
The Translatability of Multiple Sclerosis Animal Models for Biomarkers Discovery and Their Clinical Use


Abstract
1. Introduction
2. Major Pathological Hallmarks of MS
2.1. Inflammation
2.1.1. Chemokine (C-X-C Motif) Ligand 13 (CXCL13)
2.1.2. Osteopontin (OPN)
2.1.3. Interleukin-17 (IL-17)
2.2. Blood–Brain Barrier Breakdown
2.2.1. Metalloproteinase-2 and -9 (MMP2 & MMP9)
2.2.2. A Disintegrin and Metalloproteinase with a Thrombospondin Type 1 Motif, Member 13 (ADAMTS13)
2.3. Astrogliosis
2.3.1. Glial Fibrillary Acidic Protein (GFAP)
2.3.2. S100 Calcium Binding Protein B (S100B)
2.4. Myelin and Axonal Damage
2.4.1. Myelin and Oligodendrocyte Glycoprotein (MOG)
2.4.2. Myelin Basic Protein (MBP)
2.4.3. Neurofilament
2.5. Repair
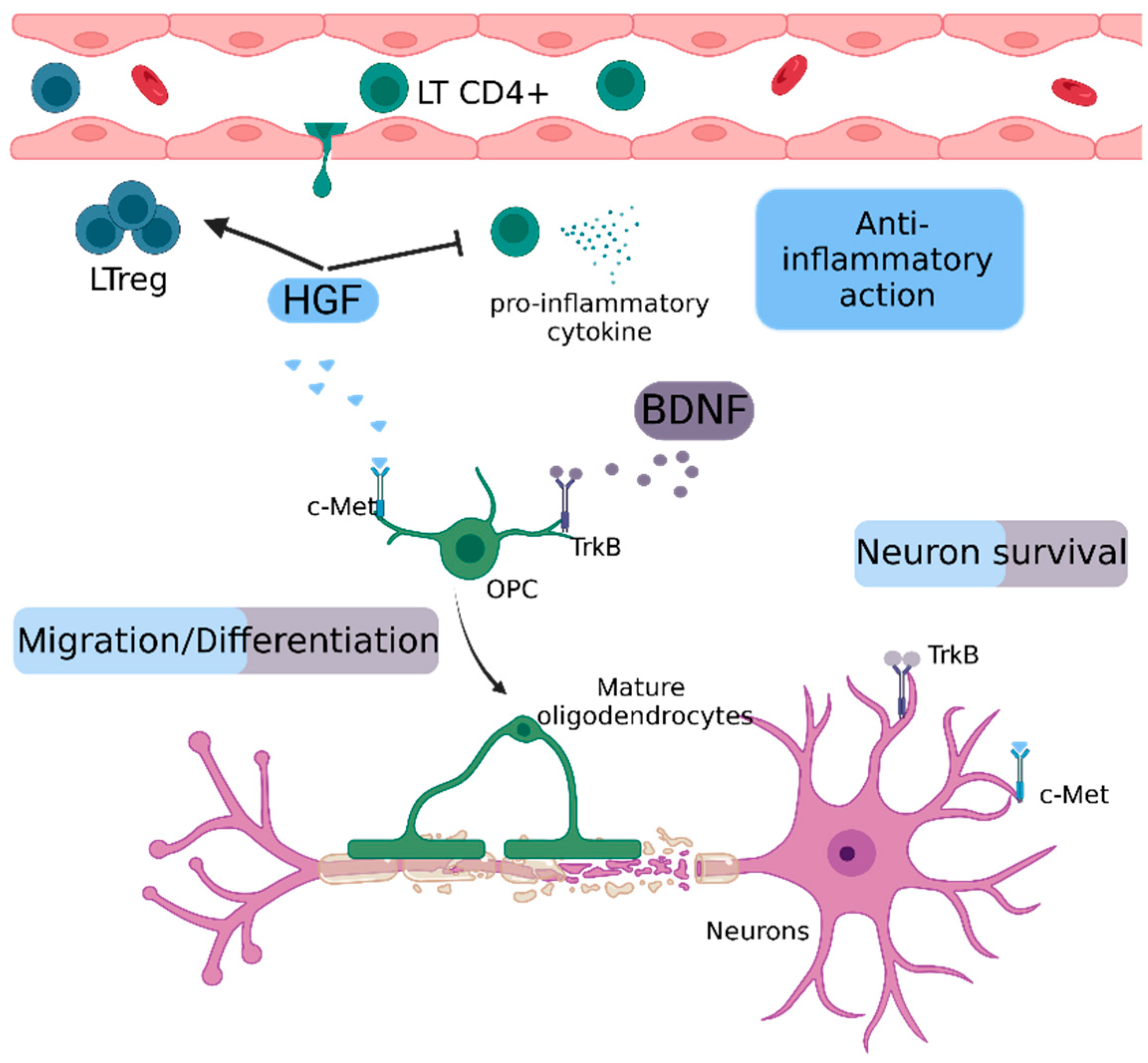
2.5.1. Hepatocyte Growth Factor (HGF)
2.5.2. Brain-Derived Neurotrophic Factor (BDNF)
3. Biomarkers Routinely Used in MS
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of Multiple Sclerosis Lesions: Implications for the Pathogenesis of Demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular Biomarkers in Multiple Sclerosis. J. Neuroinflammation 2019, 16, 272. [Google Scholar] [CrossRef] [PubMed]
- Gafson, A.; Craner, M.J.; Matthews, P.M. Personalised Medicine for Multiple Sclerosis Care. Mult. Scler. Houndmills Basingstoke Engl. 2017, 23, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Steiner, I. Experimental Allergic Encephalomyelitis: A Misleading Model of Multiple Sclerosis. Ann. Neurol. 2005, 58, 939–945. [Google Scholar] [CrossRef]
- Lassmann, H.; Bradl, M. Multiple Sclerosis: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef]
- Linker, R.A.; Brechlin, P.; Jesse, S.; Steinacker, P.; Lee, D.H.; Asif, A.R.; Jahn, O.; Tumani, H.; Gold, R.; Otto, M. Proteome Profiling in Murine Models of Multiple Sclerosis: Identification of Stage Specific Markers and Culprits for Tissue Damage. PLoS ONE 2009, 4, e7624. [Google Scholar] [CrossRef]
- Haase, S.; Linker, R.A. Inflammation in Multiple Sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007688. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS): EAE as Model for MS. Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Förster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J.G. A Chemokine-Driven Positive Feedback Loop Organizes Lymphoid Follicles. Nature 2000, 406, 309–314. [Google Scholar] [CrossRef]
- Kowarik, M.C.; Cepok, S.; Sellner, J.; Grummel, V.; Weber, M.S.; Korn, T.; Berthele, A.; Hemmer, B. CXCL13 Is the Major Determinant for B Cell Recruitment to the CSF during Neuroinflammation. J. Neuroinflammation 2012, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Gomez-Rivera, F.; Raphael, R.A.; Robinson, R.R.; Nalawade, S.; Forsthuber, T.G. TNFR2 Limits Proinflammatory Astrocyte Functions during EAE Induced by Pathogenic DR2b-Restricted T Cells. JCI Insight 2019, 4, e132527. [Google Scholar] [CrossRef] [PubMed]
- Irani, D.N. Regulated Production of CXCL13 within the Central Nervous System. J. Clin. Cell. Immunol. 2016, 7, 460. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, M.; Theil, D.; Cepok, S.; Hemmer, B.; Kivisäkk, P.; Ransohoff, R.M.; Hofbauer, M.; Farina, C.; Derfuss, T.; Hartle, C.; et al. Chemokines in Multiple Sclerosis: CXCL12 and CXCL13 up-Regulation Is Differentially Linked to CNS Immune Cell Recruitment. Brain 2006, 129, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Bagaeva, L.V.; Rao, P.; Powers, J.M.; Segal, B.M. CXC Chemokine Ligand 13 Plays a Role in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2006, 176, 7676–7685. [Google Scholar] [CrossRef]
- Rainey-Barger, E.K.; Rumble, J.M.; Lalor, S.J.; Esen, N.; Segal, B.M.; Irani, D.N. The Lymphoid Chemokine, CXCL13, Is Dispensable for the Initial Recruitment of B Cells to the Acutely Inflamed Central Nervous System. Brain Behav. Immun. 2011, 25, 922–931. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, D.; Wang, L.; Zhao, Y.; Liu, T.; Yu, Y.; Yan, T.; Cheng, Y. Cerebrospinal Fluid and Blood Cytokines as Biomarkers for Multiple Sclerosis: A Systematic Review and Meta-Analysis of 226 Studies With 13,526 Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 1026. [Google Scholar] [CrossRef]
- Harris, V.K.; Sadiq, S.A. Biomarkers of Therapeutic Response in Multiple Sclerosis: Current Status. Mol. Diagn. Ther. 2014, 18, 605–617. [Google Scholar] [CrossRef]
- Alvarez, E.; Piccio, L.; Mikesell, R.J.; Klawiter, E.C.; Parks, B.J.; Naismith, R.T.; Cross, A.H. CXCL13 Is a Biomarker of Inflammation in Multiple Sclerosis, Neuromyelitis Optica, and Other Neurological Conditions. Mult. Scler. Houndmills Basingstoke Engl. 2013, 19, 1204–1208. [Google Scholar] [CrossRef]
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The Role of Osteopontin in Inflammatory Processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef]
