

Cell-Free Filtrates (CFF) as Vectors of a Transmissible Pathologic Tissue Memory Code: A Hypothetical and Narrative Review
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Metabolic Memory Is Transferable and Induces the Recapitulation of Histopathologic Hallmarks in Normal Rats
3. Cell-Free Filtrates (CFF) from Non-Diabetic Arteriosclerotic Samples Induce the Recapitulation of Arteriolar Pathology in Host Animals
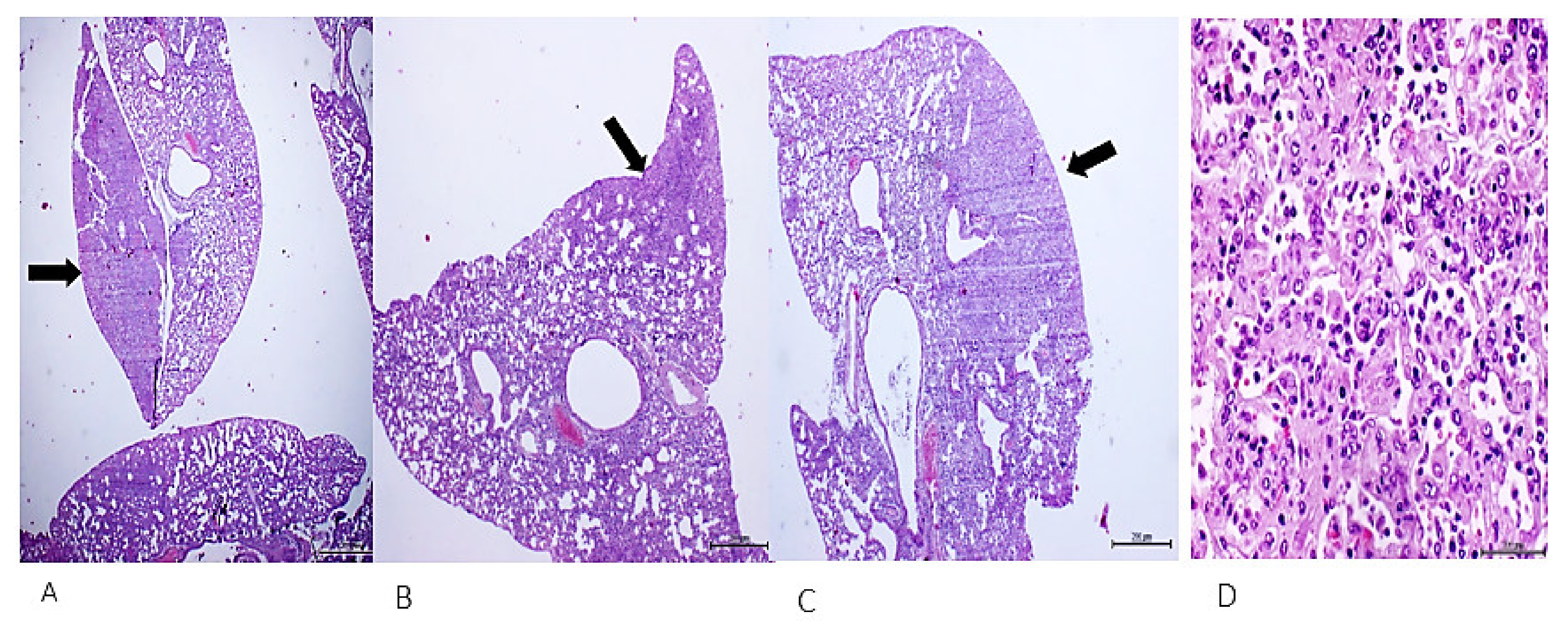
4. Cell-Free Tumor Tissue Filtrate Transforms Cells and Is Carcinogenic in Nude Mice
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CFF | cell-free filtrate |
| AGE | advanced glycation end products |
| BSA | bovine serum albumin |
| EndMT | endothelial-to-mesenchymal transition |
| NCD | non-communicable diseases |
| RAGE | receptor for AGEs |
References
- Corning, W.C. Retention of a position discrimination after regeneration in planarians. Psychon. Sci. 1966, 5, 17–18. [Google Scholar] [CrossRef]
- McConnell, J.V. Comparative Physiology: Learning in Invertebrates. Annu. Rev. Physiol. 1966, 28, 107–136. [Google Scholar] [CrossRef] [PubMed]
- Géranton, S.M. Does epigenetic ‘memory’ of early-life stress predispose to chronic pain in later life? A potential role for the stress regulator FKBP5. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190283. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.; Upadhyay, A.K.; Manni, L.; Huchon, D.; Shenkar, N. And Then There Were Three...: Extreme Regeneration Ability of the Solitary Chordate Polycarpa mytiligera. Front. Cell Dev. Biol. 2021, 9, 652466. [Google Scholar] [CrossRef]
- McCusker, C.D.; Gardiner, D.M. Understanding positional cues in salamander limb regeneration: Implications for optimizing cell-based regenerative therapies. Dis. Model. Mech. 2014, 7, 593–599. [Google Scholar] [CrossRef]
- Durso, A.; Brickner, J.H. Epigenetic transcriptional memory. Curr. Genet. 2017, 63, 435–439. [Google Scholar] [CrossRef]
- Alvarez, R.M.; Margulies, K.B. Epigenetic Memory and Cardiac Cell Therapy. J. Am. Coll. Cardiol. 2014, 64, 449–450. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Graw, F.; Magnus, C.; Regoes, R.R. Theoretical analysis of the evolution of immune memory. BMC Evol. Biol. 2010, 10, 380. [Google Scholar] [CrossRef]
- Chang, H.Y.; Chi, J.T.; Dudoit, S.; Bondre, C.; van de Rijn, M.; Botstein, D.; Brown, P.O. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. USA 2002, 99, 12877–12882. [Google Scholar] [CrossRef]
