

Neuronal Agrin Promotes Proliferation of Primary Human Myoblasts in an Age-Dependent Manner

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results
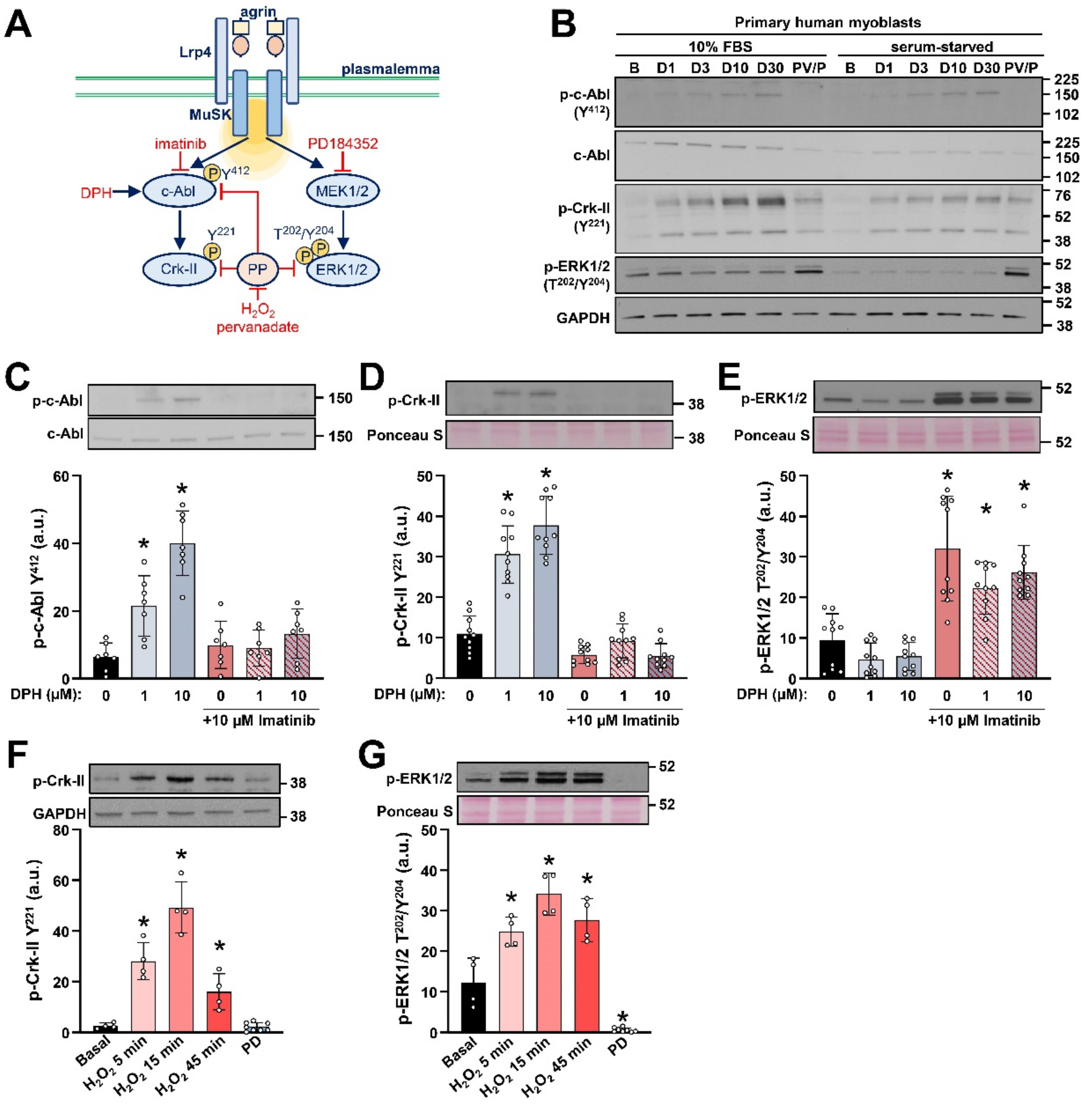
2.1. Detection of c-Abl Signalling Pathway in Primary Human Myoblasts
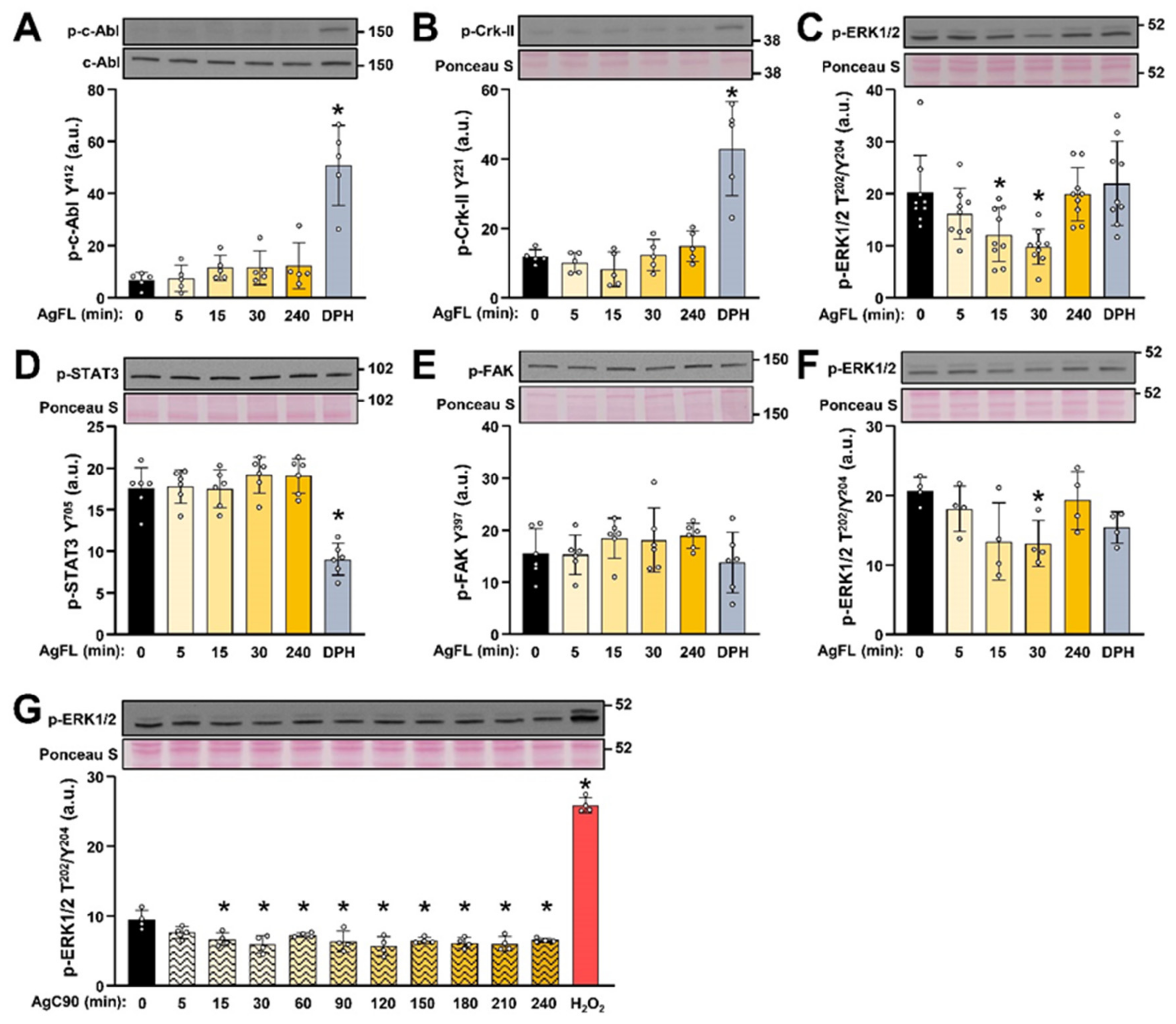
2.2. AgFL Transiently Suppresses the Phosphorylation of ERK1/2 in Primary Human Myoblasts
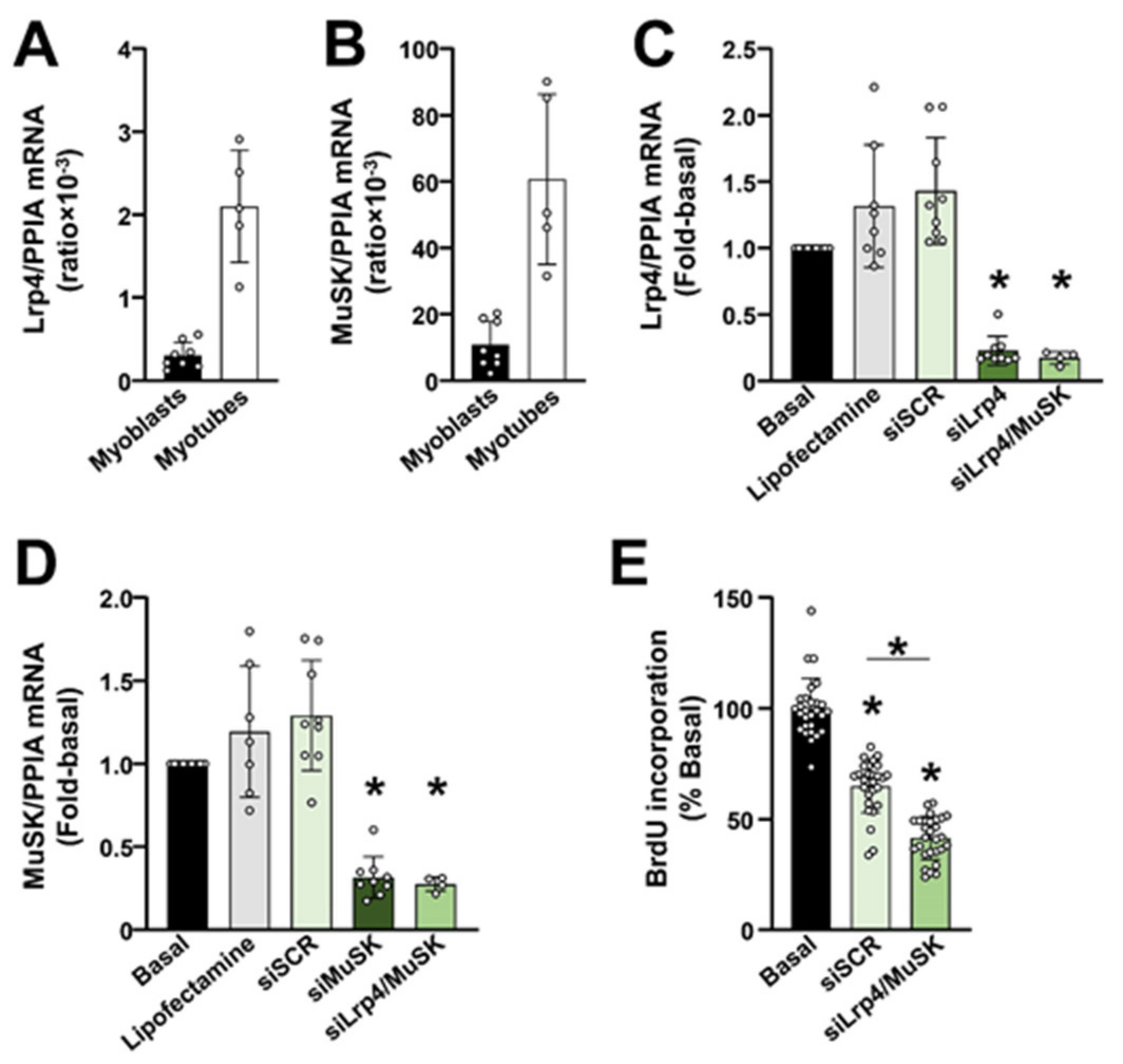