- Denhardt, D.T.; Noda, M.; O’Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a Means to Cope with Environmental Insults: Regulation of Inflammation, Tissue Remodeling, and Cell Survival. J. Clin. Investig. 2001, 107, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, K.H.; Voulgari, P.V.; Tsianos, E.V. Inflammatory Bowel Disease and Lupus: A Systematic Review of the Literature. J. Crohns Colitis 2012, 6, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Wirestam, L.; Enocsson, H.; Skogh, T.; Padyukov, L.; Jönsen, A.; Urowitz, M.B.; Gladman, D.D.; Romero-Diaz, J.; Bae, S.-C.; Fortin, P.R.; et al. Osteopontin and Disease Activity in Patients with Recent-Onset Systemic Lupus Erythematosus: Results from the SLICC Inception Cohort. J. Rheumatol. 2019, 46, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Chabas, D.; Baranzini, S.E.; Mitchell, D.; Bernard, C.C.A.; Rittling, S.R.; Denhardt, D.T.; Sobel, R.A.; Lock, C.; Karpuj, M.; Pedotti, R.; et al. The Influence of the Proinflammatory Cytokine, Osteopontin, on Autoimmune Demyelinating Disease. Science 2001, 294, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Clemente, N.; Comi, C.; Raineri, D.; Cappellano, G.; Vecchio, D.; Orilieri, E.; Gigliotti, C.L.; Boggio, E.; Dianzani, C.; Sorosina, M.; et al. Role of Anti-Osteopontin Antibodies in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2017, 8, 321. [Google Scholar] [CrossRef]
- Comabella, M.; Pericot, I.; Goertsches, R.; Nos, C.; Castillo, M.; Blas Navarro, J.; Río, J.; Montalban, X. Plasma Osteopontin Levels in Multiple Sclerosis. J. Neuroimmunol. 2005, 158, 231–239. [Google Scholar] [CrossRef]
- Börnsen, L.; Khademi, M.; Olsson, T.; Sørensen, P.S.; Sellebjerg, F. Osteopontin Concentrations Are Increased in Cerebrospinal Fluid during Attacks of Multiple Sclerosis. Mult. Scler. J. 2011, 17, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Agah, E.; Zardoui, A.; Saghazadeh, A.; Ahmadi, M.; Tafakhori, A.; Rezaei, N. Osteopontin (OPN) as a CSF and Blood Biomarker for Multiple Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2018, 13, e0190252. [Google Scholar] [CrossRef]
- Szalardy, L.; Zadori, D.; Simu, M.; Bencsik, K.; Vecsei, L.; Klivenyi, P. Evaluating Biomarkers of Neuronal Degeneration and Neuroinflammation in CSF of Patients with Multiple Sclerosis–Osteopontin as a Potential Marker of Clinical Severity. J. Neurol. Sci. 2013, 331, 38–42. [Google Scholar] [CrossRef]
- Jäger, A.; Kuchroo, V.K. Effector and Regulatory T Cell Subsets in Autoimmunity and Tissue Inflammation. Scand. J. Immunol. 2010, 72, 173–184. [Google Scholar] [CrossRef]
- Zhang, G.-X.; Gran, B.; Yu, S.; Li, J.; Siglienti, I.; Chen, X.; Kamoun, M.; Rostami, A. Induction of Experimental Autoimmune Encephalomyelitis in IL-12 Receptor-Β2-Deficient Mice: IL-12 Responsiveness Is Not Required in the Pathogenesis of Inflammatory Demyelination in the Central Nervous System. J. Immunol. 2003, 170, 2153–2160. [Google Scholar] [CrossRef] [PubMed]
- Sosa, R.A.; Murphey, C.; Robinson, R.R.; Forsthuber, T.G. IFN-γ Ameliorates Autoimmune Encephalomyelitis by Limiting Myelin Lipid Peroxidation. Proc. Natl. Acad. Sci. USA 2015, 112, E5038–E5047. [Google Scholar] [CrossRef] [PubMed]
- Arellano, G.; Ottum, P.A.; Reyes, L.I.; Burgos, P.I.; Naves, R. Stage-Specific Role of Interferon-Gamma in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front. Immunol. 2015, 6, 492. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 Cell, the New Player of Neuroinflammatory Process in Multiple Sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.; Akgün, K.; Proschmann, U.; Sellner, J.; Ziemssen, T. The Role of TH17 Cells in Multiple Sclerosis: Therapeutic Implications. Autoimmun. Rev. 2020, 19, 102647. [Google Scholar] [CrossRef] [PubMed]
- Basdeo, S.A.; Cluxton, D.; Sulaimani, J.; Moran, B.; Canavan, M.; Orr, C.; Veale, D.J.; Fearon, U.; Fletcher, J.M. Ex-Th17 (Nonclassical Th1) Cells Are Functionally Distinct from Classical Th1 and Th17 Cells and Are Not Constrained by Regulatory T Cells. J. Immunol. 2017, 198, 2249–2259. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Friese, M.A.; Craner, M.J.; Palace, J.; Newcombe, J.; Esiri, M.M.; Fugger, L. Interleukin-17 Production in Central Nervous System-Infiltrating T Cells and Glial Cells Is Associated with Active Disease in Multiple Sclerosis. Am. J. Pathol. 2008, 172, 146–155. [Google Scholar] [CrossRef]
- Zimmermann, J.; Emrich, M.; Krauthausen, M.; Saxe, S.; Nitsch, L.; Heneka, M.T.; Campbell, I.L.; Müller, M. IL-17A Promotes Granulocyte Infiltration, Myelin Loss, Microglia Activation, and Behavioral Deficits During Cuprizone-Induced Demyelination. Mol. Neurobiol. 2018, 55, 946–957. [Google Scholar] [CrossRef]
- Sarma, J.D.; Ciric, B.; Marek, R.; Sadhukhan, S.; Caruso, M.L.; Shafagh, J.; Fitzgerald, D.C.; Shindler, K.S.; Rostami, A. Functional Interleukin-17 Receptor A Is Expressed in Central Nervous System Glia and Upregulated in Experimental Autoimmune Encephalomyelitis. J. Neuroinflammation 2009, 6, 14. [Google Scholar] [CrossRef]