- Egner, I.M.; Bruusgaard, J.C.; Eftestol, E.; Gundersen, K. A cellular memory mechanism aids overload hypertrophy in muscle long after an episodic exposure to anabolic steroids. J. Physiol. 2013, 591, 6221–6330. [Google Scholar] [CrossRef]
- Gundersen, K. Muscle memory and a new cellular model for muscle atrophy and hypertrophy. J. Exp. Biol. 2016, 219, 235–242. [Google Scholar] [CrossRef]
- Naik, S.; Larsen, S.B.; Gomez, N.C.; Alaverdyan, K.; Sendoel, A.; Yuan, S.; Polak, L.; Kulukian, A.; Chai, S.; Fuchs, E. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 2017, 550, 475–480. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Dwyer, D.F.; Nyquist, S.K.; Buchheit, K.M.; Vukovic, M.; Deb, C.; Wadsworth, M.H., 2nd; Hughes, T.K.; Kazer, S.W.; Yoshimoto, E.; et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 2018, 560, 649–654. [Google Scholar] [CrossRef]
- Xu, S.; Kamato, D.; Little, P.J.; Nakagawa, S.; Pelisek, J.; Jin, Z.G. Targeting epigenetics and non-coding RNAs in atherosclerosis: From mechanisms to therapeutics. Pharmacol. Ther. 2019, 196, 15–43. [Google Scholar] [CrossRef]
- Zarzour, A.; Kim, H.W.; Weintraub, N.L. Epigenetic Regulation of Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 984–990. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Mendoza-Mari, Y.; Martinez, M.D.; Valdes-Perez, C.; Ojalvo, A.G.; Armstrong, D.G. Expression of cell proliferation cycle negative regulators in fibroblasts of an ischemic diabetic foot ulcer. A clinical case report. Int. Wound J. 2012, 10, 232–236. [Google Scholar] [CrossRef]
- Kumari, N.; Karmakar, A.; Ganesan, S.k. Targeting epigenetic modifications as a potential therapeutic option for diabetic retinopathy. J. Cell. Physiol. 2020, 235, 1933–1947. [Google Scholar] [CrossRef]
- Schisano, B.; Tripathi, G.; McGee, K.; McTernan, P.G.; Ceriello, A. Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54, 1219–1226. [Google Scholar] [CrossRef]
- Yao, Y.; Song, Q.; Hu, C.; Da, X.; Yu, Y.; He, Z.; Xu, C.; Chen, Q.; Wang, Q.K. Endothelial cell metabolic memory causes cardiovascular dysfunction in diabetes. Cardiovasc. Res. 2021, 118, 196–211. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, H.; Chen, B.-H. Reactive oxygen species mediates a metabolic memory of high glucose stress signaling in bovine retinal pericytes. Int. J. Ophthalmol. 2019, 12, 1067–1074. [Google Scholar] [CrossRef]
- WHO. Noncommunicable Diseases. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases (accessed on 24 May 2022).
