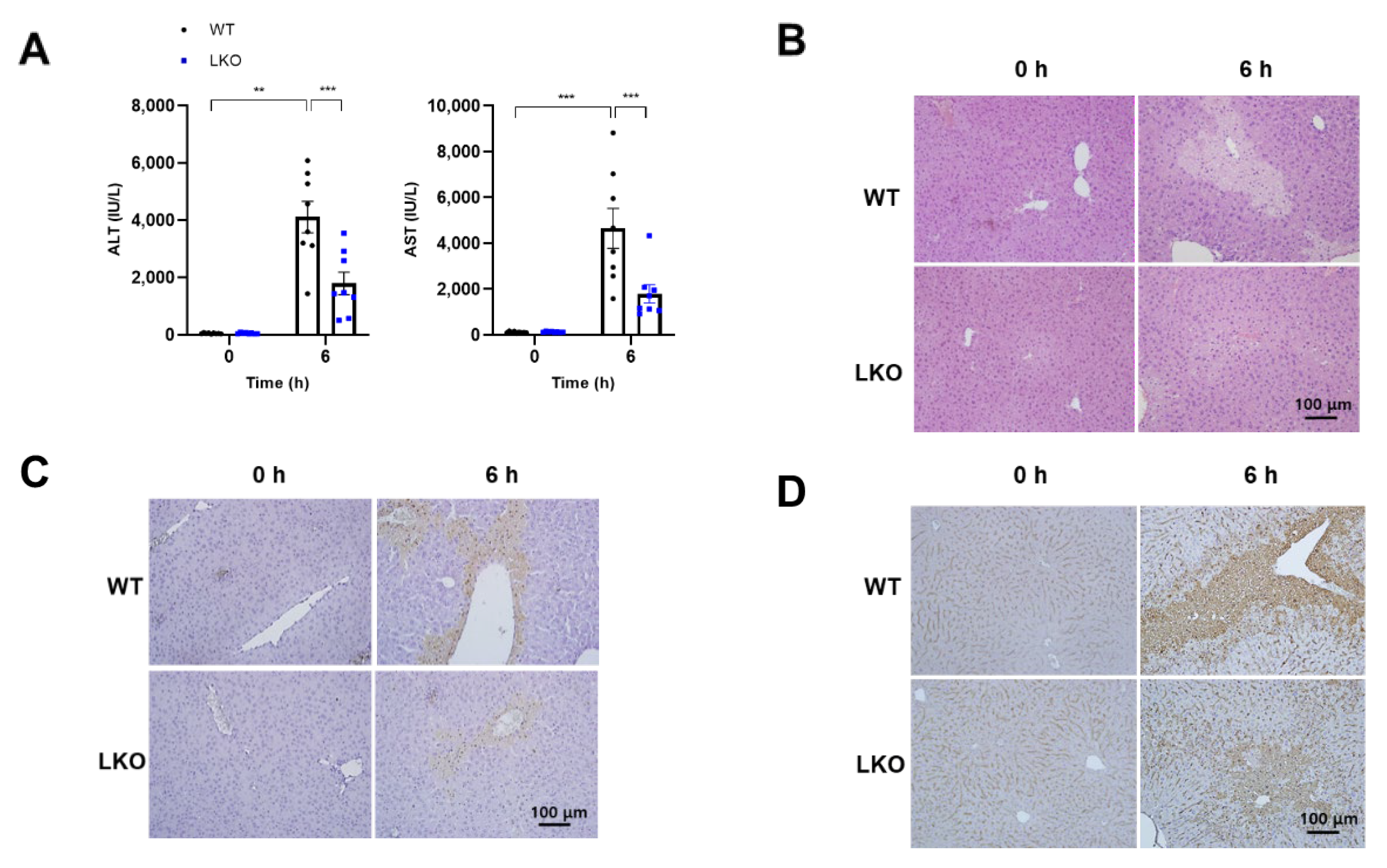
2.2. Hepatocyte-Specific Dax-1 Deficient Mice Are More Resistant to APAP-Induced Hepatotoxicity
2.3. APAP-Induced Mitochondrial Oxidative Stress Is Attenuated by Hepatocyte-Specific DAX-1 Deficiency in Mice
2.4. Hepatocyte-Specific DAX-1 Deficiency Inhibits Persistent JNK Phosphorylation in the Liver of APAP-Treated Mice
2.5. DAX-1 Deficiency in Hepatocytes Enhances the Expression of Genes Encoding Antioxidant Enzymes in the Liver following APAP Treatment
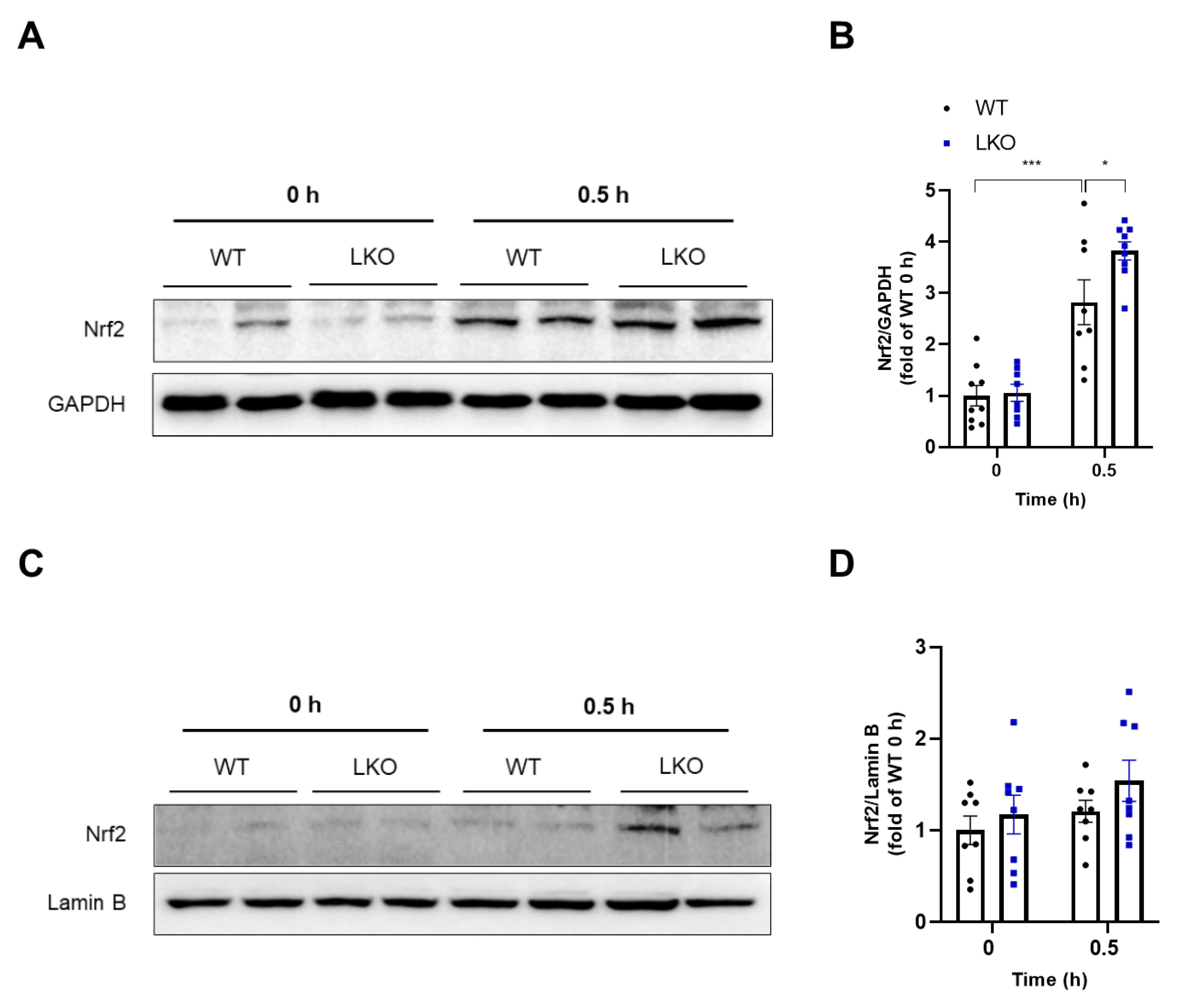
2.6. Hepatocyte-Specific DAX-1 Deficiency Increases Nrf2 Expression in the Liver of Mice Induced by APAP
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Primary Mouse Hepatocytes and In Vitro APAP Treatment
4.3. Blood Analysis
4.4. Histopathology and Immunohistochemistry
4.5. TUNEL Staining
4.6. Western Blot Analysis
4.7. RNA Extraction, Reverse Transcription, and RT-qPCR
4.8. Isolation of Subcellular Fractions
4.9. Measurements of Liver GSH Levels
4.10. Measurements of Mitochondrial ROS Levels
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoon, E.; Babar, A.; Choudhary, M.; Kutner, M.; Pyrsopoulos, N. Acetaminophen-Induced Hepatotoxicity: A Comprehensive Update. J. Clin. Transl. Hepatol. 2016, 4, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Huo, Y.; Yin, S.; Hu, H. Mechanisms of acetaminophen-induced liver injury and its implications for therapeutic interventions. Redox Biol. 2018, 17, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.D. Molecular mechanisms of the hepatotoxicity caused by acetaminophen. Semin. Liver Dis. 1990, 10, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Jaeschke, H. Acetaminophen Hepatotoxicity. Semin. Liver Dis. 2019, 39, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Gunawan, B.K.; Liu, Z.X.; Han, D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology 2006, 131, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Gum, S.I.; Cho, M.K. Recent updates on acetaminophen hepatotoxicity: The role of nrf2 in hepatoprotection. Toxicol. Res. 2013, 29, 165–172. [Google Scholar] [CrossRef]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Liu, K.H.; Owens, J.A.; Hunter-Chang, S.; Camacho, M.C.; Eboka, R.U.; Chandrasekharan, B.; Baker, N.F.; Darby, T.M.; Robinson, B.S.; et al. Gut-Resident Lactobacilli Activate Hepatic Nrf2 and Protect Against Oxidative Liver Injury. Cell Metab. 2020, 31, 956–968.e5. [Google Scholar] [CrossRef]