- Uyttenhove, C.; Van Snick, J. Development of an Anti-IL-17A Auto-Vaccine That Prevents Experimental Auto-Immune Encephalomyelitis. Eur. J. Immunol. 2006, 36, 2868–2874. [Google Scholar] [CrossRef]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-Microarray Analysis of Multiple Sclerosis Lesions Yields New Targets Validated in Autoimmune Encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Matusevicius, D.; Kivisäkk, P.; He, B.; Kostulas, N.; Özenci, V.; Fredrikson, S.; Link, H. Interleukin-17 MRNA Expression in Blood and CSF Mononuclear Cells Is Augmented in Multiple Sclerosis. Mult. Scler. J. 1999, 5, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, A.F.; Abbas, A.R.; Jeet, S.; Wong, K.; Bischof, A.; Peng, I.; Lee, J.; Bremer, M.; Eggers, E.L.; DeVoss, J.; et al. IL-17A Is Associated with the Breakdown of the Blood-Brain Barrier in Relapsing-Remitting Multiple Sclerosis. J. Neuroimmunol. 2019, 332, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, Y.; Nakae, S.; Matsuki, T.; Nambu, A.; Ishigame, H.; Kakuta, S.; Sudo, K.; Iwakura, Y. IL-17 Plays an Important Role in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2006, 177, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Rahman, M.T.; Ghosh, C.; Hossain, M.; Linfield, D.; Rezaee, F.; Janigro, D.; Marchi, N.; van Boxel-Dezaire, A.H.H. IFN-γ, IL-17A, or Zonulin Rapidly Increase the Permeability of the Blood-Brain and Small Intestinal Epithelial Barriers: Relevance for Neuro-Inflammatory Diseases. Biochem. Biophys. Res. Commun. 2018, 507, 274–279. [Google Scholar] [CrossRef]
- Sie, C.; Korn, T.; Mitsdoerffer, M. Th17 Cells in Central Nervous System Autoimmunity. Exp. Neurol. 2014, 262, 18–27. [Google Scholar] [CrossRef]
- Havrdová, E.; Belova, A.; Goloborodko, A.; Tisserant, A.; Wright, A.; Wallstroem, E.; Garren, H.; Maguire, R.P.; Johns, D.R. Activity of Secukinumab, an Anti-IL-17A Antibody, on Brain Lesions in RRMS: Results from a Randomized, Proof-of-Concept Study. J. Neurol. 2016, 263, 1287–1295. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.Á.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the Blood–Brain Barrier in Multiple Sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Tallantyre, E.C.; Dixon, J.E.; Donaldson, I.; Owens, T.; Morgan, P.S.; Morris, P.G.; Evangelou, N. Ultra-High-Field Imaging Distinguishes MS Lesions from Asymptomatic White Matter Lesions. Neurology 2011, 76, 534–539. [Google Scholar] [CrossRef]
- Maggi, P.; Absinta, M.; Grammatico, M.; Vuolo, L.; Emmi, G.; Carlucci, G.; Spagni, G.; Barilaro, A.; Repice, A.M.; Emmi, L.; et al. Central Vein Sign Differentiates Multiple Sclerosis from Central Nervous System Inflammatory Vasculopathies. Ann. Neurol. 2018, 83, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Chandler, S.; Miller, K.M.; Clements, J.M.; Lury, J.; Corkill, D.; Anthony, D.C.; Adams, S.E.; Gearing, A.J. Matrix Metalloproteinases, Tumor Necrosis Factor and Multiple Sclerosis: An Overview. J. Neuroimmunol. 1997, 72, 155–161. [Google Scholar] [CrossRef]
- Benešová, Y.; Vašků, A.; Novotná, H.; Litzman, J.; Štourač, P.; Beránek, M.; Kadaňka, Z.; Bednařík, J. Matrix Metalloproteinase-9 and Matrix Metalloproteinase-2 as Biomarkers of Various Courses in Multiple Sclerosis. Mult. Scler. J. 2009, 15, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Leppert, D.; Ford, J.; Stabler, G.; Grygar, C.; Lienert, C.; Huber, S.; Miller, K.M.; Hauser, S.L.; Kappos, L. Matrix Metalloproteinase-9 (Gelatinase B) Is Selectively Elevated in CSF during Relapses and Stable Phases of Multiple Sclerosis. Brain J. Neurol. 1998, 121 Pt 12, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E.; Goodkin, D.E.; Gee, L.; Bacchetti, P.; Sloan, R.; Stewart, T.; Andersson, P.B.; Stabler, G.; Miller, K. Serum MMP-9 and TIMP-1 Levels Are Related to MRI Activity in Relapsing Multiple Sclerosis. Neurology 1999, 53, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W.; Zabad, R.K.; Agrawal, S.; Goncalves DaSilva, A.; Metz, L.M. Elevation of Matrix Metalloproteinases (MMPs) in Multiple Sclerosis and Impact of Immunomodulators. J. Neurol. Sci. 2007, 259, 79–84. [Google Scholar] [CrossRef]
- Kieseier, B.C.; Kiefer, R.; Clements, J.M.; Miller, K.; Wells, G.M.; Schweitzer, T.; Gearing, A.J.; Hartung, H.P. Matrix Metalloproteinase-9 and -7 Are Regulated in Experimental Autoimmune Encephalomyelitis. Brain 1998, 121, 159–166. [Google Scholar] [CrossRef]
- Onwuha-Ekpete, L.C.; Tokmina-Roszyk, D.; Fields, G.B. Selective Inhibition of Matrix Metalloproteinase-9 Reduces Clinical Severity in a Murine Model of Multiple Sclerosis via a Reduced Immune Response. J. Immunol. 2016, 196, 139.12. [Google Scholar]
- Sang, Q.-X.; Muroski, M.; Roycik, M.; Newcomer, R.; Van den Steen, P.; Opdenakker, G.; Monroe, H.; Sahab, Z. Matrix Metalloproteinase-9/Gelatinase B Is a Putative Therapeutic Target of Chronic Obstructive Pulmonary Disease and Multiple Sclerosis. Curr. Pharm. Biotechnol. 2008, 9, 34–46. [Google Scholar] [CrossRef]
- Agrawal, S.; Anderson, P.; Durbeej, M.; van Rooijen, N.; Ivars, F.; Opdenakker, G.; Sorokin, L.M. Dystroglycan Is Selectively Cleaved at the Parenchymal Basement Membrane at Sites of Leukocyte Extravasation in Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 2006, 203, 1007–1019. [Google Scholar] [CrossRef]