- Hancock, C.; Kingo, L.; Raynaud, O. The private sector, international development and NCDs. Glob. Health 2011, 7, 23. [Google Scholar] [CrossRef]
- Kunugi, H.; Ali, A.M. Royal Jelly and Its Components Promote Healthy Aging and Longevity: From Animal Models to Humans. Int. J. Mol. Sci. 2019, 20, 4662. [Google Scholar] [CrossRef]
- Ferrari, L.; Pavanello, S.; Bollati, V. Molecular and epigenetic markers as promising tools to quantify the effect of occupational exposures and the risk of developing non-communicable diseases. La Med. Lav. 2019, 110, 168. [Google Scholar]
- Mampay, M.; Sheridan, G.K. REST: An epigenetic regulator of neuronal stress responses in the young and ageing brain. Front. Neuroendocrinol. 2019, 53, 100744. [Google Scholar] [CrossRef] [PubMed]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenet. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- El-Osta, A.; Brasacchio, D.; Yao, D.; Pocai, A.; Jones, P.L.; Roeder, R.G.; Cooper, M.E.; Brownlee, M. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J. Exp. Med. 2008, 205, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.; Plutzky, J.; El-Osta, A. Epigenetic Changes in Diabetes and Cardiovascular Risk. Circ. Res. 2016, 118, 1706–1722. [Google Scholar] [CrossRef]
- Lu, H.-C.; Dai, W.-N.; He, L.-Y. Epigenetic Histone Modifications in the Pathogenesis of Diabetic Kidney Disease. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 329–344. [Google Scholar] [CrossRef]
- Paneni, F.; Volpe, M.; Lüscher, T.F.; Cosentino, F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: Bringing all the strands together. Diabetes 2013, 62, 1800–1807. [Google Scholar] [CrossRef]
- Khan, A.W.; Paneni, F.; Jandeleit-Dahm, K.A. Cell-specific epigenetic changes in atherosclerosis. Clin. Sci. 2021, 135, 1165–1187. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Chiu, J.-J. Atherosclerosis and flow: Roles of epigenetic modulation in vascular endothelium. J. Biomed. Sci. 2019, 26, 56. [Google Scholar] [CrossRef]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, M.-Y. Cancer epigenetics: Past, present and future. Semin. Cancer Biol. 2022, 83, 4–14. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Esteller, M. Non-coding RNAs, epigenetics, and cancer: Tying it all together. Cancer Metastasis Rev. 2018, 37, 55–73. [Google Scholar] [CrossRef]
- Park, J.W.; Han, J.-W. Targeting epigenetics for cancer therapy. Arch. Pharmacal Res. 2019, 42, 159–170. [Google Scholar] [CrossRef]
- Wang, Y.-P.; Lei, Q.-Y. Metabolic recoding of epigenetics in cancer. Cancer Commun. 2018, 38, 25. [Google Scholar] [CrossRef]