2.3. The Lrp4/MuSK Agrin Receptor Complex Is Required for Proliferation of Primary Human Myoblasts
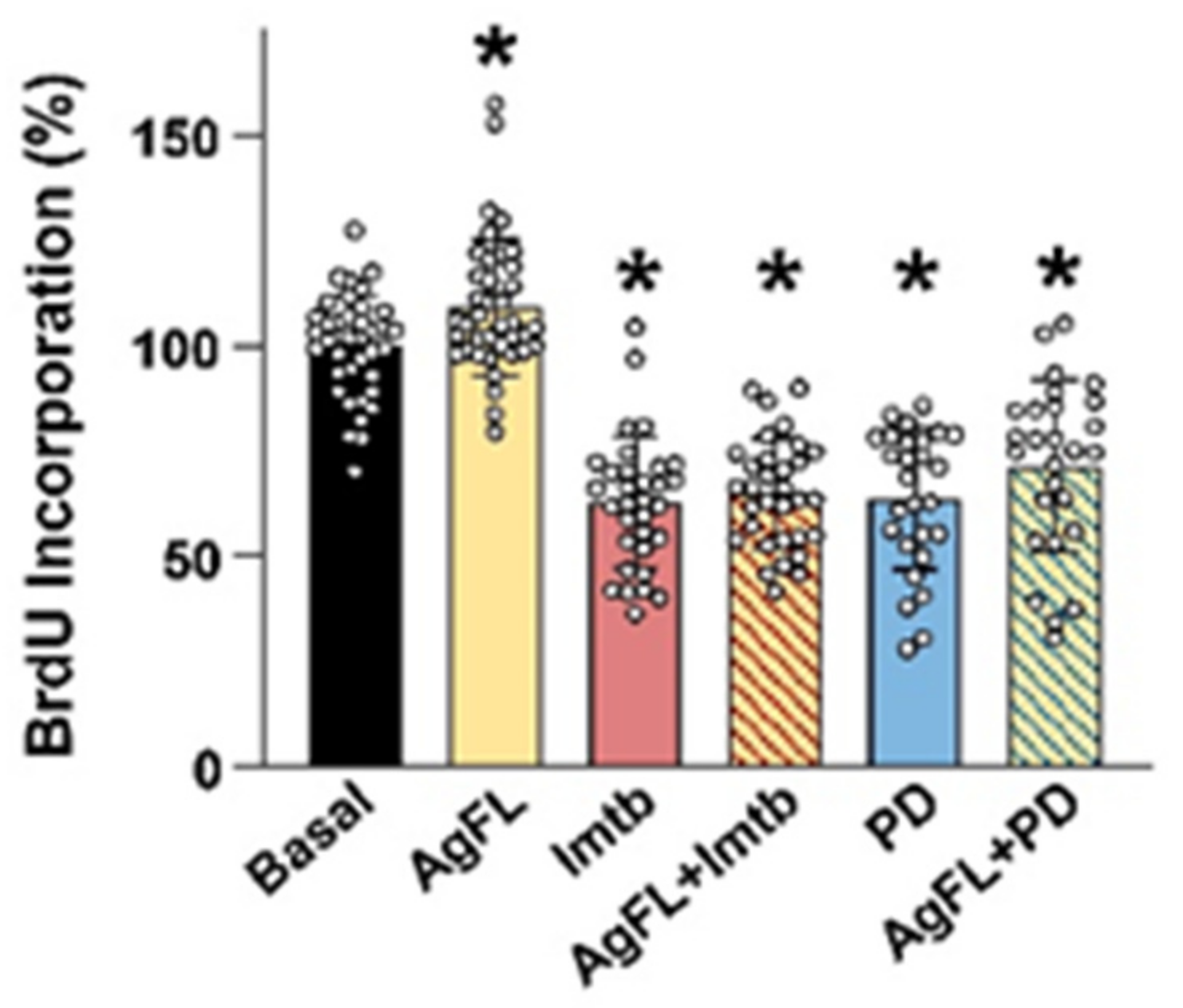
2.4. Effect of AgFL on Proliferation of Primary Human Myoblasts
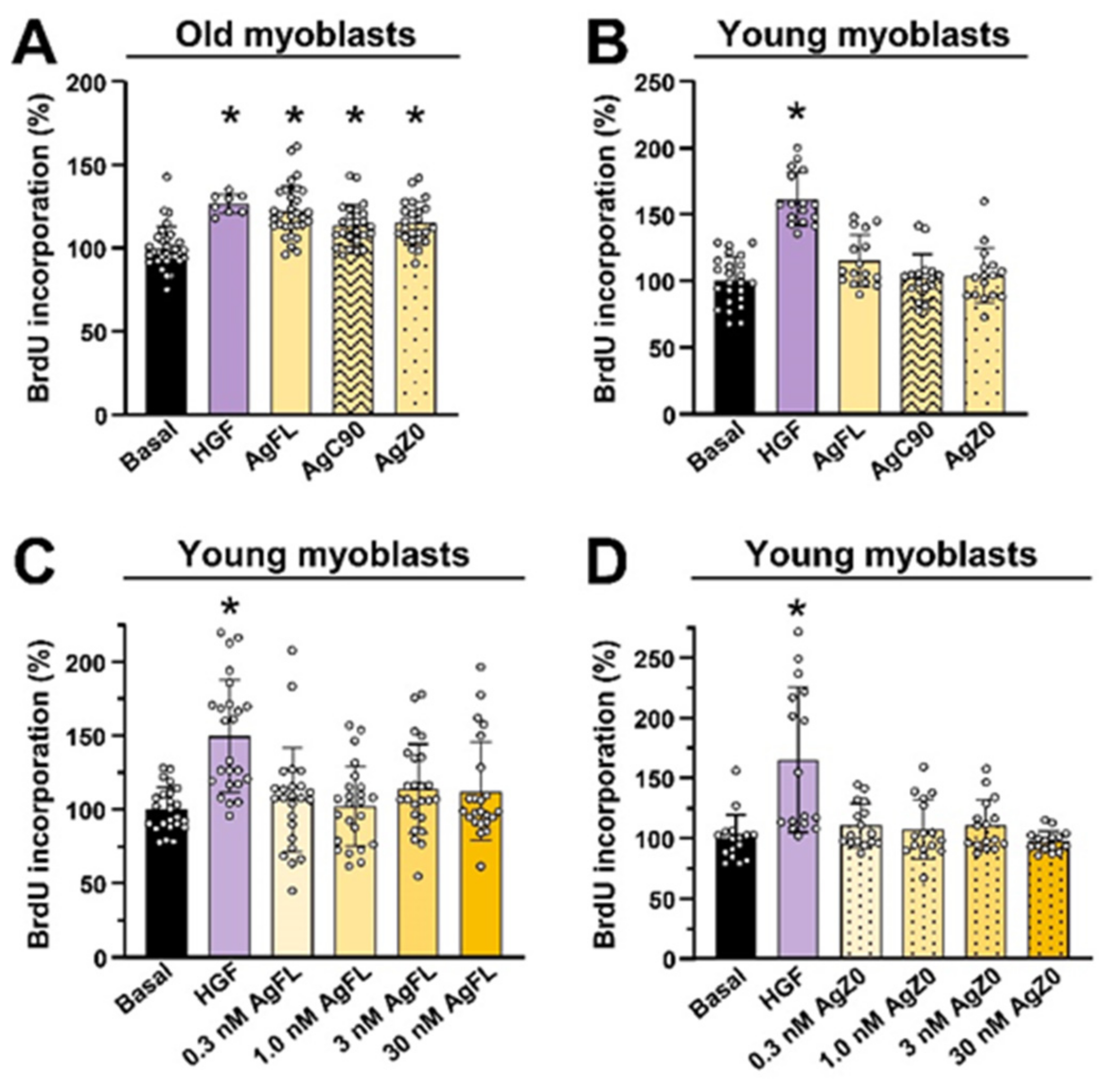
2.5. Effect of AgFL and AgZ0 on Proliferation of Primary Human Myoblasts Is Age-Dependent
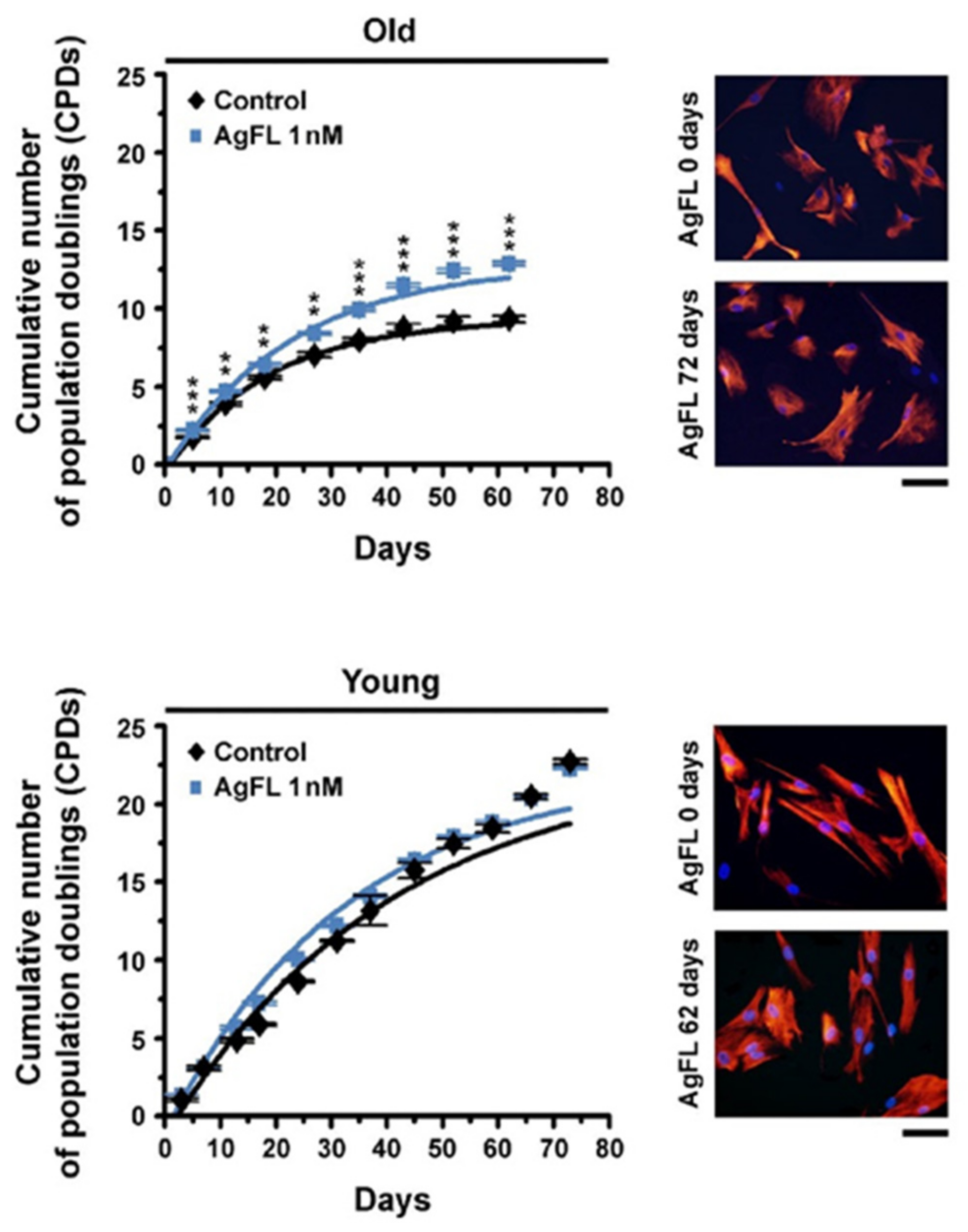