- Waubant, E.; Goodkin, D.; Bostrom, A.; Bacchetti, P.; Hietpas, J.; Lindberg, R.; Leppert, D. IFN Lowers MMP-9/TIMP-1 Ratio, Which Predicts New Enhancing Lesions in Patients with SPMS. Neurology 2003, 60, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Avolio, C.; Ruggieri, M.; Giuliani, F.; Liuzzi, G.M.; Leante, R.; Riccio, P.; Livrea, P.; Trojano, M. Serum MMP-2 and MMP-9 Are Elevated in Different Multiple Sclerosis Subtypes. J. Neuroimmunol. 2003, 136, 46–53. [Google Scholar] [CrossRef]
- Castellazzi, M.; Ligi, D.; Contaldi, E.; Quartana, D.; Fonderico, M.; Borgatti, L.; Bellini, T.; Trentini, A.; Granieri, E.; Fainardi, E.; et al. Multiplex Matrix Metalloproteinases Analysis in the Cerebrospinal Fluid Reveals Potential Specific Patterns in Multiple Sclerosis Patients. Front. Neurol. 2018, 9, 1080. [Google Scholar] [CrossRef] [PubMed]
- Fainardi, E.; Castellazzi, M.; Bellini, T.; Manfrinato, M.C.; Baldi, E.; Casetta, I.; Paolino, E.; Granieri, E.; Dallocchio, F. Cerebrospinal Fluid and Serum Levels and Intrathecal Production of Active Matrix Metalloproteinase-9 (MMP-9) as Markers of Disease Activity in Patients with Multiple Sclerosis. Mult. Scler. J. 2006, 12, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Yong, V.W. Differential Mechanisms of Action of Interferon-Beta and Glatiramer Aetate in MS. Neurology 2002, 59, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, I.; Martens, E.; Van Den Steen, P.E.; Proost, P.; Ronsse, I.; Opdenakker, G. Gelatinase B/Matrix Metalloproteinase-9 Cleaves Interferon-β and Is a Target for Immunotherapy. Brain 2003, 126, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S.; Schwendimann, R.N.; Kelley, R.E.; Gonzalez-Toledo, E.; Jimenez, J.J.; Mauro, L.; Jy, W.; Smith, S.J. Combination Therapy with Interferon Beta-1a and Doxycycline in Multiple Sclerosis: An Open-Label Trial. Arch. Neurol. 2008, 65, 199–204. [Google Scholar] [CrossRef]
- Donadelli, R.; Orje, J.N.; Capoferri, C.; Remuzzi, G.; Ruggeri, Z.M. Size Regulation of von Willebrand Factor–Mediated Platelet Thrombi by ADAMTS13 in Flowing Blood. Blood 2006, 107, 1943–1950. [Google Scholar] [CrossRef]
- Cai, P.; Luo, H.; Xu, H.; Zhu, X.; Xu, W.; Dai, Y.; Xiao, J.; Cao, Y.; Zhao, Y.; Zhao, B.-Q.; et al. Recombinant ADAMTS 13 Attenuates Brain Injury After Intracerebral Hemorrhage. Stroke 2015, 46, 2647–2653. [Google Scholar] [CrossRef]
- Ziliotto, N.; Bernardi, F.; Jakimovski, D.; Baroni, M.; Marchetti, G.; Bergsland, N.; Ramasamy, D.P.; Weinstock-Guttman, B.; Schweser, F.; Zamboni, P.; et al. Hemostasis Biomarkers in Multiple Sclerosis. Eur. J. Neurol. 2018, 25, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Stolz, L.; Derouiche, A.; Devraj, K.; Weber, F.; Brunkhorst, R.; Foerch, C. Anticoagulation with Warfarin and Rivaroxaban Ameliorates Experimental Autoimmune Encephalomyelitis. J. Neuroinflammation 2017, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Liu, L.; Xu, X.; Zhao, F.; Deng, J.; Tang, X.; Wang, X.; Zhao, B.-Q.; Zhang, X.; Zhao, Y. ADAMTS13 Ameliorates Inflammatory Responses in Experimental Autoimmune Encephalomyelitis. J. Neuroinflammation 2020, 17, 67. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Perez-Catalan, N.A.; Doe, C.Q.; Ackerman, S.D. The Role of Astrocyte-Mediated Plasticity in Neural Circuit Development and Function. Neural Dev. 2021, 16, 1. [Google Scholar] [CrossRef]
- Pérez-Alvarez, A.; Araque, A. Astrocyte-Neuron Interaction at Tripartite Synapses. Curr. Drug Targets 2013, 14, 1220–1224. [Google Scholar] [CrossRef]
- Garden, G.A.; Campbell, B.M. Glial Biomarkers in Human Central Nervous System Disease. Glia 2016, 64, 1755–1771. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Reactive Gliosis in the Pathogenesis of CNS Diseases. Biochim. Biophys. Acta 2016, 1862, 483–491. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180. [Google Scholar] [CrossRef]
- Matute, C.; Alberdi, E.; Domercq, M.; Sánchez-Gómez, M.-V.; Pérez-Samartín, A.; Rodríguez-Antigüedad, A.; Pérez-Cerdá, F. Excitotoxic Damage to White Matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef]
- Middeldorp, J.; Hol, E.M. GFAP in Health and Disease. Prog. Neurobiol. 2011, 93, 421–443. [Google Scholar] [CrossRef]
- Diaz-Arrastia, R.; Wang, K.K.W.; Papa, L.; Sorani, M.D.; Yue, J.K.; Puccio, A.M.; McMahon, P.J.; Inoue, T.; Yuh, E.L.; Lingsma, H.F.; et al. Acute Biomarkers of Traumatic Brain Injury: Relationship between Plasma Levels of Ubiquitin C-Terminal Hydrolase-L1 and Glial Fibrillary Acidic Protein. J. Neurotrauma 2014, 31, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Jukkola, P.; Guerrero, T.; Gray, V.; Gu, C. Astrocytes Differentially Respond to Inflammatory Autoimmune Insults and Imbalances of Neural Activity. Acta Neuropathol. Commun. 2013, 1, 70. [Google Scholar] [CrossRef] [PubMed]