- Bataille, A.M.; Manautou, J.E. Nrf2: A potential target for new therapeutics in liver disease. Clin. Pharmacol. Ther. 2012, 92, 340–348. [Google Scholar] [CrossRef]
- Niakan, K.K.; McCabe, E.R. DAX1 origin, function, and novel role. Mol. Genet. Metab. 2005, 86, 70–83. [Google Scholar] [CrossRef]
- Swain, A.; Zanaria, E.; Hacker, A.; Lovell-Badge, R.; Camerino, G. Mouse Dax1 expression is consistent with a role in sex determination as well as in adrenal and hypothalamus function. Nat. Genet. 1996, 12, 404–409. [Google Scholar] [CrossRef]
- Nedumaran, B.; Kim, G.S.; Hong, S.; Yoon, Y.S.; Kim, Y.H.; Lee, C.H.; Lee, Y.C.; Koo, S.H.; Choi, H.S. Orphan nuclear receptor DAX-1 acts as a novel corepressor of liver X receptor alpha and inhibits hepatic lipogenesis. J. Biol. Chem. 2010, 285, 9221–9232. [Google Scholar] [CrossRef]
- Chanda, D.; Park, J.H.; Choi, H.S. Molecular basis of endocrine regulation by orphan nuclear receptor Small Heterodimer Partner. Endocr. J. 2008, 55, 253–268. [Google Scholar] [CrossRef]
- Kim, Y.H.; Noh, J.R.; Hwang, J.H.; Kim, K.S.; Choi, D.H.; Kim, J.H.; Moon, S.J.; Choi, J.H.; Herault, Y.; Lee, T.G.; et al. Hepatocyte SHP deficiency protects mice from acetaminophen-evoked liver injury in a JNK-signaling regulation and GADD45beta-dependent manner. Arch. Toxicol. 2018, 92, 2563–2572. [Google Scholar] [CrossRef]
- Noh, J.R.; Kim, Y.H.; Kim, D.K.; Hwang, J.H.; Kim, K.S.; Choi, D.H.; Lee, S.J.; Lee, H.G.; Lee, T.G.; Weng, H.L.; et al. Small Heterodimer Partner Deficiency Increases Inflammatory Liver Injury Through C-X-C motif chemokine ligand 2-Driven Neutrophil Recruitment in Mice. Toxicol. Sci. 2018, 163, 254–264. [Google Scholar] [CrossRef]
- Yuk, J.M.; Shin, D.M.; Lee, H.M.; Kim, J.J.; Kim, S.W.; Jin, H.S.; Yang, C.S.; Park, K.A.; Chanda, D.; Kim, D.K.; et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 2011, 12, 742–751. [Google Scholar] [CrossRef]