2.6. Long-Term Exposure to AgFL Increases Proliferation of Old Myoblasts
2.7. Long-Term Exposure to AgFL Does Not Impair Myotube Formation and Establishment of the E–C Coupling Mechanism in Myotubes
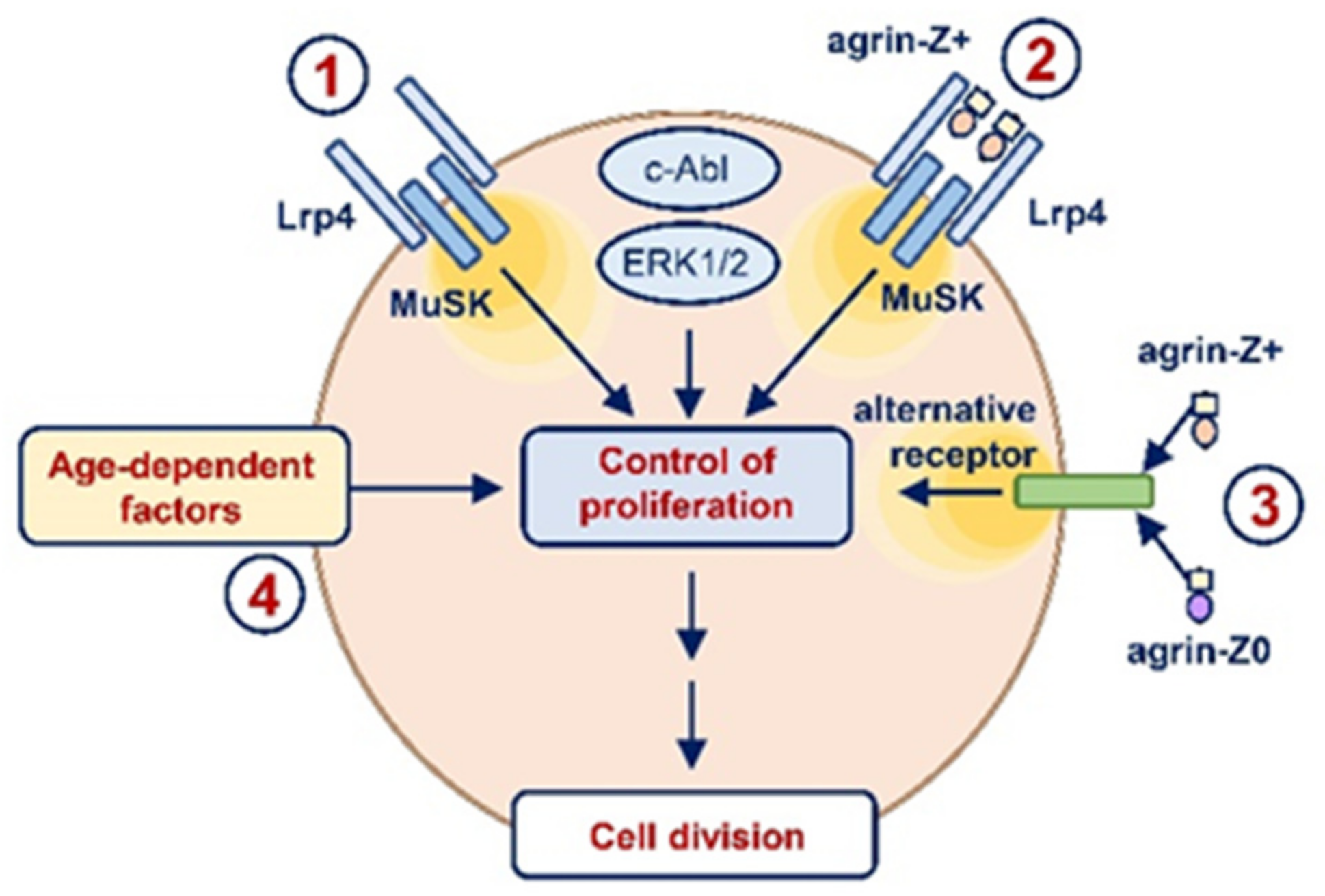
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Antibodies
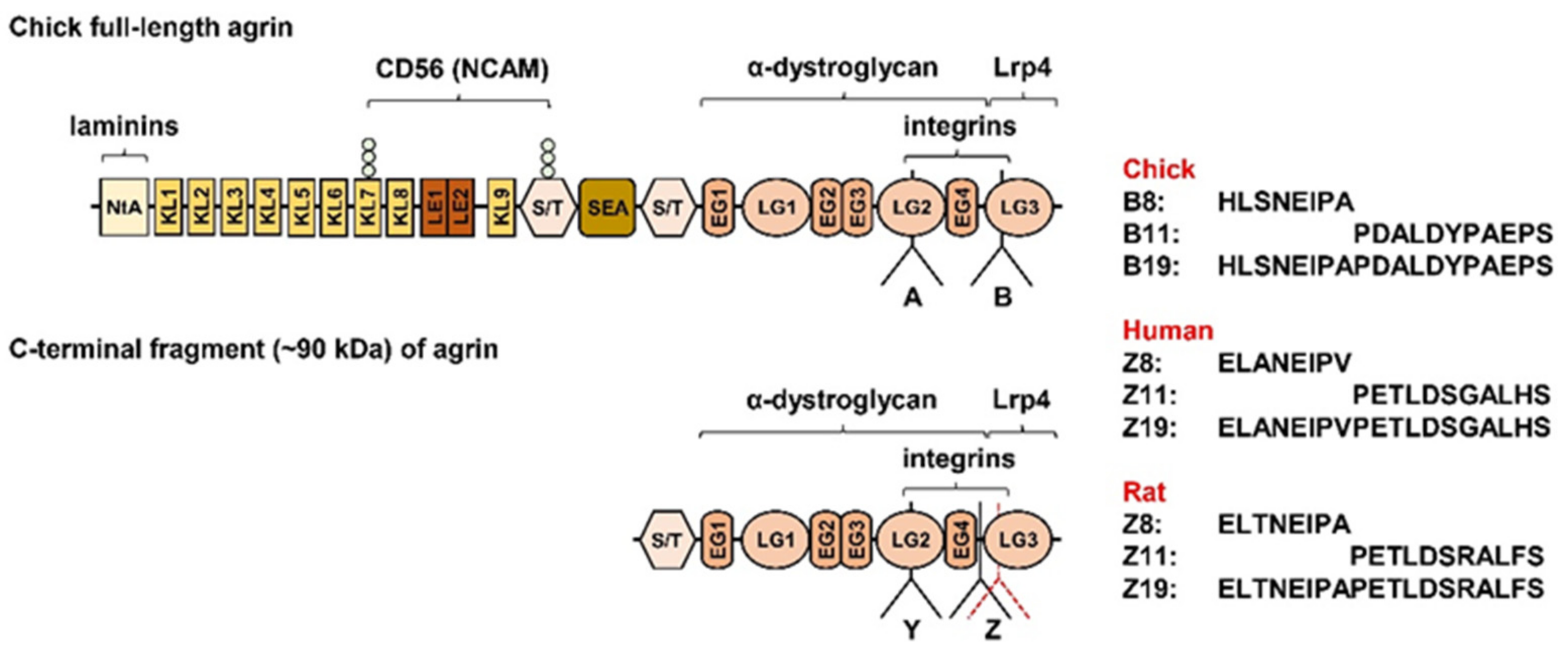
4.3. Agrin

4.4. Isolation and Purification of Human Skeletal Myoblasts
4.5. Desmin Staining
4.6. C2C12 Cell Culture
4.7. Pervanadate Treatment
4.8. Growth Curves and Cumulative Number of Population Doublings
4.9. BrdU Incorporation
4.10. Quantitative PCR
4.11. Western Blot
4.12. Myoblast Fusion Efficiency
4.13. Ca2+ Imaging in Cultured Myotubes
4.14. Gene Silencing of Lrp4 and MuSK
4.15. Statistical Analysis
5. Conclusions
- Neuronal agrin induced transient dephosphorylation of ERK1/2 in primary human myoblasts.