- Voskuhl, R.R.; Peterson, R.S.; Song, B.; Ao, Y.; Morales, L.B.J.; Tiwari-Woodruff, S.; Sofroniew, M.V. Reactive Astrocytes Form Scar-like Perivascular Barriers to Leukocytes during Adaptive Immune Inflammation of the CNS. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 11511–11522. [Google Scholar] [CrossRef]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of Astrocyte Activation by Glycolipids Drives Chronic CNS Inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Eikelenboom, M.J.; Gveric, D.; Keir, G.; Chapman, M.; Lazeron, R.H.C.; Cuzner, M.L.; Polman, C.H.; Uitdehaag, B.M.J.; Thompson, E.J.; et al. Markers for Different Glial Cell Responses in Multiple Sclerosis: Clinical and Pathological Correlations. Brain 2002, 125, 1462–1473. [Google Scholar] [CrossRef]
- Momtazmanesh, S.; Shobeiri, P.; Saghazadeh, A.; Teunissen, C.E.; Burman, J.; Szalardy, L.; Klivenyi, P.; Bartos, A.; Fernandes, A.; Rezaei, N. Neuronal and Glial CSF Biomarkers in Multiple Sclerosis: A Systematic Review and Meta-Analysis. Rev. Neurosci. 2021, 32, 573–595. [Google Scholar] [CrossRef]
- Abdelhak, A.; Huss, A.; Kassubek, J.; Tumani, H.; Otto, M. Serum GFAP as a Biomarker for Disease Severity in Multiple Sclerosis. Sci. Rep. 2018, 8, 14798. [Google Scholar] [CrossRef]
- Michetti, F.; Corvino, V.; Geloso, M.C.; Lattanzi, W.; Bernardini, C.; Serpero, L.; Gazzolo, D. The S100B Protein in Biological Fluids: More than a Lifelong Biomarker of Brain Distress. J. Neurochem. 2012, 120, 644–659. [Google Scholar] [CrossRef]
- Barateiro, A.; Afonso, V.; Santos, G.; Cerqueira, J.J.; Brites, D.; van Horssen, J.; Fernandes, A. S100B as a Potential Biomarker and Therapeutic Target in Multiple Sclerosis. Mol. Neurobiol. 2016, 53, 3976–3991. [Google Scholar] [CrossRef]
- Santos, G.; Barateiro, A.; Gomes, C.M.; Brites, D.; Fernandes, A. Impaired Oligodendrogenesis and Myelination by Elevated S100B Levels during Neurodevelopment. Neuropharmacology 2018, 129, 69–83. [Google Scholar] [CrossRef]
- Yan, S.S.; Wu, Z.-Y.; Zhang, H.P.; Furtado, G.; Chen, X.; Yan, S.F.; Schmidt, A.M.; Brown, C.; Stern, A.; LaFaille, J.; et al. Suppression of Experimental Autoimmune Encephalomyelitis by Selective Blockade of Encephalitogenic T-Cell Infiltration of the Central Nervous System. Nat. Med. 2003, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.E.; Mok, T.; Sweeney, B.; Ryan, A.M.; Dev, K.K. The Use of Cytokine Signature Patterns: Separating Drug Naïve, Interferon and Natalizumab-Treated Multiple Sclerosis Patients. Autoimmunity 2014, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Di Sante, G.; Amadio, S.; Sampaolese, B.; Clementi, M.E.; Valentini, M.; Volonté, C.; Casalbore, P.; Ria, F.; Michetti, F. The S100B Inhibitor Pentamidine Ameliorates Clinical Score and Neuropathology of Relapsing—Remitting Multiple Sclerosis Mouse Model. Cells 2020, 9, 748. [Google Scholar] [CrossRef] [PubMed]
- Traugott, U.; Reinherz, E.L.; Raine, C.S. Multiple Sclerosis: Distribution of T Cell Subsets within Active Chronic Lesions. Science 1983, 219, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, Y.; Jin, S.; Wang, J.; Kawanokuchi, J.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Chronological Changes of CD4+ and CD8+ T Cell Subsets in the Experimental Autoimmune Encephalomyelitis, a Mouse Model of Multiple Sclerosis. Tohoku J. Exp. Med. 2007, 213, 329–339. [Google Scholar] [CrossRef][Green Version]
- Bevan, M.J. Helping the CD8+ T-Cell Response. Nat. Rev. Immunol. 2004, 4, 595–602. [Google Scholar] [CrossRef]
- Pitt, D.; Werner, P.; Raine, C.S. Glutamate Excitotoxicity in a Model of Multiple Sclerosis. Nat. Med. 2000, 6, 67–70. [Google Scholar] [CrossRef]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.M.; Linington, C. Differential Ultrastructural Localization of Myelin Basic Protein, Myelin/Oligodendroglial Glycoprotein, and 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase in the CNS of Adult Rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef]
- Matthieu, J.M.; Amiguet, P. Myelin/Oligodendrocyte Glycoprotein Expression during Development in Normal and Myelin-Deficient Mice. Dev. Neurosci. 1990, 12, 293–302. [Google Scholar] [CrossRef]
- Solly, S.K.; Thomas, J.L.; Monge, M.; Demerens, C.; Lubetzki, C.; Gardinier, M.V.; Matthieu, J.M.; Zalc, B. Myelin/Oligodendrocyte Glycoprotein (MOG) Expression Is Associated with Myelin Deposition. Glia 1996, 18, 39–48. [Google Scholar] [CrossRef]
- Peschl, P.; Bradl, M.; Höftberger, R.; Berger, T.; Reindl, M. Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front. Immunol. 2017, 8, 529. [Google Scholar] [CrossRef] [PubMed]
- Lebar, R.; Lubetzki, C.; Vincent, C. The M2 Autoantigen of Central Nervous System Myelin, a Glycoprotein Present in Oligodendrocyte Membrane. Clin. Exp. Immunol. 1986, 66, 423. [Google Scholar] [PubMed]
- Schluesener, H.J.; Sobel, R.A.; Linington, C.; Weiner, H.L. A Monoclonal Antibody against a Myelin Oligodendrocyte Glycoprotein Induces Relapses and Demyelination in Central Nervous System Autoimmune Disease. J. Immunol. 1987, 139, 4016–4021. [Google Scholar] [PubMed]
- Linington, C.; Lassmann, H. Antibody Responses in Chronic Relapsing Experimental Allergic Encephalomyelitis: Correlation of Serum Demyelinating Activity with Antibody Titre to the Myelin/Oligodendrocyte Glycoprotein (MOG). J. Neuroimmunol. 1987, 17, 61–69. [Google Scholar] [CrossRef]
