

Comparative Metagenomics and Metabolomes Reveals Abnormal Metabolism Activity Is Associated with Gut Microbiota in Alzheimer’s Disease Mice

Abstract
1. Introduction
2. Results
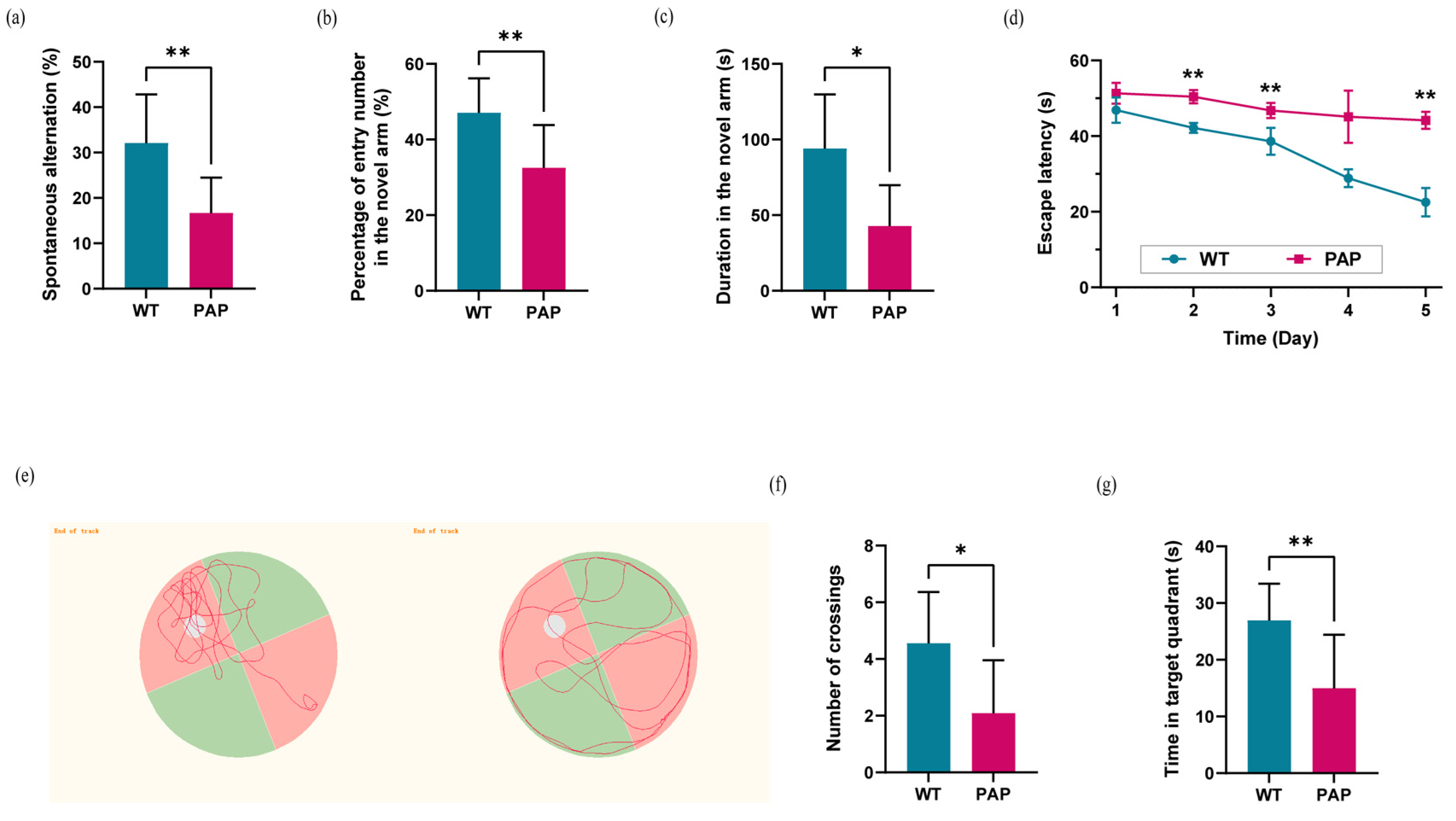
2.1. Evaluation of Learning and Memory Capability in PAP Mice
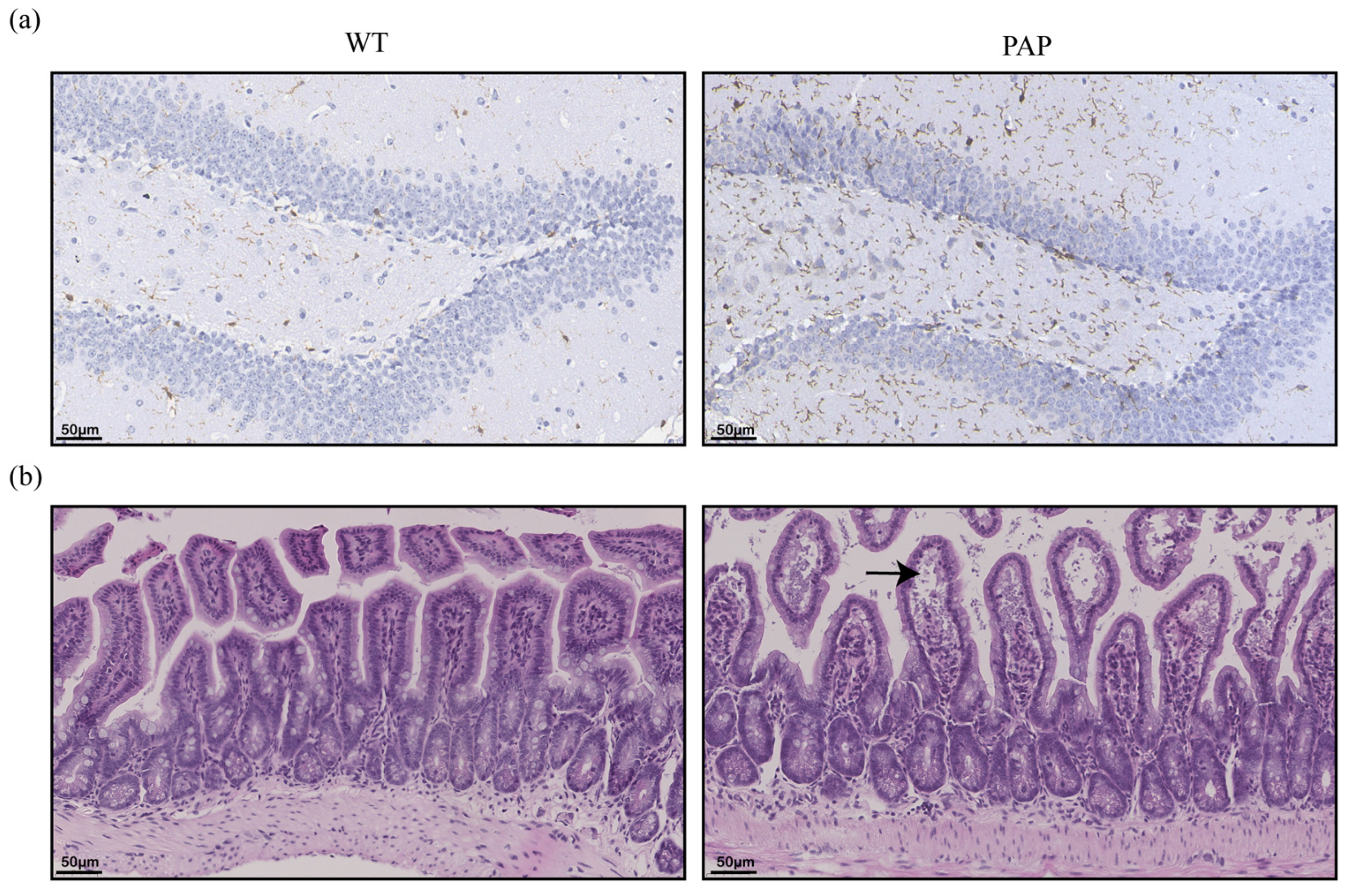
2.2. Pathological Changes of Brain and Intestinal Tissues in PAP Mice
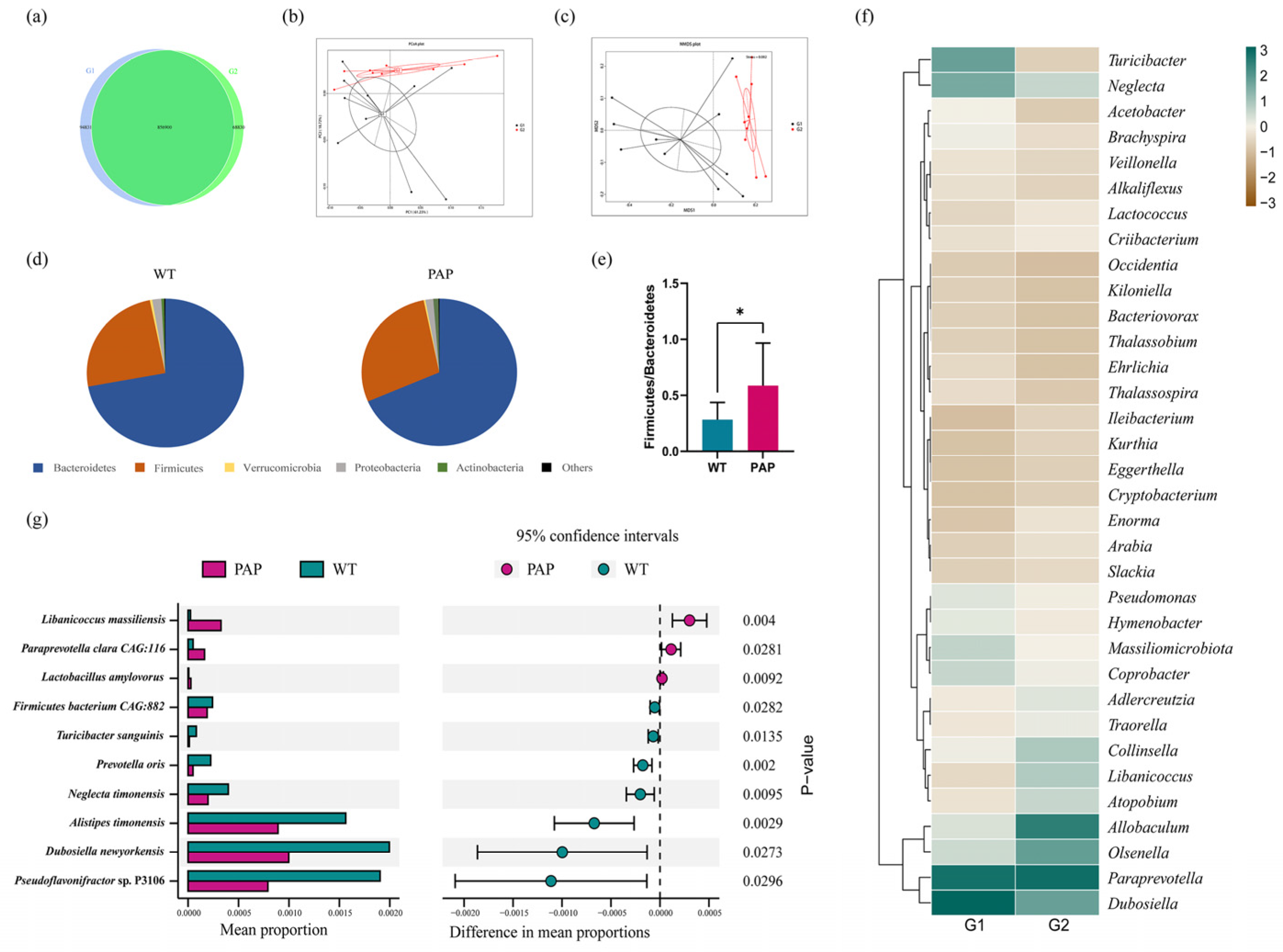
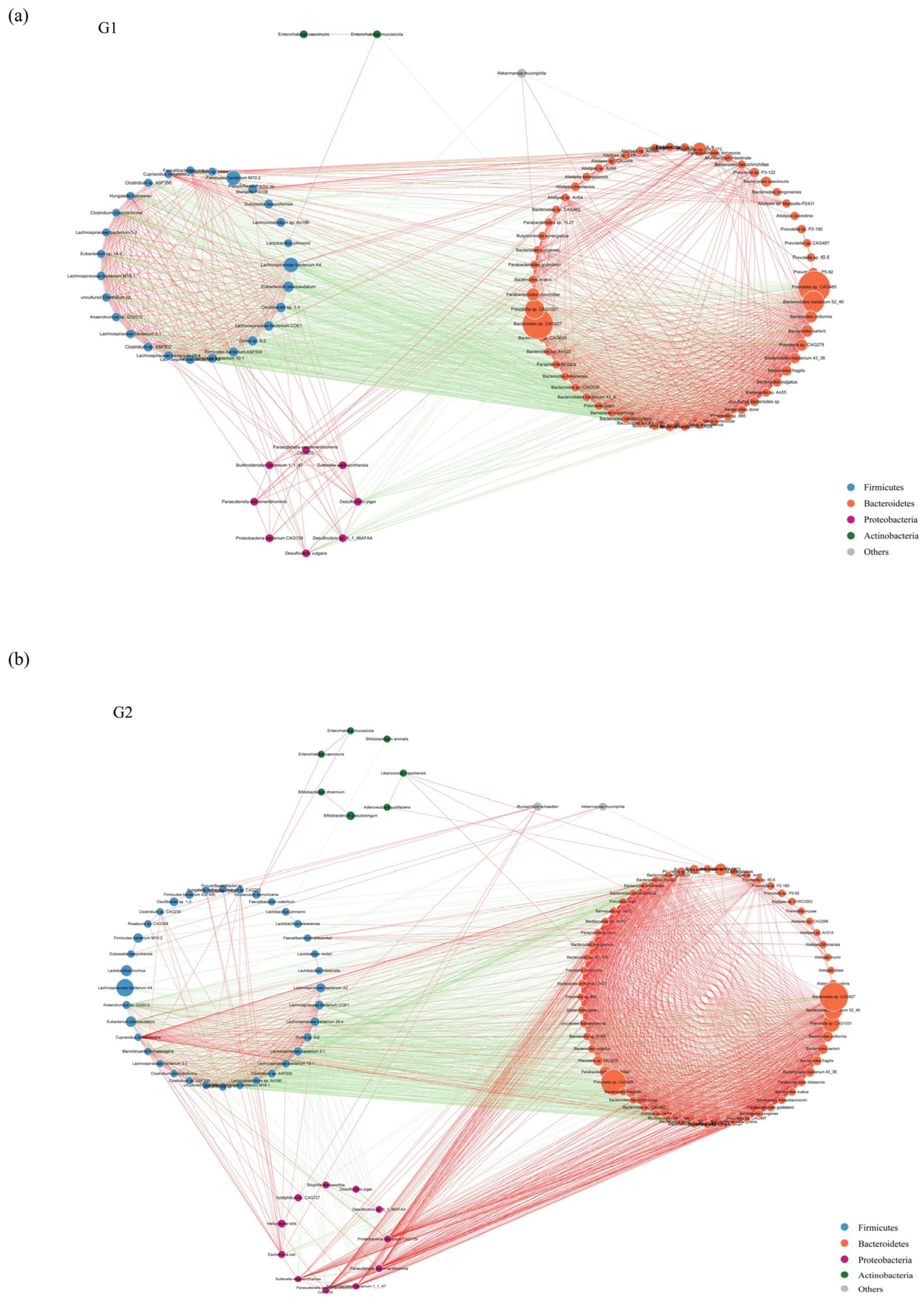
2.3. Metagenomic Sequencing Revealed Significant Differences of Gut Microbiota between PAP Mice and WT Mice
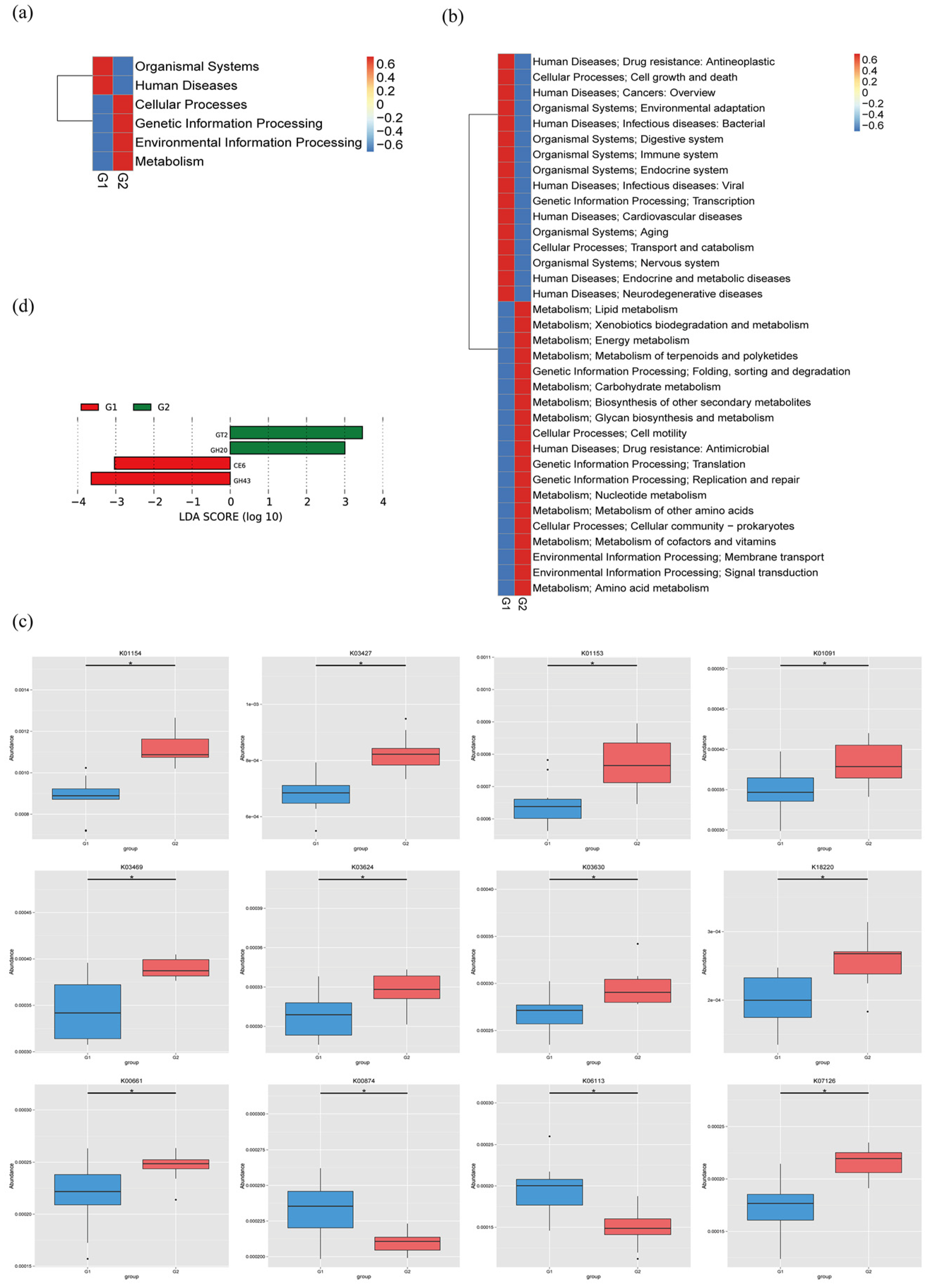
2.4. Functional Analysis of Metagenomic Sequencing Revealed Disrupted Bacteria Functions in PAP Mice
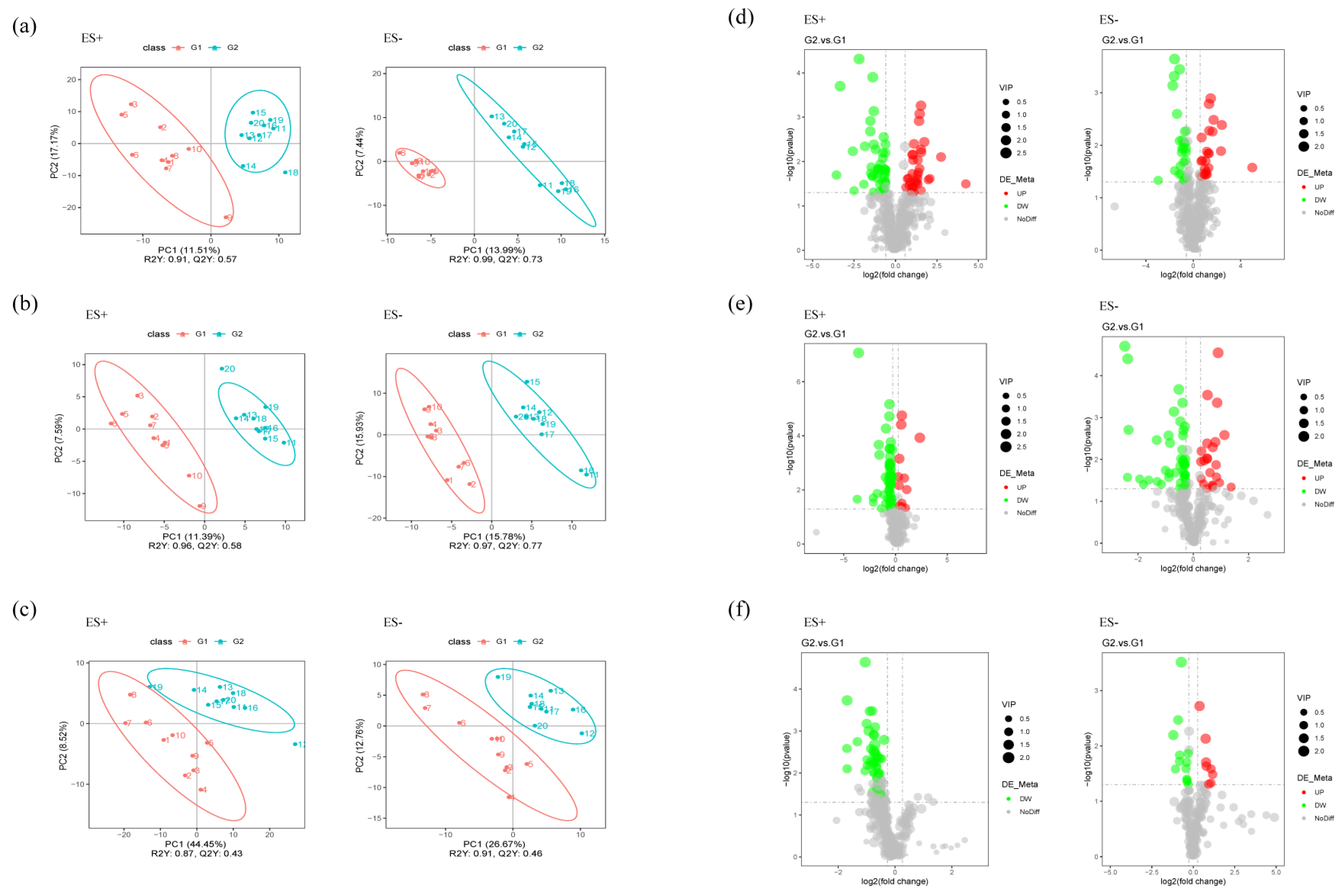
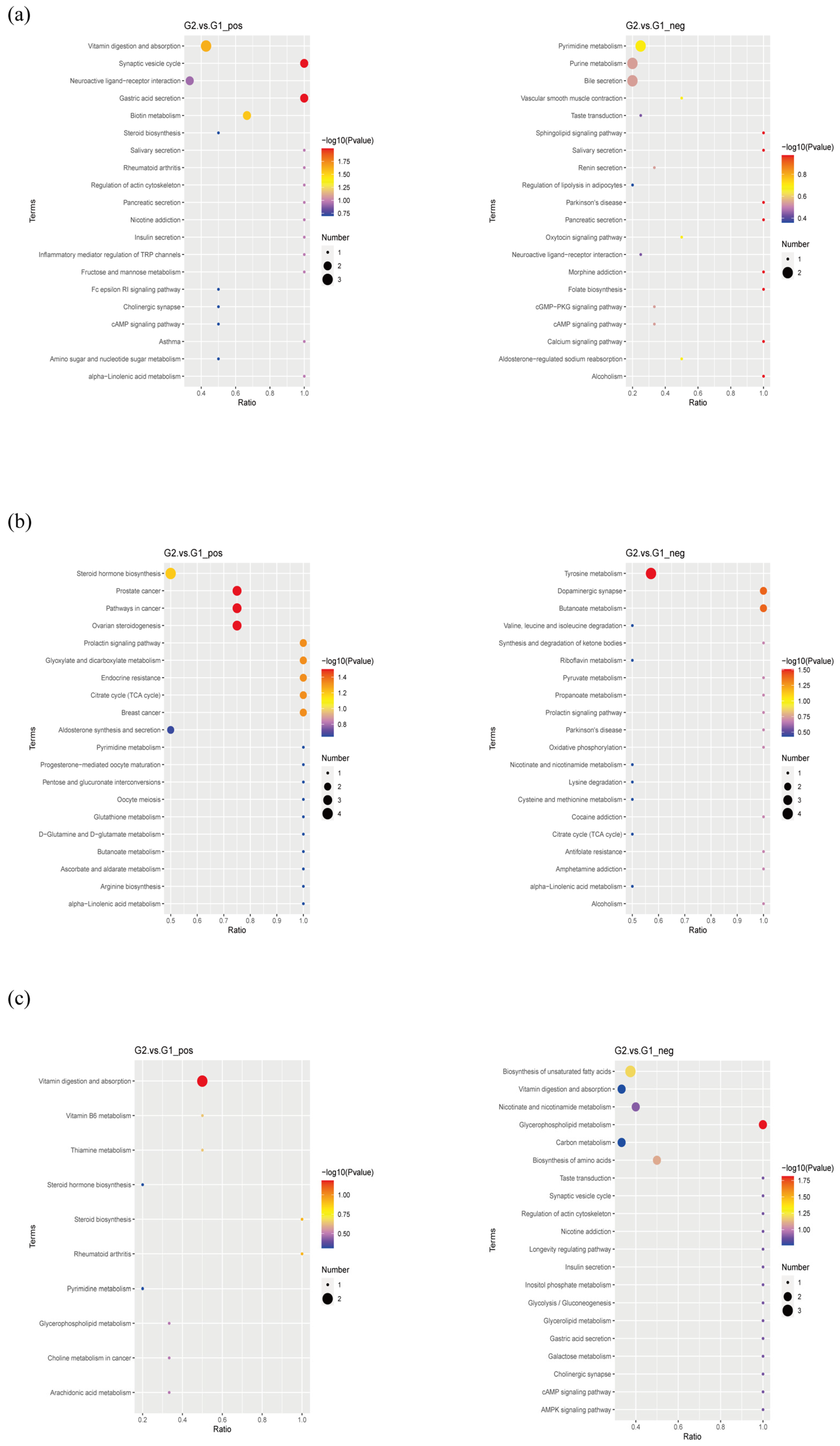
2.5. Metabolomics Analysis Revealed Aberrant Metabolic Patterns in PAP Mice
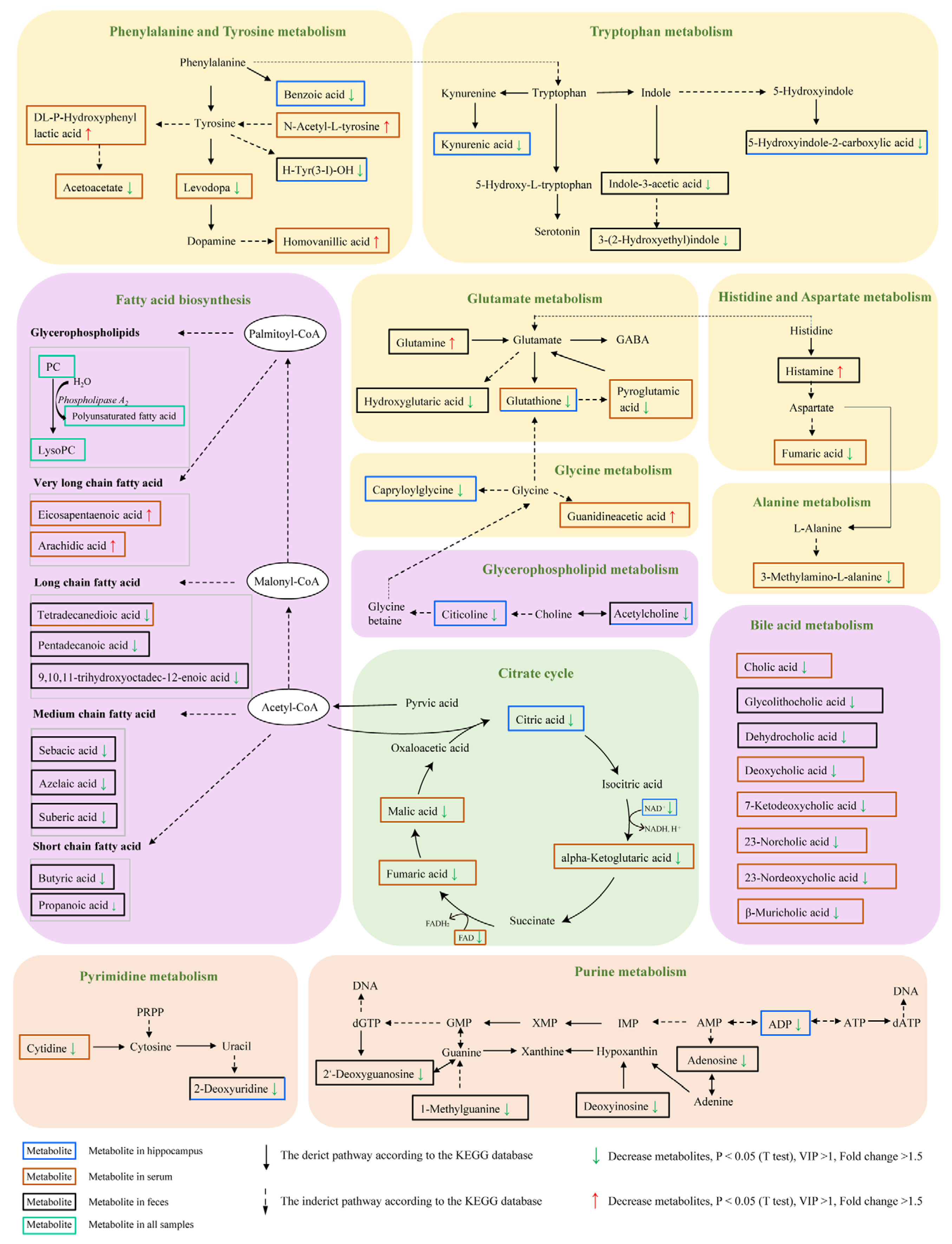
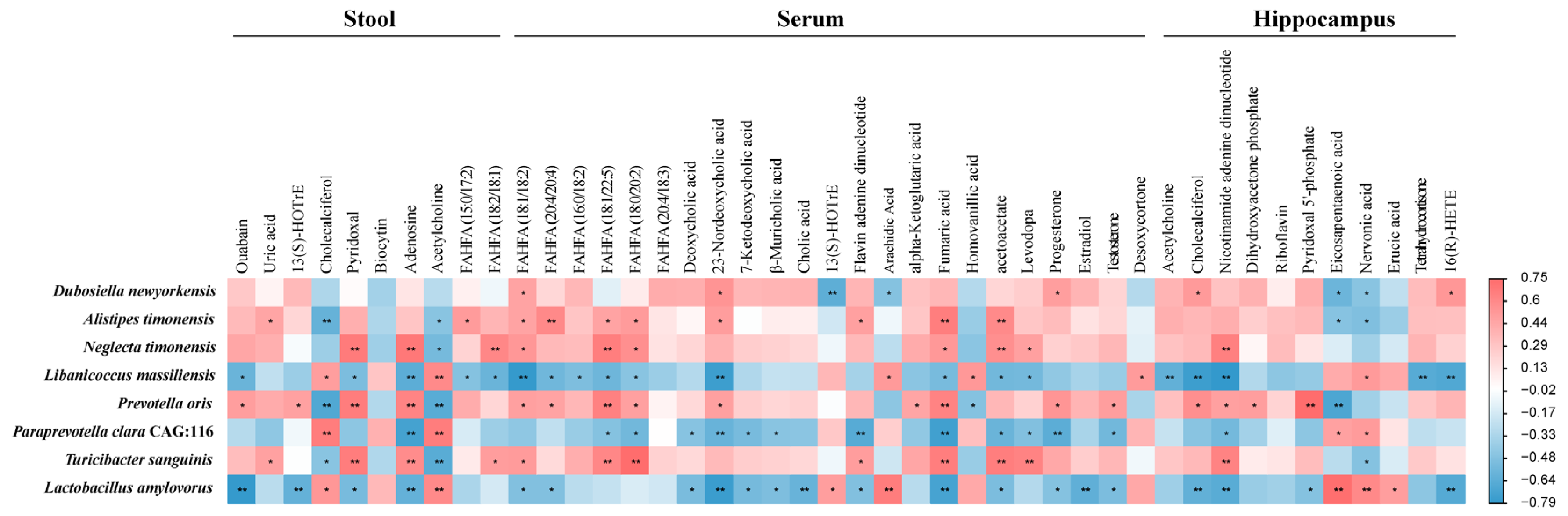
2.6. Microbiota–Host Metabolic Interaction
3. Discussion
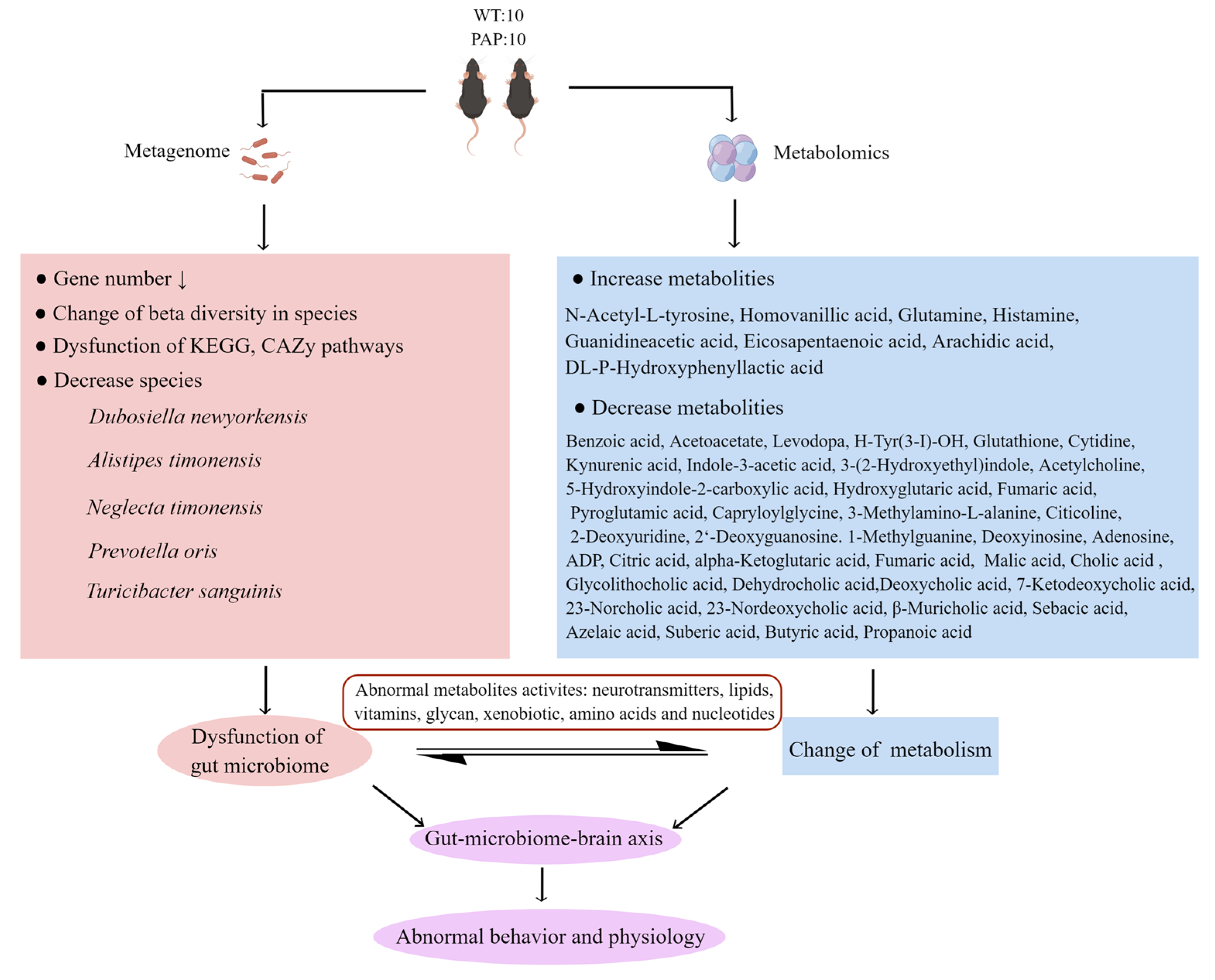
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Behavioral Tests
5.2.1. Y Maze
5.2.2. MWM Test
5.3. Morphological Examination
5.4. Sample Collection and Preparation
5.5. Metagenomic Analysis
5.6. Metabonomic Analysis Based on Liquid Chromatography-Mass Spectrometry (LC/MS)
5.7. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Podcasy, J.L.; Epperson, C.N. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 2016, 18, 437–446. [Google Scholar] [CrossRef]