- Mendoza-Mari, Y.; Valdés-Pérez, C.; Rodríguez-Corrales, E.; Suárez-Alba, J.; Garcia-Ojalvo, A.; Guillén-Nieto, G. Histological and transcriptional expression differences between diabetic foot and pressure ulcers. J. Diabetes Metab. 2013, 4, 296. [Google Scholar]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- Su, L.; Patti, M.E. Paternal Nongenetic Intergenerational Transmission of Metabolic Disease Risk. Curr. Diabetes Rep. 2019, 19, 38. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Fernández-Mayola, M.; Mendoza-Mari, Y.; Garcia-Ojalvo, A.; Playford, R.J.; Guillén-Nieto, G. Intralesional Infiltrations of Cell-Free Filtrates Derived from Human Diabetic Tissues Delay the Healing Process and Recreate Diabetes Histopathological Changes in Healthy Rats. Front. Clin. Diabetes Health 2021, 2, 617741. [Google Scholar] [CrossRef]
- Shahab, U.; Ahmad, M.K.; Mahdi, A.A.; Waseem, M.; Arif, B.; Moinuddin, A.S. The receptor for advanced glycation end products: A fuel to pancreatic cancer. Semin. Cancer Biol. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Akhter, F.; Khan, M.S.; Singh, S.; Ahmad, S. An immunohistochemical analysis to validate the rationale behind the enhanced immunogenicity of D-ribosylated low density lipo-protein. PLoS ONE 2014, 9, e113144. [Google Scholar] [CrossRef]
- Ahmad, S.; Akhter, F.; Shahab, U.; Rafi, Z.; Khan, M.S.; Nabi, R.; Khan, M.S.; Ahmad, K.; Ashraf, J.M.; Moinuddin. Do all roads lead to the Rome? The glycation perspective! Semin. Cancer Biol. 2018, 49, 9–19. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Fernandez-Mayola, M.; Mendoza-Mari, Y.; Garcia-Ojalvo, A.; Martinez-Jimenez, I.; Rodriguez-Rodriguez, N.; Playford, R.J.; Reyes-Acosta, O.; Lopez-Marin, L.; Guillén-Nieto, G. Intralesional Infiltrations of Arteriosclerotic Tissue Cells-Free Filtrate Reproduce Vascular Pathology in Healthy Recipient Rats. Int. J. Mol. Sci. 2022, 23, 1511. [Google Scholar] [CrossRef]
- Chevalier, J.; Yin, H.; Arpino, J.M.; O’Neil, C.; Nong, Z.; Gilmore, K.J.; Lee, J.J.; Prescott, E.; Hewak, M.; Rice, C.L.; et al. Obstruction of Small Arterioles in Patients with Critical Limb Ischemia due to Partial Endothelial-to-Mesenchymal Transition. iScience 2020, 23, 101251. [Google Scholar] [CrossRef]
- Karvinen, H.; Pasanen, E.; Rissanen, T.T.; Korpisalo, P.; Vahakangas, E.; Jazwa, A.; Giacca, M.; Yla-Herttuala, S. Long-term VEGF-A expression promotes aberrant angiogenesis and fibrosis in skeletal muscle. Gene Ther. 2011, 18, 1166–1172. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Mendoza-Mari, Y.; Martinez-Jimenez, I.; Suarez-Alba, J.; Rodriguez-Rodriguez, N.; Garcia Ojalvo, A.; Fernandez-Mayola, M.; Rosales, P.; Arteaga, E.; Duvergel-Calderin, D.; et al. Induction of Premalignant and Malignant Changes in Nude Mice by Human Tumors-Derived Cell-Free Filtrates. Ann. Hematol. Oncol. 2022, 9, 1387. [Google Scholar]
- Rous, P. Transmission of a malignant new growth by means of a cell-free filtrate. JAMA 1983, 250, 1445–1449. [Google Scholar] [CrossRef]
- Elemento, O. The road from Rous sarcoma virus to precision medicine. J. Exp. Med. 2021, 218, e20201754. [Google Scholar] [CrossRef] [PubMed]
- Berns, A. We Are Standing on the Shoulders of Giants. Cancer Res. 2016, 76, 4307–4308. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lipsick, J. A History of Cancer Research: Tumor Viruses. Cold Spring Harb. Perspect. Biol. 2021, 13, a035774. [Google Scholar] [CrossRef] [PubMed]