- Ichikawa, M.; Johns, T.G.; Liu, J.; Bernard, C.C. Analysis of the Fine B Cell Specificity during the Chronic/Relapsing Course of a Multiple Sclerosis-like Disease in Lewis Rats Injected with the Encephalitogenic Myelin Oligodendrocyte Glycoprotein Peptide 35–55. J. Immunol. 1996, 157, 919–926. [Google Scholar]
- Bittner, S.; Afzali, A.M.; Wiendl, H.; Meuth, S.G. Myelin Oligodendrocyte Glycoprotein (MOG35-55) Induced Experimental Autoimmune Encephalomyelitis (EAE) in C57BL/6 Mice. J. Vis. Exp. JoVE 2014, 86, 51275. [Google Scholar] [CrossRef]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin Antibodies as a Predictor of Clinically Definite Multiple Sclerosis after a First Demyelinating Event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef]
- Kuhle, J.; Pohl, C.; Mehling, M.; Edan, G.; Freedman, M.S.; Hartung, H.-P.; Polman, C.H.; Miller, D.H.; Montalban, X.; Barkhof, F.; et al. Lack of Association between Antimyelin Antibodies and Progression to Multiple Sclerosis. N. Engl. J. Med. 2007, 356, 371–378. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Stellmann, J.P.; Huss, A.; Ayzenberg, I.; Willing, A.; Trebst, C.; Pawlitzki, M.; Abdelhak, A.; Grüter, T.; et al. MOG-IgG in Primary and Secondary Chronic Progressive Multiple Sclerosis: A Multicenter Study of 200 Patients and Review of the Literature. J. Neuroinflammation 2018, 15, 88. [Google Scholar] [CrossRef]
- Cobo-Calvo, Á.; d’Indy, H.; Ruiz, A.; Collongues, N.; Kremer, L.; Durand-Dubief, F.; Rollot, F.; Casey, R.; Vukusic, S.; Seze, J.D.; et al. Frequency of Myelin Oligodendrocyte Glycoprotein Antibody in Multiple Sclerosis: A Multicenter Cross-Sectional Study. Neurol.—Neuroimmunol. Neuroinflammation 2020, 7, e649. [Google Scholar] [CrossRef]
- Kitley, J.; Woodhall, M.; Waters, P.; Leite, M.I.; Devenney, E.; Craig, J.; Palace, J.; Vincent, A. Myelin-Oligodendrocyte Glycoprotein Antibodies in Adults with a Neuromyelitis Optica Phenotype. Neurology 2012, 79, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Sato, D.K.; Callegaro, D.; Lana-Peixoto, M.A.; Waters, P.J.; de Haidar Jorge, F.M.; Takahashi, T.; Nakashima, I.; Apostolos-Pereira, S.L.; Talim, N.; Simm, R.F.; et al. Distinction between MOG Antibody-Positive and AQP4 Antibody-Positive NMO Spectrum Disorders. Neurology 2014, 82, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Armangue, T.; Olivé-Cirera, G.; Martínez-Hernandez, E.; Sepulveda, M.; Ruiz-Garcia, R.; Muñoz-Batista, M.; Ariño, H.; González-Álvarez, V.; Felipe-Rucián, A.; Martínez-González, M.J.; et al. Associations of Paediatric Demyelinating and Encephalitic Syndromes with Myelin Oligodendrocyte Glycoprotein Antibodies: A Multicentre Observational Study. Lancet Neurol. 2020, 19, 234–246. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of Tissue-Specific Cell Death Using Methylation Patterns of Circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.A.; Kenna, L.A.; Tipon, R.C.; Spelios, M.G.; Stecker, M.M.; Akirav, E.M. A Minimally-Invasive Blood-Derived Biomarker of Oligodendrocyte Cell-Loss in Multiple Sclerosis. eBioMedicine 2016, 10, 227–235. [Google Scholar] [CrossRef][Green Version]
- Braun, P.E.; Pereyra, P.M.; Greenfield, S. Myelin Organization and Development: A Biochemical Perspective. Prog. Clin. Biol. Res. 1980, 49, 1–17. [Google Scholar]
- Readhead, C.; Hood, L. The Dysmyelinating Mouse Mutations Shiverer (Shi) and Myelin Deficient (Shimld). Behav. Genet. 1990, 20, 213–234. [Google Scholar] [CrossRef]
- Takahashi, N.; Roach, A.; Teplow, D.B.; Prusiner, S.B.; Hood, L. Cloning and Characterization of the Myelin Basic Protein Gene from Mouse: One Gene Can Encode Both 14 Kd and 18.5 Kd MBPs by Alternate Use of Exons. Cell 1985, 42, 139–148. [Google Scholar] [CrossRef]
- Delaney, K.H.; Kwiecien, J.M.; Wegiel, J.; Wisniewski, H.M.; Percy, D.H.; Fletch, A.L. Familial Dysmyelination in a Long Evans Rat Mutant. Lab. Anim. Sci. 1995, 45, 547–553. [Google Scholar]
- Van Haren, K.; Tomooka, B.H.; Kidd, B.A.; Banwell, B.; Bar-Or, A.; Chitnis, T.; Tenembaum, S.N.; Pohl, D.; Rostasy, K.; Dale, R.C.; et al. Serum Autoantibodies to Myelin Peptides Distinguish Acute Disseminated Encephalomyelitis from Relapsing-Remitting Multiple Sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2013, 19, 1726–1733. [Google Scholar] [CrossRef]
- Lamers, K.J.; Uitdehaag, B.M.; Hommes, O.R.; Doesburg, W.; Wevers, R.A.; von Geel, W.J. The Short-Term Effect of an Immunosuppressive Treatment on CSF Myelin Basic Protein in Chronic Progressive Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 1988, 51, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Sellebjerg, F.; Börnsen, L.; Ammitzbøll, C.; Nielsen, J.E.; Vinther-Jensen, T.; Hjermind, L.E.; von Essen, M.; Ratzer, R.L.; Soelberg Sørensen, P.; Romme Christensen, J. Defining Active Progressive Multiple Sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2017, 23, 1727–1735. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.R.; Herndon, R.M.; McKhann, G.M. Radioimmunoassay of Myelin Basic Protein in Spinal Fluid. An Index of Active Demyelination. N. Engl. J. Med. 1976, 295, 1455–1457. [Google Scholar] [CrossRef]