- Yang, D.; Zhu, W.; Wang, Y.; Tan, F.; Ma, Z.; Gao, J.; Lin, X. Selection of mutant microplasmin for amyloid-beta cleavage in vivo. Sci. Rep. 2020, 10, 12117. [Google Scholar] [CrossRef]
- Bellia, F.; Lanza, V.; Garcia-Vinuales, S.; Ahmed, I.M.M.; Pietropaolo, A.; Iacobucci, C.; Malgieri, G.; D’Abrosca, G.; Fattorusso, R.; Nicoletti, V.G.; et al. Ubiquitin binds the amyloid beta peptide and interferes with its clearance pathways. Chem. Sci. 2019, 10, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wu, L.; Peng, G.; Han, Y.; Tang, R.; Ge, J.; Zhang, L.; Jia, L.; Yue, S.; Zhou, K.; et al. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav. Immun. 2019, 80, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fak, F.; Jucker, M.; Lasser, T.; et al. Erratum: Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 46856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.F.; Huang, L.; et al. Altered Gut Microbiota in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, H.; Guo, Y.; Du, X.; Qin, C. Gut microbiota regulate cognitive deficits and amyloid deposition in a model of Alzheimer’s disease. J. Neurochem. 2020, 155, 448–461. [Google Scholar] [CrossRef]
- Koh, A.; Backhed, F. From Association to Causality: The Role of the Gut Microbiota and Its Functional Products on Host Metabolism. Mol. Cell 2020, 78, 584–596. [Google Scholar] [CrossRef]