- Lrp4/MuSK was required for proliferation of primary human myoblasts.
- Acute and chronic treatment with exogenous neuronal agrin increased the proliferation of primary human myoblasts of old donors but did not affect the proliferation of myoblasts of young donors.
- Stimulation of proliferation by neuronal agrin likely involved Lrp4/MuSK-dependent as well as Lrp4/MuSK-independent pathways.
- Collectively, our results highlight an age-dependent function for neuronal agrin in the modulation of human myoblast proliferation. Furthermore, they exclude a direct negative effect of neuronal agrin on the regenerative potential of myoblasts in humans.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nitkin, R.M.; Smith, M.A.; Magill, C.; Fallon, J.R.; Yao, Y.M.; Wallace, B.G.; McMahan, U.J. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J. Cell Biol. 1987, 105, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.W.; Nitkin, R.M.; Wallace, B.G.; Rubin, L.L.; McMahan, U.J. Components of Torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J. Cell Biol. 1984, 99, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Bezakova, G.; Ruegg, M.A. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003, 4, 295–308. [Google Scholar] [CrossRef] [PubMed]
- McMahan, U.J.; Horton, S.E.; Werle, M.J.; Honig, L.S.; Kroger, S.; Ruegg, M.A.; Escher, G. Agrin isoforms and their role in synaptogenesis. Curr. Opin. Cell Biol. 1992, 4, 869–874. [Google Scholar] [CrossRef]
- Burden, S.J.; Huijbers, M.G.; Remedio, L. Fundamental Molecules and Mechanisms for Forming and Maintaining Neuromuscular Synapses. Int. J. Mol. Sci. 2018, 19, 490. [Google Scholar] [CrossRef]
- Jennings, C.G.; Dyer, S.M.; Burden, S.J. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc. Natl. Acad. Sci. USA 1993, 90, 2895–2899. [Google Scholar] [CrossRef]
- Valenzuela, D.M.; Stitt, T.N.; DiStefano, P.S.; Rojas, E.; Mattsson, K.; Compton, D.L.; Nunez, L.; Park, J.S.; Stark, J.L.; Gies, D.R.; et al. Receptor tyrosine kinase specific for the skeletal muscle lineage: Expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron 1995, 15, 573–584. [Google Scholar] [CrossRef]
- Glass, D.J.; Bowen, D.C.; Stitt, T.N.; Radziejewski, C.; Bruno, J.; Ryan, T.E.; Gies, D.R.; Shah, S.; Mattsson, K.; Burden, S.J.; et al. Agrin acts via a MuSK receptor complex. Cell 1996, 85, 513–523. [Google Scholar] [CrossRef]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. LRP4 serves as a coreceptor of agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef]
- Fish, L.A.; Fallon, J.R. Multiple MuSK signaling pathways and the aging neuromuscular junction. Neurosci. Lett. 2020, 731, 135014. [Google Scholar] [CrossRef]
- Zhang, W.; Coldefy, A.S.; Hubbard, S.R.; Burden, S.J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 2011, 286, 40624–40630. [Google Scholar] [CrossRef]
- Ruegg, M.A.; Tsim, K.W.; Horton, S.E.; Kroger, S.; Escher, G.; Gensch, E.M.; McMahan, U.J. The agrin gene codes for a family of basal lamina proteins that differ in function and distribution. Neuron 1992, 8, 691–699. [Google Scholar] [CrossRef]
- Burgess, R.W.; Nguyen, Q.T.; Son, Y.J.; Lichtman, J.W.; Sanes, J.R. Alternatively spliced isoforms of nerve- and muscle-derived agrin: Their roles at the neuromuscular junction. Neuron 1999, 23, 33–44. [Google Scholar] [CrossRef]
- Hettwer, S.; Dahinden, P.; Kucsera, S.; Farina, C.; Ahmed, S.; Fariello, R.; Drey, M.; Sieber, C.C.; Vrijbloed, J.W. Elevated levels of a C-terminal agrin fragment identifies a new subset of sarcopenia patients. Exp. Gerontol. 2013, 48, 69–75. [Google Scholar] [CrossRef]
- Landi, F.; Calvani, R.; Lorenzi, M.; Martone, A.M.; Tosato, M.; Drey, M.; D’Angelo, E.; Capoluongo, E.; Russo, A.; Bernabei, R.; et al. Serum levels of C-terminal agrin fragment (CAF) are associated with sarcopenia in older multimorbid community-dwellers: Results from the ilSIRENTE study. Exp. Gerontol. 2016, 79, 31–36. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Lorenzi, M.; Marini, F.; D’Angelo, E.; Martone, A.M.; Celi, M.; Tosato, M.; Bernabei, R.; Landi, F. Serum levels of C-terminal agrin fragment (CAF) are associated with sarcopenia in older hip fractured patients. Exp. Gerontol. 2014, 60, 79–82. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Bolliger, M.F.; Zurlinden, A.; Luscher, D.; Butikofer, L.; Shakhova, O.; Francolini, M.; Kozlov, S.V.; Cinelli, P.; Stephan, A.; Kistler, A.D.; et al. Specific proteolytic cleavage of agrin regulates maturation of the neuromuscular junction. J. Cell Sci. 2010, 123, 3944–3955. [Google Scholar] [CrossRef]
- Butikofer, L.; Zurlinden, A.; Bolliger, M.F.; Kunz, B.; Sonderegger, P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 2011, 25, 4378–4393. [Google Scholar] [CrossRef]
- Reif, R.; Sales, S.; Hettwer, S.; Dreier, B.; Gisler, C.; Wolfel, J.; Luscher, D.; Zurlinden, A.; Stephan, A.; Ahmed, S.; et al. Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J. 2007, 21, 3468–3478. [Google Scholar] [CrossRef]
- Hettwer, S.; Lin, S.; Kucsera, S.; Haubitz, M.; Oliveri, F.; Fariello, R.G.; Ruegg, M.A.; Vrijbloed, J.W. Injection of a soluble fragment of neural agrin (NT-1654) considerably improves the muscle pathology caused by the disassembly of the neuromuscular junction. PLoS ONE 2014, 9, e88739. [Google Scholar] [CrossRef]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Chen, W.; Datzkiw, D.; Rudnicki, M.A. Satellite cells in ageing: Use it or lose it. Open Biol. 2020, 10, 200048. [Google Scholar] [CrossRef]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef]
- Ciciliot, S.; Schiaffino, S. Regeneration of mammalian skeletal muscle. Basic mechanisms and clinical implications. Curr. Pharm. Des. 2010, 16, 906–914. [Google Scholar] [CrossRef]
- Bandi, E.; Jevsek, M.; Mars, T.; Jurdana, M.; Formaggio, E.; Sciancalepore, M.; Fumagalli, G.; Grubic, Z.; Ruzzier, F.; Lorenzon, P. Neural agrin controls maturation of the excitation-contraction coupling mechanism in human myotubes developing in vitro. Am. J. Physiol. Cell Physiol. 2008, 294, C66–C73. [Google Scholar] [CrossRef]
- Jurdana, M.; Fumagalli, G.; Grubic, Z.; Lorenzon, P.; Mars, T.; Sciancalepore, M. Neural agrin changes the electrical properties of developing human skeletal muscle cells. Cell. Mol. Neurobiol. 2009, 29, 123–131. [Google Scholar] [CrossRef]