- Liu, Q.; Xie, F.; Siedlak, S.L.; Nunomura, A.; Honda, K.; Moreira, P.I.; Zhua, X.; Smith, M.A.; Perry, G. Neurofilament Proteins in Neurodegenerative Diseases. Cell. Mol. Life Sci. CMLS 2004, 61, 3057–3075. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Lee, V.M.-Y.; Trojanowski, J.Q. The Cytoskeleton in Neurodegenerative Diseases. J. Pathol. 2004, 204, 438–449. [Google Scholar] [CrossRef]
- Perrot, R.; Eyer, J. Neuronal Intermediate Filaments and Neurodegenerative Disorders. Brain Res. Bull. 2009, 80, 282–295. [Google Scholar] [CrossRef]
- Szaro, B.G.; Strong, M.J. Post-Transcriptional Control of Neurofilaments: New Roles in Development, Regeneration and Neurodegenerative Disease. Trends Neurosci. 2010, 33, 27–37. [Google Scholar] [CrossRef]
- Gnanapavan, S.; Grant, D.; Pryce, G.; Jackson, S.; Baker, D.; Giovannoni, G. Neurofilament a Biomarker of Neurodegeneration in Autoimmune Encephalomyelitis. Autoimmunity 2012, 45, 298–303. [Google Scholar] [CrossRef]
- Varhaug, K.N.; Torkildsen, Ø.; Myhr, K.-M.; Vedeler, C.A. Neurofilament Light Chain as a Biomarker in Multiple Sclerosis. Front. Neurol. 2019, 10, 338. [Google Scholar] [CrossRef]
- Kuhle, J.; Leppert, D.; Petzold, A.; Regeniter, A.; Schindler, C.; Mehling, M.; Anthony, D.C.; Kappos, L.; Lindberg, R.L.P. Neurofilament Heavy Chain in CSF Correlates with Relapses and Disability in Multiple Sclerosis. Neurology 2011, 76, 1206–1213. [Google Scholar] [CrossRef]
- Kuhle, J.; Plattner, K.; Bestwick, J.P.; Lindberg, R.L.; Ramagopalan, S.V.; Norgren, N.; Nissim, A.; Malaspina, A.; Leppert, D.; Giovannoni, G.; et al. A Comparative Study of CSF Neurofilament Light and Heavy Chain Protein in MS. Mult. Scler. Houndmills Basingstoke Engl. 2013, 19, 1597–1603. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Wang, B. Neurofilament Light Chain in Blood as a Diagnostic and Predictive Biomarker for Multiple Sclerosis: A Systematic Review and Meta-Analysis. PLoS ONE 2022, 17, e0274565. Available online: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0274565 (accessed on 19 September 2022). [CrossRef] [PubMed]
- Martínez, M.A.M.; Olsson, B.; Bau, L.; Matas, E.; Cobo Calvo, Á.; Andreasson, U.; Blennow, K.; Romero-Pinel, L.; Martínez-Yélamos, S.; Zetterberg, H. Glial and Neuronal Markers in Cerebrospinal Fluid Predict Progression in Multiple Sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2015, 21, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Piehl, F.; Kockum, I.; Khademi, M.; Blennow, K.; Lycke, J.; Zetterberg, H.; Olsson, T. Plasma Neurofilament Light Chain Levels in Patients with MS Switching from Injectable Therapies to Fingolimod. Mult. Scler. Houndmills Basingstoke Engl. 2018, 24, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Delcoigne, B.; Manouchehrinia, A.; Barro, C.; Benkert, P.; Michalak, Z.; Kappos, L.; Leppert, D.; Tsai, J.A.; Plavina, T.; Kieseier, B.C.; et al. Blood Neurofilament Light Levels Segregate Treatment Effects in Multiple Sclerosis. Neurology 2020, 94, e1201–e1212. [Google Scholar] [CrossRef]
- Axelsson, M.; Malmeström, C.; Nilsson, S.; Haghighi, S.; Rosengren, L.; Lycke, J. Glial Fibrillary Acidic Protein: A Potential Biomarker for Progression in Multiple Sclerosis. J. Neurol. 2011, 258, 882–888. [Google Scholar] [CrossRef]
- Boyd, A.; Zhang, H.; Williams, A. Insufficient OPC Migration into Demyelinated Lesions Is a Cause of Poor Remyelination in MS and Mouse Models. Acta Neuropathol. 2013, 125, 841–859. [Google Scholar] [CrossRef]
- Binamé, F.; Pham-Van, L.D.; Bagnard, D. Manipulating Oligodendrocyte Intrinsic Regeneration Mechanism to Promote Remyelination. Cell. Mol. Life Sci. CMLS 2021, 78, 5257–5273. [Google Scholar] [CrossRef]
- Chari, D.M. Remyelination in Multiple Sclerosis. Int. Rev. Neurobiol. 2007, 79, 589–620. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Santiago-Raber, M.-L.; Schneiter, G.; Chofflon, M.; Funakoshi, H.; Nakamura, T.; Lalive, P.H. Hepatocyte Growth Factor Inhibits CNS Autoimmunity by Inducing Tolerogenic Dendritic Cells and CD25+ Foxp3+ Regulatory T Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6424–6429. [Google Scholar] [CrossRef]
- Bai, L.; Lennon, D.P.; Caplan, A.I.; DeChant, A.; Hecker, J.; Kranso, J.; Zaremba, A.; Miller, R.H. Hepatocyte Growth Factor Mediates Mesenchymal Stem Cell–Induced Recovery in Multiple Sclerosis Models. Nat. Neurosci. 2012, 15, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Rivkees, S.A. Hepatocyte Growth Factor Stimulates the Proliferation and Migration of Oligodendrocyte Precursor Cells. J. Neurosci. Res. 2002, 69, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Lalive, P.; Paglinawan, R.; Biollaz, G.; Kappos, E.; Leone, D.; Malipiero, U.; Relvas, J.; Moransard, M.; Suter, T.; Fontana, A. TGF-β-Treated Microglia Induce Oligodendrocyte Precursor Cell Chemotaxis through the HGF-c-Met Pathway. Eur. J. Immunol. 2005, 35, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Iwanami, A.; Nakamura, M.; Yamane, J.; Watanabe, K.; Suzuki, Y.; Miyazawa, D.; Shibata, S.; Funakoshi, H.; Miyatake, S.; et al. Hepatocyte Growth Factor Promotes Endogenous Repair and Functional Recovery after Spinal Cord Injury. J. Neurosci. Res. 2007, 85, 2332–2342. [Google Scholar] [CrossRef]