- Ansoleaga, B.; Jove, M.; Schluter, A.; Garcia-Esparcia, P.; Moreno, J.; Pujol, A.; Pamplona, R.; Portero-Otin, M.; Ferrer, I. Deregulation of purine metabolism in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 68–80. [Google Scholar] [CrossRef]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Vitorica, J.; Gomez-Ariza, J.L. Metabolomic screening of regional brain alterations in the APP/PS1 transgenic model of Alzheimer’s disease by direct infusion mass spectrometry. J. Pharm. Biomed. Anal. 2015, 102, 425–435. [Google Scholar] [CrossRef]
- Lin, S.; Liu, H.; Kanawati, B.; Liu, L.; Dong, J.; Li, M.; Huang, J.; Schmitt-Kopplin, P.; Cai, Z. Hippocampal metabolomics using ultrahigh-resolution mass spectrometry reveals neuroinflammation from Alzheimer’s disease in CRND8 mice. Anal. Bioanal. Chem. 2013, 405, 5105–5117. [Google Scholar] [CrossRef]
- Luan, H.; Wang, X.; Cai, Z. Mass spectrometry-based metabolomics: Targeting the crosstalk between gut microbiota and brain in neurodegenerative disorders. Mass Spectrom. Rev. 2019, 38, 22–33. [Google Scholar] [CrossRef]
- Zhang, F.; Shi, J.S.; Zhou, H.; Wilson, B.; Hong, J.S.; Gao, H.M. Resveratrol protects dopamine neurons against lipopolysaccharide-induced neurotoxicity through its anti-inflammatory actions. Mol. Pharmacol. 2010, 78, 466–477. [Google Scholar] [CrossRef]
- Treangen, T.J.; Wagner, J.; Burns, M.P.; Villapol, S. Traumatic Brain Injury in Mice Induces Acute Bacterial Dysbiosis Within the Fecal Microbiome. Front. Immunol. 2018, 9, 2757. [Google Scholar] [CrossRef]