- Bhushan, B.; Apte, U. Acetaminophen Test Battery (ATB): A Comprehensive Method to Study Acetaminophen-Induced Acute Liver Injury. Gene Expr. 2020, 20, 125–138. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Metabolism and disposition of acetaminophen: Recent advances in relation to hepatotoxicity and diagnosis. Pharm. Res. 2013, 30, 2174–2187. [Google Scholar] [CrossRef]
- Bae, G.H.; Lee, S.K.; Kim, H.S.; Lee, M.; Lee, H.Y.; Bae, Y.S. Lysophosphatidic acid protects against acetaminophen-induced acute liver injury. Exp. Mol. Med. 2017, 49, e407. [Google Scholar] [CrossRef]
- Liu, H.; Lo, C.R.; Czaja, M.J. NF-kappaB inhibition sensitizes hepatocytes to TNF-induced apoptosis through a sustained activation of JNK and c-Jun. Hepatology 2002, 35, 772–778. [Google Scholar] [CrossRef]
- Du, K.; Ramachandran, A.; Jaeschke, H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol. 2016, 10, 148–156. [Google Scholar] [CrossRef]
- Enomoto, A.; Itoh, K.; Nagayoshi, E.; Haruta, J.; Kimura, T.; O’Connor, T.; Harada, T.; Yamamoto, M. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol. Sci. 2001, 59, 169–177. [Google Scholar] [CrossRef]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef]
- Copple, I.M.; Goldring, C.E.; Jenkins, R.E.; Chia, A.J.; Randle, L.E.; Hayes, J.D.; Kitteringham, N.R.; Park, B.K. The hepatotoxic metabolite of acetaminophen directly activates the Keap1-Nrf2 cell defense system. Hepatology 2008, 48, 1292–1301. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species (Apex) 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Chao, X.; Wang, H.; Jaeschke, H.; Ding, W.X. Role and mechanisms of autophagy in acetaminophen-induced liver injury. Liver Int. 2018, 38, 1363–1374. [Google Scholar] [CrossRef]
- Wang, D.S.; Kobayashi, T.; Senthilkumaran, B.; Sakai, F.; Sudhakumari, C.C.; Suzuki, T.; Yoshikuni, M.; Matsuda, M.; Morohashi, K.; Nagahama, Y. Molecular cloning of DAX1 and SHP cDNAs and their expression patterns in the Nile tilapia, Oreochromis niloticus. Biochem. Biophys Res. Commun. 2002, 297, 632–640. [Google Scholar] [CrossRef]
- Walesky, C.M.; Kolb, K.E.; Winston, C.L.; Henderson, J.; Kruft, B.; Fleming, I.; Ko, S.; Monga, S.P.; Mueller, F.; Apte, U.; et al. Functional compensation precedes recovery of tissue mass following acute liver injury. Nat. Commun. 2020, 11, 5785. [Google Scholar] [CrossRef]