- Bezakova, G.; Lomo, T. Muscle activity and muscle agrin regulate the organization of cytoskeletal proteins and attached acetylcholine receptor (AchR) aggregates in skeletal muscle fibers. J. Cell Biol. 2001, 153, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Frump, D.; Lin, M.; Caiozzo, V.J.; Mozaffar, T.; Steward, O.; Gupta, R. Matrix metalloproteinase 3 deletion preserves denervated motor endplates after traumatic nerve injury. Ann. Neurol. 2013, 73, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.S.; Conboy, I.M.; Conboy, M.J.; Shen, J.; Rando, T.A. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell 2008, 2, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Clegg, C.H.; Linkhart, T.A.; Olwin, B.B.; Hauschka, S.D. Growth factor control of skeletal muscle differentiation: Commitment to terminal differentiation occurs in G1 phase and is repressed by fibroblast growth factor. J. Cell Biol. 1987, 105, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Scata, K.A.; Bernard, D.W.; Fox, J.; Swain, J.L. FGF receptor availability regulates skeletal myogenesis. Exp. Cell Res. 1999, 250, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef]
- Anastasi, S.; Giordano, S.; Sthandier, O.; Gambarotta, G.; Maione, R.; Comoglio, P.; Amati, P. A natural hepatocyte growth factor/scatter factor autocrine loop in myoblast cells and the effect of the constitutive Met kinase activation on myogenic differentiation. J. Cell Biol. 1997, 137, 1057–1068. [Google Scholar] [CrossRef]
- Gal-Levi, R.; Leshem, Y.; Aoki, S.; Nakamura, T.; Halevy, O. Hepatocyte growth factor plays a dual role in regulating skeletal muscle satellite cell proliferation and differentiation. Biochim. Biophys. Acta 1998, 1402, 39–51. [Google Scholar] [CrossRef]
- Mazzon, C.; Anselmo, A.; Cibella, J.; Soldani, C.; Destro, A.; Kim, N.; Roncalli, M.; Burden, S.J.; Dustin, M.L.; Sarukhan, A.; et al. The critical role of agrin in the hematopoietic stem cell niche. Blood 2011, 118, 2733–2742. [Google Scholar] [CrossRef]
- Mazzon, C.; Anselmo, A.; Soldani, C.; Cibella, J.; Ploia, C.; Moalli, F.; Burden, S.J.; Dustin, M.L.; Sarukhan, A.; Viola, A. Agrin is required for survival and function of monocytic cells. Blood 2012, 119, 5502–5511. [Google Scholar] [CrossRef]
- Chakraborty, S.; Lakshmanan, M.; Swa, H.L.; Chen, J.; Zhang, X.; Ong, Y.S.; Loo, L.S.; Akincilar, S.C.; Gunaratne, J.; Tergaonkar, V.; et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat. Commun. 2015, 6, 6184. [Google Scholar] [CrossRef]
- Steiner, E.; Enzmann, G.U.; Lyck, R.; Lin, S.; Ruegg, M.A.; Kroger, S.; Engelhardt, B. The heparan sulfate proteoglycan agrin contributes to barrier properties of mouse brain endothelial cells by stabilizing adherens junctions. Cell Tissue Res. 2014, 358, 465–479. [Google Scholar] [CrossRef]
- Finn, A.J.; Feng, G.; Pendergast, A.M. Postsynaptic requirement for Abl kinases in assembly of the neuromuscular junction. Nat. Neurosci. 2003, 6, 717–723. [Google Scholar] [CrossRef]
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef]
- Khatri, A.; Wang, J.; Pendergast, A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016, 129, 9–16. [Google Scholar] [CrossRef]
- Rimer, M. Modulation of agrin-induced acetylcholine receptor clustering by extracellular signal-regulated kinases 1 and 2 in cultured myotubes. J. Biol. Chem. 2010, 285, 32370–32377. [Google Scholar] [CrossRef]
- Rimer, M. Extracellular signal-regulated kinases 1 and 2 regulate neuromuscular junction and myofiber phenotypes in mammalian skeletal muscle. Neurosci. Lett. 2020, 715, 134671. [Google Scholar] [CrossRef]
- Boulton, T.G.; Yancopoulos, G.D.; Gregory, J.S.; Slaughter, C.; Moomaw, C.; Hsu, J.; Cobb, M.H. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990, 249, 64–67. [Google Scholar] [CrossRef]
- Ray, L.B.; Sturgill, T.W. Rapid stimulation by insulin of a serine/threonine kinase in 3T3-L1 adipocytes that phosphorylates microtubule-associated protein 2 in vitro. Proc. Natl. Acad. Sci. USA 1987, 84, 1502–1506. [Google Scholar] [CrossRef]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Eigler, T.; Zarfati, G.; Amzallag, E.; Sinha, S.; Segev, N.; Zabary, Y.; Zaritsky, A.; Shakked, A.; Umansky, K.B.; Schejter, E.D.; et al. ERK1/2 inhibition promotes robust myotube growth via CaMKII activation resulting in myoblast-to-myotube fusion. Dev. Cell 2021, 56, 3349–3363. [Google Scholar] [CrossRef]
- Montecino, F.; Gonzalez, N.; Blanco, N.; Ramirez, M.J.; Gonzalez-Martin, A.; Alvarez, A.R.; Olguin, H. c-Abl Kinase Is Required for Satellite Cell Function Through Pax7 Regulation. Front. Cell Dev. Biol. 2021, 9, 606403. [Google Scholar] [CrossRef]
- Bae, G.U.; Kim, B.G.; Lee, H.J.; Oh, J.E.; Lee, S.J.; Zhang, W.; Krauss, R.S.; Kang, J.S. Cdo binds Abl to promote p38alpha/beta mitogen-activated protein kinase activity and myogenic differentiation. Mol. Cell. Biol. 2009, 29, 4130–4143. [Google Scholar] [CrossRef]
- Puri, P.L.; Bhakta, K.; Wood, L.D.; Costanzo, A.; Zhu, J.; Wang, J.Y. A myogenic differentiation checkpoint activated by genotoxic stress. Nat. Genet. 2002, 32, 585–593. [Google Scholar] [CrossRef]
- Pendergast, A.M.; Muller, A.J.; Havlik, M.H.; Clark, R.; McCormick, F.; Witte, O.N. Evidence for regulation of the human ABL tyrosine kinase by a cellular inhibitor. Proc. Natl. Acad. Sci. USA 1991, 88, 5927–5931. [Google Scholar] [CrossRef]
- Yang, J.; Campobasso, N.; Biju, M.P.; Fisher, K.; Pan, X.Q.; Cottom, J.; Galbraith, S.; Ho, T.; Zhang, H.; Hong, X.; et al. Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem. Biol. 2011, 18, 177–186. [Google Scholar] [CrossRef][Green Version]
- Brasher, B.B.; Van Etten, R.A. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J. Biol. Chem. 2000, 275, 35631–35637. [Google Scholar] [CrossRef]
- Feller, S.M.; Knudsen, B.; Hanafusa, H. c-Abl kinase regulates the protein binding activity of c-Crk. EMBO J. 1994, 13, 2341–2351. [Google Scholar] [CrossRef]
- Sattler, M.; Verma, S.; Shrikhande, G.; Byrne, C.H.; Pride, Y.B.; Winkler, T.; Greenfield, E.A.; Salgia, R.; Griffin, J.D. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J. Biol. Chem. 2000, 275, 24273–24278. [Google Scholar] [CrossRef]
- Huyer, G.; Liu, S.; Kelly, J.; Moffat, J.; Payette, P.; Kennedy, B.; Tsaprailis, G.; Gresser, M.J.; Ramachandran, C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997, 272, 843–851. [Google Scholar] [CrossRef]
- Heffetz, D.; Bushkin, I.; Dror, R.; Zick, Y. The insulinomimetic agents H2O2 and vanadate stimulate protein tyrosine phosphorylation in intact cells. J. Biol. Chem. 1990, 265, 2896–2902. [Google Scholar] [CrossRef]
- Pirkmajer, S.; Chibalin, A.V. Serum starvation: Caveat emptor. Am. J. Physiol. Cell Physiol. 2011, 301, C272–C279. [Google Scholar] [CrossRef] [PubMed]