- Antonissen, G.; Croubels, S.; Pasmans, F.; Ducatelle, R.; Eeckhaut, V.; Devreese, M.; Verlinden, M.; Haesebrouck, F.; Eeckhout, M.; De Saeger, S.; et al. Fumonisins affect the intestinal microbial homeostasis in broiler chickens, predisposing to necrotic enteritis. Vet. Res. 2015, 46, 98. [Google Scholar] [CrossRef]
- Li, Y.; Ning, L.; Yin, Y.; Wang, R.; Zhang, Z.; Hao, L.; Wang, B.; Zhao, X.; Yang, X.; Yin, L.; et al. Age-related shifts in gut microbiota contribute to cognitive decline in aged rats. Aging 2020, 12, 7801–7817. [Google Scholar] [CrossRef]
- Wu, M.L.; Yang, X.Q.; Xue, L.; Duan, W.; Du, J.R. Age-related cognitive decline is associated with microbiota-gut-brain axis disorders and neuroinflammation in mice. Behav. Brain Res. 2021, 402, 113125. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef]
- Yang, L.; Cui, Y.; Liang, H.; Li, Z.; Wang, N.; Wang, Y.; Zheng, G. Multifunctional Selenium Nanoparticles with Different Surface Modifications Ameliorate Neuroinflammation through the Gut Microbiota-NLRP3 Inflammasome-Brain Axis in APP/PS1 Mice. ACS Appl. Mater Interfaces 2022, 14, 30557–30570. [Google Scholar] [CrossRef]
- New, F.N.; Brito, I.L. What Is Metagenomics Teaching Us, and What Is Missed? Annu. Rev. Microbiol. 2020, 74, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Licht, T.R.; Madsen, B.; Wilcks, A. Selection of bacteria originating from a human intestinal microbiota in the gut of previously germ-free rats. FEMS Microbiol. Lett. 2007, 277, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Daza, M.C.; Roquim, M.; Dudonne, S.; Pilon, G.; Levy, E.; Marette, A.; Roy, D.; Desjardins, Y. Berry Polyphenols and Fibers Modulate Distinct Microbial Metabolic Functions and Gut Microbiota Enterotype-Like Clustering in Obese Mice. Front. Microbiol. 2020, 11, 2032. [Google Scholar] [CrossRef]
- Finamore, A.; Roselli, M.; Imbinto, A.; Seeboth, J.; Oswald, I.P.; Mengheri, E. Lactobacillus amylovorus inhibits the TLR4 inflammatory signaling triggered by enterotoxigenic Escherichia coli via modulation of the negative regulators and involvement of TLR2 in intestinal Caco-2 cells and pig explants. PLoS ONE 2014, 9, e94891. [Google Scholar] [CrossRef]
- Huo, Z.; Yu, L.; Yang, J.; Zhu, Y.; Bennett, D.A.; Zhao, J. Brain and blood metabolome for Alzheimer’s dementia: Findings from a targeted metabolomics analysis. Neurobiol. Aging 2020, 86, 123–133. [Google Scholar] [CrossRef]
- Hurtado, M.O.; Kohler, I.; de Lange, E.C. Next-generation biomarker discovery in Alzheimer’s disease using metabolomics—From animal to human studies. Bioanalysis 2018, 10, 1525–1546. [Google Scholar] [CrossRef]
- Whiley, L.; Sen, A.; Heaton, J.; Proitsi, P.; Garcia-Gomez, D.; Leung, R.; Smith, N.; Thambisetty, M.; Kloszewska, I.; Mecocci, P.; et al. Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 271–278. [Google Scholar] [CrossRef]