- Vasishta, S.; Umakanth, S.; Adiga, P.; Joshi, M.B. Extrinsic and intrinsic factors influencing metabolic memory in type 2 diabetes. Vasc. Pharmacol. 2021, 142, 106933. [Google Scholar] [CrossRef]
- Chawla, D.; Tripathi, A.K. Role of advanced glycation end products (AGEs) and its receptor (RAGE)-mediated diabetic vascular complications. Integr. Food Nutr. Metab. 2019, 6, 1–6. [Google Scholar] [CrossRef]
- Berlanga-Acosta, J.; Mendoza-Mari, Y.; Garcia-Ojalvo, A.; Fernandez-Mayola, M.; Guillén-Nieto, G. Epidermal Growth Factor Therapy Impact on Scar Tissue Resilience of Diabetic Lower Limbs Ulcers-An Enlightening Hypothesis. J. Diabetes Metab. 2021, 9, 1000798. [Google Scholar]
- Berlanga-Acosta, J.; Guillén-Nieto, G.; Rodriguez-Rodriguez, N.; Mendoza-Mari, Y.; Bringas-Vega, M.L.; Berlanga-Saez, J.O.; García del Barco Herrera, D.; Martinez-Jimenez, I.; Hernandez-Gutierrez, S.; Valdés-Sosa, P.A. Cellular Senescence as the Pathogenic Hub of Diabetes-Related Wound Chronicity. Front. Endocrinol. 2020, 11, 573032. [Google Scholar] [CrossRef]
- Kovacic, J.C.; Dimmeler, S.; Harvey, R.P.; Finkel, T.; Aikawa, E.; Krenning, G.; Baker, A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 190–209. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Patil, A.A.; Rhee, W.J. Exosomes: Biogenesis, Composition, Functions, and Their Role in Pre-metastatic Niche Formation. Biotechnol. Bioprocess Eng. 2019, 24, 689–701. [Google Scholar] [CrossRef]
- Shu, S.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic reprogramming of stromal fibroblasts by melanoma exosome microRNA favours a pre-metastatic microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef]
- Castano, C.; Novials, A.; Parrizas, M. Exosomes and diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3107. [Google Scholar] [CrossRef]
- Mosquera-Heredia, M.I.; Morales, L.C.; Vidal, O.M.; Barcelo, E.; Silvera-Redondo, C.; Velez, J.I.; Garavito-Galofre, P. Exosomes: Potential Disease Biomarkers and New Therapeutic Targets. Biomedicines 2021, 9, 1061. [Google Scholar] [CrossRef]
- Paskeh, M.D.A.; Entezari, M.; Mirzaei, S.; Zabolian, A.; Saleki, H.; Naghdi, M.J.; Sabet, S.; Khoshbakht, M.A.; Hashemi, M.; Hushmandi, K.; et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J. Hematol. Oncol. 2022, 15, 1–39. [Google Scholar] [CrossRef]
- Stefanius, K.; Servage, K.; Orth, K. Exosomes in cancer development. Curr. Opin. Genet. Dev. 2021, 66, 83–92. [Google Scholar] [CrossRef]
- Jawaid, A.; Jehle, K.-L.; Mansuy, I.M. Impact of Parental Exposure on Offspring Health in Humans. Trends Genet. 2020, 37, 373–388. [Google Scholar] [CrossRef]
- Ren, J.; Cheng, Y.; Ming, Z.H.; Dong, X.Y.; Zhou, Y.Z.; Ding, G.L.; Pang, H.Y.; Rahman, T.U.; Akbar, R.; Huang, H.F.; et al. Intrauterine hyperglycemia exposure results in intergenerational inheritance via DNA methylation reprogramming on F1 PGCs. Epigenet. Chromatin 2018, 11, 20. [Google Scholar] [CrossRef]