- Burden, S.J.; Fuhrer, C.; Hubbard, S.R. Agrin/MuSK signaling: Willing and Abl. Nat. Neurosci. 2003, 6, 653–654. [Google Scholar] [CrossRef] [PubMed]
- Burden, S.J.; Yumoto, N.; Zhang, W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a009167. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Andreolotti, A.G.; Bragado, M.J.; Tapia, J.A.; Jensen, R.T.; Garcia-Marin, L.J. Adapter protein CRKII signaling is involved in the rat pancreatic acini response to reactive oxygen species. J. Cell. Biochem. 2006, 97, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.A.; Pollock, S.S.; Coleman, C.N.; Calderwood, S.K. X-irradiation, phorbol esters, and H2O2 stimulate mitogen-activated protein kinase activity in NIH-3T3 cells through the formation of reactive oxygen intermediates. Cancer Res. 1994, 54, 12–15. [Google Scholar]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar] [CrossRef]
- Milligan, S.A.; Owens, M.W.; Grisham, M.B. Differential regulation of extracellular signal-regulated kinase and nuclear factor-kappa B signal transduction pathways by hydrogen peroxide and tumor necrosis factor. Arch. Biochem. Biophys. 1998, 352, 255–262. [Google Scholar] [CrossRef]
- Sebolt-Leopold, J.S.; Dudley, D.T.; Herrera, R.; Van Becelaere, K.; Wiland, A.; Gowan, R.C.; Tecle, H.; Barrett, S.D.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef]
- Sun, L.; Ma, K.; Wang, H.; Xiao, F.; Gao, Y.; Zhang, W.; Wang, K.; Gao, X.; Ip, N.; Wu, Z. JAK1-STAT1-STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 2007, 179, 129–138. [Google Scholar] [CrossRef]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardi, M.; Munoz-Canoves, P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef]
- Wang, K.; Wang, C.; Xiao, F.; Wang, H.; Wu, Z. JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 2008, 283, 34029–34036. [Google Scholar] [CrossRef]
- Snyder, M.; Huang, X.Y.; Zhang, J.J. Identification of novel direct Stat3 target genes for control of growth and differentiation. J. Biol. Chem. 2008, 283, 3791–3798. [Google Scholar] [CrossRef]
- Njah, K.; Chakraborty, S.; Qiu, B.; Arumugam, S.; Raju, A.; Pobbati, A.V.; Lakshmanan, M.; Tergaonkar, V.; Thibault, G.; Wang, X.; et al. A Role of Agrin in Maintaining the Stability of Vascular Endothelial Growth Factor Receptor-2 during Tumor Angiogenesis. Cell Rep. 2019, 28, 949–965. [Google Scholar] [CrossRef]
- Quach, N.L.; Biressi, S.; Reichardt, L.F.; Keller, C.; Rando, T.A. Focal adhesion kinase signaling regulates the expression of caveolin 3 and beta1 integrin, genes essential for normal myoblast fusion. Mol. Biol. Cell 2009, 20, 3422–3435. [Google Scholar] [CrossRef]
- Clemente, C.F.; Corat, M.A.; Saad, S.T.; Franchini, K.G. Differentiation of C2C12 myoblasts is critically regulated by FAK signaling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R862–R870. [Google Scholar] [CrossRef]
- Sastry, S.K.; Lakonishok, M.; Wu, S.; Truong, T.Q.; Huttenlocher, A.; Turner, C.E.; Horwitz, A.F. Quantitative changes in integrin and focal adhesion signaling regulate myoblast cell cycle withdrawal. J. Cell Biol. 1999, 144, 1295–1309. [Google Scholar] [CrossRef]
- Tiffin, N.; Adi, S.; Stokoe, D.; Wu, N.Y.; Rosenthal, S.M. Akt phosphorylation is not sufficient for insulin-like growth factor-stimulated myogenin expression but must be accompanied by down-regulation of mitogen-activated protein kinase/extracellular signal-regulated kinase phosphorylation. Endocrinology 2004, 145, 4991–4996. [Google Scholar] [CrossRef][Green Version]
- Allen, R.E.; Sheehan, S.M.; Taylor, R.G.; Kendall, T.L.; Rice, G.M. Hepatocyte growth factor activates quiescent skeletal muscle satellite cells in vitro. J. Cell. Physiol. 1995, 165, 307–312. [Google Scholar] [CrossRef]
- Tatsumi, R.; Anderson, J.E.; Nevoret, C.J.; Halevy, O.; Allen, R.E. HGF/SF is present in normal adult skeletal muscle and is capable of activating satellite cells. Dev. Biol. 1998, 194, 114–128. [Google Scholar] [CrossRef]
- Melzer, W.; Herrmann-Frank, A.; Luttgau, H.C. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta 1995, 1241, 59–116. [Google Scholar] [CrossRef]
- Rios, E.; Ma, J.J.; Gonzalez, A. The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J. Muscle Res. Cell Motil. 1991, 12, 127–135. [Google Scholar] [CrossRef]
- Lorenzon, P.; Giovannelli, A.; Ragozzino, D.; Eusebi, F.; Ruzzier, F. Spontaneous and repetitive calcium transients in C2C12 mouse myotubes during in vitro myogenesis. Eur. J. Neurosci. 1997, 9, 800–808. [Google Scholar] [CrossRef]
- Engert, J.C.; Berglund, E.B.; Rosenthal, N. Proliferation precedes differentiation in IGF-I-stimulated myogenesis. J. Cell Biol. 1996, 135, 431–440. [Google Scholar] [CrossRef]
- Rosenthal, S.M.; Cheng, Z.Q. Opposing early and late effects of insulin-like growth factor I on differentiation and the cell cycle regulatory retinoblastoma protein in skeletal myoblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 10307–10311. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, R.; Tang, D.D. Abl regulates smooth muscle cell proliferation by modulating actin dynamics and ERK1/2 activation. Am. J. Physiol. Cell Physiol. 2012, 302, C1026–C1034. [Google Scholar] [CrossRef] [PubMed]
- Halevy, O.; Cantley, L.C. Differential regulation of the phosphoinositide 3-kinase and MAP kinase pathways by hepatocyte growth factor vs. insulin-like growth factor-I in myogenic cells. Exp. Cell Res. 2004, 297, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Jones, N.C.; Fedorov, Y.V.; Rosenthal, R.S.; Olwin, B.B. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J. Cell. Physiol. 2001, 186, 104–115. [Google Scholar] [CrossRef]
- Abou-Khalil, R.; Le Grand, F.; Pallafacchina, G.; Valable, S.; Authier, F.J.; Rudnicki, M.A.; Gherardi, R.K.; Germain, S.; Chretien, F.; Sotiropoulos, A.; et al. Autocrine and paracrine angiopoietin 1/Tie-2 signaling promotes muscle satellite cell self-renewal. Cell Stem Cell 2009, 5, 298–309. [Google Scholar] [CrossRef]
- Reed, S.A.; Ouellette, S.E.; Liu, X.; Allen, R.E.; Johnson, S.E. E2F5 and LEK1 translocation to the nucleus is an early event demarcating myoblast quiescence. J. Cell. Biochem. 2007, 101, 1394–1408. [Google Scholar] [CrossRef]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Gutarra, S.; Garcia-Prat, L.; Rodriguez-Ubreva, J.; Ortet, L.; Ruiz-Bonilla, V.; Jardi, M.; Ballestar, E.; Gonzalez, S.; Serrano, A.L.; et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 2014, 506, 316–321. [Google Scholar] [CrossRef]