- Simpson, B.N.; Kim, M.; Chuang, Y.F.; Beason-Held, L.; Kitner-Triolo, M.; Kraut, M.; Lirette, S.T.; Windham, B.G.; Griswold, M.E.; Legido-Quigley, C.; et al. Blood metabolite markers of cognitive performance and brain function in aging. J. Cereb. Blood Flow Metab. 2016, 36, 1212–1223. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, R.; Wang, W.; Qi, L.; Huang, T. Associations between gut microbiota and Alzheimer’s disease, major depressive disorder, and schizophrenia. J. Neuroinflamm. 2020, 17, 288. [Google Scholar] [CrossRef]
- Iaccarino, L.; Sala, A.; Caminiti, S.P.; Presotto, L.; Perani, D.; Alzheimer’s Disease Neuroimaging, I. In vivo MRI Structural and PET Metabolic Connectivity Study of Dopamine Pathways in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 75, 1003–1016. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef]
- Hatano, T.; Ohnuma, T.; Sakai, Y.; Shibata, N.; Maeshima, H.; Hanzawa, R.; Suzuki, T.; Arai, H. Plasma alanine levels increase in patients with schizophrenia as their clinical symptoms improve-Results from the Juntendo University Schizophrenia Projects (JUSP). Psychiatry Res. 2010, 177, 27–31. [Google Scholar] [CrossRef]
- Tsai, G.E.; Yang, P.; Chang, Y.C.; Chong, M.Y. D-alanine added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 2006, 59, 230–234. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J. Alzheimers Dis. 2020, 78, 683–697. [Google Scholar] [CrossRef]
- Doifode, T.; Giridharan, V.V.; Generoso, J.S.; Bhatti, G.; Collodel, A.; Schulz, P.E.; Forlenza, O.V.; Barichello, T. The impact of the microbiota-gut-brain axis on Alzheimer’s disease pathophysiology. Pharmacol. Res. 2021, 164, 105314. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, C.; Wu, J.; Tao, J.J.; Sui, X.L.; Yao, Z.G.; Xu, Y.F.; Huang, L.; Zhu, H.; Sheng, S.L.; et al. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J. Alzheimers Dis. 2014, 41, 1193–1205. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Gorska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Barri, T.; Dragsted, L.O. UPLC-ESI-QTOF/MS and multivariate data analysis for blood plasma and serum metabolomics: Effect of experimental artefacts and anticoagulant. Anal. Chim. Acta 2013, 768, 118–128. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr. Metabolomics 2013, 1, 92–107. [Google Scholar] [CrossRef]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Date | Clean Date | GC(%) | Effective (%) |
|---|---|---|---|---|
| WT1 | 6248.17 | 6238.41 | 47.17 | 99.844 |
| WT2 | 6055.95 | 6048.28 | 47.76 | 99.873 |
| WT3 | 6852.46 | 6840.39 | 45.97 | 99.824 |
| WT4 | 6040.52 | 6031.72 | 45.10 | 99.854 |
| WT5 | 6205.41 | 6196.22 | 44.73 | 99.852 |
| WT6 | 6419.97 | 6411.56 | 48.30 | 99.869 |
| WT7 | 6037.43 | 6027.86 | 49.06 | 99.841 |
| WT8 | 6528.44 | 6521.04 | 49.18 | 99.887 |
| WT9 | 6666.75 | 6649.82 | 48.47 | 99.746 |
| WT10 | 6504.01 | 6489.21 | 47.29 | 99.772 |
| PAP1 | 6182.81 | 6167.85 | 47.14 | 99.758 |
| PAP2 | 5993.42 | 5982.55 | 48.96 | 99.819 |
| PAP3 | 6212.57 | 6199.63 | 48.05 | 99.792 |
| PAP4 | 6600.03 | 6574.39 | 47.45 | 99.612 |
| PAP5 | 6887.99 | 6871.95 | 47.65 | 99.767 |
| PAP6 | 6285.34 | 6270.28 | 48.46 | 99.760 |