- Kim, Y.H.; Hwang, J.H.; Kim, K.S.; Noh, J.R.; Choi, D.H.; Kim, D.K.; Tadi, S.; Yim, Y.H.; Choi, H.S.; Lee, C.H. Metformin ameliorates acetaminophen hepatotoxicity via Gadd45beta-dependent regulation of JNK signaling in mice. J. Hepatol. 2015, 63, 75–82. [Google Scholar] [CrossRef]
- Iyer, A.K.; Zhang, Y.H.; McCabe, E.R. Dosage-sensitive sex reversal adrenal hypoplasia congenita critical region on the X chromosome, gene 1 (DAX1) (NR0B1) and small heterodimer partner (SHP) (NR0B2) form homodimers individually, as well as DAX1-SHP heterodimers. Mol. Endocrinol. 2006, 20, 2326–2342. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, H.; McGill, M.R.; Williams, C.D.; Ramachandran, A. Current issues with acetaminophen hepatotoxicity--a clinically relevant model to test the efficacy of natural products. Life Sci. 2011, 88, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Saito, C.; Zwingmann, C.; Jaeschke, H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology 2010, 51, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Lu, M.C.; Zhang, X.; Wu, F.; Tan, S.J.; Zhao, J.; You, Q.D.; Jiang, Z.Y. Discovery of a Potent Kelch-Like ECH-Associated Protein 1-Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) Protein-Protein Interaction Inhibitor with Natural Proline Structure as a Cytoprotective Agent against Acetaminophen-Induced Hepatotoxicity. J. Med. Chem. 2019, 62, 6796–6813. [Google Scholar] [CrossRef]
- Hanawa, N.; Shinohara, M.; Saberi, B.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J. Biol. Chem. 2008, 283, 13565–13577. [Google Scholar] [CrossRef]
- Saito, C.; Lemasters, J.J.; Jaeschke, H. c-Jun N-terminal kinase modulates oxidant stress and peroxynitrite formation independent of inducible nitric oxide synthase in acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2010, 246, 8–17. [Google Scholar] [CrossRef]
- Williams, C.D.; McGill, M.R.; Lebofsky, M.; Bajt, M.L.; Jaeschke, H. Protection against acetaminophen-induced liver injury by allopurinol is dependent on aldehyde oxidase-mediated liver preconditioning. Toxicol. Appl. Pharmacol. 2014, 274, 417–424. [Google Scholar] [CrossRef]