- Renault, V.; Thornell, L.E.; Eriksson, P.O.; Butler-Browne, G.; Mouly, V. Regenerative potential of human skeletal muscle during aging. Aging Cell 2002, 1, 132–139. [Google Scholar] [CrossRef]
- Sugiyama, J.; Bowen, D.C.; Hall, Z.W. Dystroglycan binds nerve and muscle agrin. Neuron 1994, 13, 103–115. [Google Scholar] [CrossRef]
- O’Toole, J.J.; Deyst, K.A.; Bowe, M.A.; Nastuk, M.A.; McKechnie, B.A.; Fallon, J.R. Alternative splicing of agrin regulates its binding to heparin alpha-dystroglycan, and the cell surface. Proc. Natl. Acad. Sci. USA 1996, 93, 7369–7374. [Google Scholar] [CrossRef]
- Burgess, R.W.; Dickman, D.K.; Nunez, L.; Glass, D.J.; Sanes, J.R. Mapping sites responsible for interactions of agrin with neurons. J. Neurochem. 2002, 83, 271–284. [Google Scholar] [CrossRef]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch-Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Bassat, D.R.; et al. The extracellular matrix protein Agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef]
- Bezakova, G.; Helm, J.P.; Francolini, M.; Lomo, T. Effects of purified recombinant neural and muscle agrin on skeletal muscle fibers in vivo. J. Cell Biol. 2001, 153, 1441–1452. [Google Scholar] [CrossRef]
- Stetefeld, J.; Alexandrescu, A.T.; Maciejewski, M.W.; Jenny, M.; Rathgeb-Szabo, K.; Schulthess, T.; Landwehr, R.; Frank, S.; Ruegg, M.A.; Kammerer, R.A. Modulation of agrin function by alternative splicing and Ca2+ binding. Structure 2004, 12, 503–515. [Google Scholar] [CrossRef]
- Mars, T.; King, M.P.; Miranda, A.F.; Walker, W.F.; Mis, K.; Grubic, Z. Functional innervation of cultured human skeletal muscle proceeds by two modes with regard to agrin effects. Neuroscience 2003, 118, 87–97. [Google Scholar] [CrossRef]
- Jan, V.; Mis, K.; Nikolic, N.; Dolinar, K.; Petric, M.; Bone, A.; Thoresen, G.H.; Rustan, A.C.; Mars, T.; Chibalin, A.V.; et al. Effect of differentiation, de novo innervation, and electrical pulse stimulation on mRNA and protein expression of Na+,K+-ATPase, FXYD1, and FXYD5 in cultured human skeletal muscle cells. PLoS ONE 2021, 16, e0247377. [Google Scholar] [CrossRef]
- Pirkmajer, S.; Bezjak, K.; Matkovic, U.; Dolinar, K.; Jiang, L.Q.; Mis, K.; Gros, K.; Milovanova, K.; Pirkmajer, K.P.; Mars, T.; et al. Ouabain Suppresses IL-6/STAT3 Signaling and Promotes Cytokine Secretion in Cultured Skeletal Muscle Cells. Front. Physiol. 2020, 11, 566584. [Google Scholar] [CrossRef]
- Kaufman, S.J.; Foster, R.F. Replicating myoblasts express a muscle-specific phenotype. Proc. Natl. Acad. Sci. USA 1988, 85, 9606–9610. [Google Scholar] [CrossRef]
- Behr, T.; Fischer, P.; Muller-Felber, W.; Schmidt-Achert, M.; Pongratz, D. Myofibrillogenesis in primary tissue cultures of adult human skeletal muscle: Expression of desmin, titin, and nebulin. Clin. Investig. 1994, 72, 150–155. [Google Scholar] [CrossRef]
- Derkinderen, P.; Scales, T.M.; Hanger, D.P.; Leung, K.Y.; Byers, H.L.; Ward, M.A.; Lenz, C.; Price, C.; Bird, I.N.; Perera, T.; et al. Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J. Neurosci. 2005, 25, 6584–6593. [Google Scholar] [CrossRef]
- Tuomi, J.M.; Voorbraak, F.; Jones, D.L.; Ruijter, J.M. Bias in the Cq value observed with hydrolysis probe based quantitative PCR can be corrected with the estimated PCR efficiency value. Methods 2010, 50, 313–322. [Google Scholar] [CrossRef]
- Skorja Milic, N.; Dolinar, K.; Mis, K.; Matkovic, U.; Bizjak, M.; Pavlin, M.; Podbregar, M.; Pirkmajer, S. Suppression of Pyruvate Dehydrogenase Kinase by Dichloroacetate in Cancer and Skeletal Muscle Cells Is Isoform Specific and Partially Independent of HIF-1alpha. Int. J. Mol. Sci. 2021, 22, 8610. [Google Scholar] [CrossRef]
- Mendham, A.E.; Goedecke, J.H.; Zeng, Y.; Larsen, S.; George, C.; Hauksson, J.; Fortuin-de Smidt, M.C.; Chibalin, A.V.; Olsson, T.; Chorell, E. Exercise training improves mitochondrial respiration and is associated with an altered intramuscular phospholipid signature in women with obesity. Diabetologia 2021, 64, 1642–1659. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cumulative Number of Population Doublings | ||
|---|---|---|
| Age of Donor (Years) | Control | 1 nM AgFL |
| 66 | 1.63 ± 0.02 (n = 5) | 2.38 ± 0.09 *** (n = 5) |
| 60 | 4.24 ± 0.09 (n = 4) | 4.83 ± 0.08 ** (n = 4) |
| 58 | 9.95 ± 0.32 (n = 4) | 13.07 ± 0.14 *** (n = 4) |
| 7 | 22.70 ± 0.15 (n = 3) | 22.28 ± 0.08 (n = 3) |
| 5 | 12.21 ± 0.09 (n = 4) | 12.33 ± 0.07 (n = 4) |
| Primary Antibody | Secondary Antibody | ||||
|---|---|---|---|---|---|
| Target | Catalogue No. | Dilution | Source | Source/Target | Dilution |
| c-Abl | CTS #2862 | 1:1000 | rabbit | GAR | 1:25,000 |
| p-c-Abl (Tyr412) | CST #2865 | 1:1000 | rabbit | GAR | 1:8000–1:25,000 |
| p-Crk-II (Tyr221) | CST #3491 | 1:1000 | rabbit | GAR | 1:12,500–1:25,000 |
| desmin | Dako #M076029 | 1:50 | mouse | GAM | 1:200 |
| p-ERK1/2 (Thr202/Tyr204) | CST #4370 | 1:20,000 | rabbit | GAR | 1:25,000–1:50,000 |
| total ERK1/2 | CST #4695 | 1:1000 | rabbit | GAR | 1:25,000 |
| p-FAK (Tyr397) | CST #8556 | 1:1000 | rabbit | GAR | 1:8000–1:12,500 |
| GAPDH | CST #5174 | 1:1000 | rabbit | GAR | 1:25,000–1:50,000 |
| p-STAT3 (Tyr705) | CST #9145 | 1:2000 | rabbit | GAR | 1:4000–1:25,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gros, K.; Matkovič, U.; Parato, G.; Miš, K.; Luin, E.; Bernareggi, A.; Sciancalepore, M.; Marš, T.; Lorenzon, P.; Pirkmajer, S. Neuronal Agrin Promotes Proliferation of Primary Human Myoblasts in an Age-Dependent Manner. Int. J. Mol. Sci. 2022, 23, 11784. https://doi.org/10.3390/ijms231911784
Gros K, Matkovič U, Parato G, Miš K, Luin E, Bernareggi A, Sciancalepore M, Marš T, Lorenzon P, Pirkmajer S. Neuronal Agrin Promotes Proliferation of Primary Human Myoblasts in an Age-Dependent Manner. International Journal of Molecular Sciences. 2022; 23(19):11784. https://doi.org/10.3390/ijms231911784
Chicago/Turabian StyleGros, Katarina, Urška Matkovič, Giulia Parato, Katarina Miš, Elisa Luin, Annalisa Bernareggi, Marina Sciancalepore, Tomaž Marš, Paola Lorenzon, and Sergej Pirkmajer. 2022. "Neuronal Agrin Promotes Proliferation of Primary Human Myoblasts in an Age-Dependent Manner" International Journal of Molecular Sciences 23, no. 19: 11784. https://doi.org/10.3390/ijms231911784
APA StyleGros, K., Matkovič, U., Parato, G., Miš, K., Luin, E., Bernareggi, A., Sciancalepore, M., Marš, T., Lorenzon, P., & Pirkmajer, S. (2022). Neuronal Agrin Promotes Proliferation of Primary Human Myoblasts in an Age-Dependent Manner. International Journal of Molecular Sciences, 23(19), 11784. https://doi.org/10.3390/ijms231911784