| PAP7 | 6665.45 | 6651.59 | 48.60 | 99.792 |
| PAP8 | 6831.09 | 6814.69 | 45.81 | 99.760 |
| PAP9 | 6142.11 | 6124.22 | 48.05 | 99.709 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, P.; Zhu, H.; Li, X.; Shi, W.; Guo, Y.; Du, X.; Zhang, L.; Su, L.; Qin, C. Comparative Metagenomics and Metabolomes Reveals Abnormal Metabolism Activity Is Associated with Gut Microbiota in Alzheimer’s Disease Mice. Int. J. Mol. Sci. 2022, 23, 11560. https://doi.org/10.3390/ijms231911560
Sun P, Zhu H, Li X, Shi W, Guo Y, Du X, Zhang L, Su L, Qin C. Comparative Metagenomics and Metabolomes Reveals Abnormal Metabolism Activity Is Associated with Gut Microbiota in Alzheimer’s Disease Mice. International Journal of Molecular Sciences. 2022; 23(19):11560. https://doi.org/10.3390/ijms231911560
Chicago/Turabian StyleSun, Peilin, Hua Zhu, Xue Li, Weixiong Shi, Yaxi Guo, Xiaopeng Du, Ling Zhang, Lei Su, and Chuan Qin. 2022. "Comparative Metagenomics and Metabolomes Reveals Abnormal Metabolism Activity Is Associated with Gut Microbiota in Alzheimer’s Disease Mice" International Journal of Molecular Sciences 23, no. 19: 11560. https://doi.org/10.3390/ijms231911560
APA StyleSun, P., Zhu, H., Li, X., Shi, W., Guo, Y., Du, X., Zhang, L., Su, L., & Qin, C. (2022). Comparative Metagenomics and Metabolomes Reveals Abnormal Metabolism Activity Is Associated with Gut Microbiota in Alzheimer’s Disease Mice. International Journal of Molecular Sciences, 23(19), 11560. https://doi.org/10.3390/ijms231911560