- Du, K.; Williams, C.D.; McGill, M.R.; Jaeschke, H. Lower susceptibility of female mice to acetaminophen hepatotoxicity: Role of mitochondrial glutathione, oxidant stress and c-jun N-terminal kinase. Toxicol. Appl. Pharmacol. 2014, 281, 58–66. [Google Scholar] [CrossRef]
- Tong, S.J.; Liu, J.; Wang, X.; Qu, L.X. microRNA-181 promotes prostate cancer cell proliferation by regulating DAX-1 expression. Exp. Ther. Med. 2014, 8, 1296–1300. [Google Scholar] [CrossRef]
- Du, X.; Yang, Y.; Xu, C.; Peng, Z.; Zhang, M.; Lei, L.; Gao, W.; Dong, Y.; Shi, Z.; Sun, X.; et al. Upregulation of miR-181a impairs hepatic glucose and lipid homeostasis. Oncotarget 2017, 8, 91362–91378. [Google Scholar] [CrossRef]
- Weston, C.J.; Zimmermann, H.W.; Adams, D.H. The Role of Myeloid-Derived Cells in the Progression of Liver Disease. Front. Immunol. 2019, 10, 893. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suh, Y.-J.; Yun, H.-J.; Kim, Y.-B.; Kang, E.-J.; Choi, J.H.; Choi, Y.-K.; Lee, I.-B.; Choi, D.-H.; Seo, Y.J.; Noh, J.-R.; et al. Hepatocyte-Specific Deficiency of DAX-1 Protects Mice from Acetaminophen-Induced Hepatotoxicity by Activating NRF2 Signaling. Int. J. Mol. Sci. 2022, 23, 11786. https://doi.org/10.3390/ijms231911786
Suh Y-J, Yun H-J, Kim Y-B, Kang E-J, Choi JH, Choi Y-K, Lee I-B, Choi D-H, Seo YJ, Noh J-R, et al. Hepatocyte-Specific Deficiency of DAX-1 Protects Mice from Acetaminophen-Induced Hepatotoxicity by Activating NRF2 Signaling. International Journal of Molecular Sciences. 2022; 23(19):11786. https://doi.org/10.3390/ijms231911786
Chicago/Turabian StyleSuh, Young-Joo, Hyo-Jeong Yun, Yu-Bin Kim, Eun-Jung Kang, Jung Hyeon Choi, Young-Keun Choi, In-Bok Lee, Dong-Hee Choi, Yun Jeong Seo, Jung-Ran Noh, and et al. 2022. "Hepatocyte-Specific Deficiency of DAX-1 Protects Mice from Acetaminophen-Induced Hepatotoxicity by Activating NRF2 Signaling" International Journal of Molecular Sciences 23, no. 19: 11786. https://doi.org/10.3390/ijms231911786
APA StyleSuh, Y.-J., Yun, H.-J., Kim, Y.-B., Kang, E.-J., Choi, J. H., Choi, Y.-K., Lee, I.-B., Choi, D.-H., Seo, Y. J., Noh, J.-R., Lee, J.-S., Kim, Y.-H., & Lee, C.-H. (2022). Hepatocyte-Specific Deficiency of DAX-1 Protects Mice from Acetaminophen-Induced Hepatotoxicity by Activating NRF2 Signaling. International Journal of Molecular Sciences, 23(19), 11786. https://doi.org/10.3390/ijms231911786