- Entringer, S.; De Punder, K.; Buss, C.; Wadhwa, P.D. The fetal programming of telomere biology hypothesis: An update. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170151. [Google Scholar] [CrossRef]
- Dhawan, S.; Natarajan, R. Epigenetics and Type 2 Diabetes Risk. Curr. Diabetes Rep. 2019, 19, 47. [Google Scholar] [CrossRef]
- Kereliuk, S.M.; Dolinsky, V.W. Recent Experimental Studies of Maternal Obesity, Diabetes during Pregnancy and the Developmental Origins of Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 4467. [Google Scholar] [CrossRef]
- Bianco, M.E.; Josefson, J.L. Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes. Curr. Diabetes Rep. 2019, 19, 143. [Google Scholar] [CrossRef] [PubMed]
- Rehan, V.K.; Liu, J.; Naeem, E.; Tian, J.; Sakurai, R.; Kwong, K.; Akbari, O.; Torday, J.S. Perinatal nicotine exposure induces asthma in second generation offspring. BMC Med. 2012, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Weber-Stadlbauer, U.; Richetto, J.; Labouesse, M.A.; Bohacek, J.; Mansuy, I.M.; Meyer, U. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol. Psychiatry 2017, 22, 102–112. [Google Scholar] [CrossRef]
- Lehrner, A.; Yehuda, R. Cultural trauma and epigenetic inheritance. Dev. Psychopathol. 2018, 30, 1763–1777. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berlanga-Acosta, J.; Fernandez-Mayola, M.; Mendoza-Mari, Y.; Garcia-Ojalvo, A.; Martinez-Jimenez, I.; Rodriguez-Rodriguez, N.; Garcia del Barco Herrera, D.; Guillén-Nieto, G. Cell-Free Filtrates (CFF) as Vectors of a Transmissible Pathologic Tissue Memory Code: A Hypothetical and Narrative Review. Int. J. Mol. Sci. 2022, 23, 11575. https://doi.org/10.3390/ijms231911575
Berlanga-Acosta J, Fernandez-Mayola M, Mendoza-Mari Y, Garcia-Ojalvo A, Martinez-Jimenez I, Rodriguez-Rodriguez N, Garcia del Barco Herrera D, Guillén-Nieto G. Cell-Free Filtrates (CFF) as Vectors of a Transmissible Pathologic Tissue Memory Code: A Hypothetical and Narrative Review. International Journal of Molecular Sciences. 2022; 23(19):11575. https://doi.org/10.3390/ijms231911575
Chicago/Turabian StyleBerlanga-Acosta, Jorge, Maday Fernandez-Mayola, Yssel Mendoza-Mari, Ariana Garcia-Ojalvo, Indira Martinez-Jimenez, Nadia Rodriguez-Rodriguez, Diana Garcia del Barco Herrera, and Gerardo Guillén-Nieto. 2022. "Cell-Free Filtrates (CFF) as Vectors of a Transmissible Pathologic Tissue Memory Code: A Hypothetical and Narrative Review" International Journal of Molecular Sciences 23, no. 19: 11575. https://doi.org/10.3390/ijms231911575
APA StyleBerlanga-Acosta, J., Fernandez-Mayola, M., Mendoza-Mari, Y., Garcia-Ojalvo, A., Martinez-Jimenez, I., Rodriguez-Rodriguez, N., Garcia del Barco Herrera, D., & Guillén-Nieto, G. (2022). Cell-Free Filtrates (CFF) as Vectors of a Transmissible Pathologic Tissue Memory Code: A Hypothetical and Narrative Review. International Journal of Molecular Sciences, 23(19), 11575. https://doi.org/10.3390/ijms231911575