- Sun, W.; Funakoshi, H.; Nakamura, T. Overexpression of HGF Retards Disease Progression and Prolongs Life Span in a Transgenic Mouse Model of ALS. J Neurosci 2002, 22, 6537–6548. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.M.; Jun, E.; Conlon, H.; Sadiq, S.A. Cerebrospinal Hepatocyte Growth Factor Levels Correlate Negatively with Disease Activity in Multiple Sclerosis. J. Neuroimmunol. 2012, 251, 80–86. [Google Scholar] [CrossRef]
- Xiao, J. Neuroprotection on Multiple Sclerosis: A BDNF Perspective. J. Neurol. Neurophysiol. 2012, 3, 108. [Google Scholar] [CrossRef]
- Burbach, G.J.; Hellweg, R.; Haas, C.A.; Del Turco, D.; Deicke, U.; Abramowski, D.; Jucker, M.; Staufenbiel, M.; Deller, T. Induction of Brain-Derived Neurotrophic Factor in Plaque-Associated Glial Cells of Aged APP23 Transgenic Mice. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 2421–2430. [Google Scholar] [CrossRef]
- Stadelmann, C.; Kerschensteiner, M.; Misgeld, T.; Brück, W.; Hohlfeld, R.; Lassmann, H. BDNF and Gp145trkB in Multiple Sclerosis Brain Lesions: Neuroprotective Interactions between Immune and Neuronal Cells? Brain 2002, 125, 75–85. [Google Scholar] [CrossRef]
- Linker, R.A.; Lee, D.-H.; Demir, S.; Wiese, S.; Kruse, N.; Siglienti, I.; Gerhardt, E.; Neumann, H.; Sendtner, M.; Luhder, F.; et al. Functional Role of Brain-Derived Neurotrophic Factor in Neuroprotective Autoimmunity: Therapeutic Implications in a Model of Multiple Sclerosis. Brain 2010, 133, 2248–2263. [Google Scholar] [CrossRef]
- Makar, T.K.; Bever, C.T.; Singh, I.S.; Royal, W.; Sahu, S.N.; Sura, T.P.; Sultana, S.; Sura, K.T.; Patel, N.; Dhib-Jalbut, S.; et al. Brain-Derived Neurotrophic Factor Gene Delivery in an Animal Model of Multiple Sclerosis Using Bone Marrow Stem Cells as a Vehicle. J. Neuroimmunol. 2009, 210, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Wens, I.; Keytsman, C.; Deckx, N.; Cools, N.; Dalgas, U.; Eijnde, B.O. Brain Derived Neurotrophic Factor in Multiple Sclerosis: Effect of 24 Weeks Endurance and Resistance Training. Eur. J. Neurol. 2016, 23, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Islas-Hernandez, A.; Aguilar-Talamantes, H.S.; Bertado-Cortes, B.; de Jesus Mejia-delCastillo, G.; Carrera-Pineda, R.; Cuevas-Garcia, C.F.; Garcia-delaTorre, P. BDNF and Tau as Biomarkers of Severity in Multiple Sclerosis. Biomark. Med. 2018, 12, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, D.; Urshansky, N.; Karni, A. Low and Dysregulated BDNF Secretion from Immune Cells of MS Patients Is Related to Reduced Neuroprotection. J. Neuroimmunol. 2008, 195, 186–193. [Google Scholar] [CrossRef]
- Caggiula, M.; Batocchi, A.P.; Frisullo, G.; Angelucci, F.; Patanella, A.K.; Sancricca, C.; Nociti, V.; Tonali, P.A.; Mirabella, M. Neurotrophic Factors and Clinical Recovery in Relapsing-Remitting Multiple Sclerosis. Scand. J. Immunol. 2005, 62, 176–182. [Google Scholar] [CrossRef]
- Kopec, B.M.; Kiptoo, P.; Zhao, L.; Rosa-Molinar, E.; Siahaan, T.J. Noninvasive Brain Delivery and Efficacy of BDNF to Stimulate Neuroregeneration and Suppression of Disease Relapse in EAE Mice. Mol. Pharm. 2019, 17, 404–416. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of Multiple Sclerosis: 2017 Revisions of the McDonald Criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Filippi, M.; Preziosa, P.; Banwell, B.L.; Barkhof, F.; Ciccarelli, O.; De Stefano, N.; Geurts, J.J.G.; Paul, F.; Reich, D.S.; Toosy, A.T.; et al. Assessment of Lesions on Magnetic Resonance Imaging in Multiple Sclerosis: Practical Guidelines. Brain J. Neurol. 2019, 142, 1858–1875. [Google Scholar] [CrossRef]
- Saccà, F.; Lanzillo, R.; Signori, A.; Maniscalco, G.T.; Signoriello, E.; Lo Fermo, S.; Repice, A.; Annovazzi, P.; Baroncini, D.; Clerico, M.; et al. Determinants of Therapy Switch in Multiple Sclerosis Treatment-Naïve Patients: A Real-Life Study. Mult. Scler. Houndmills Basingstoke Engl. 2019, 25, 1263–1272. [Google Scholar] [CrossRef]
- Ayrignac, X.; Bigaut, K.; Pelletier, J.; de Seze, J.; Demortiere, S.; Collongues, N.; Maarouf, A.; Pinna, F.; Aouinti, S.; Carra Dallière, C.; et al. First Line Treatment Failure: Predictive Factors in a Cohort of 863 Relapsing Remitting MS Patients. Mult. Scler. Relat. Disord. 2021, 48, 102686. [Google Scholar] [CrossRef]
- Tintore, M.; Rovira, À.; Río, J.; Otero-Romero, S.; Arrambide, G.; Tur, C.; Comabella, M.; Nos, C.; Arévalo, M.J.; Negrotto, L.; et al. Defining High, Medium and Low Impact Prognostic Factors for Developing Multiple Sclerosis. Brain J. Neurol. 2015, 138, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, W.J.; Altmann, D.R.; Prados, F.; Miszkiel, K.A.; Eshaghi, A.; Gandini Wheeler-Kingshott, C.A.M.; Barkhof, F.; Ciccarelli, O. Early Imaging Predictors of Long-Term Outcomes in Relapse-Onset Multiple Sclerosis. Brain J. Neurol. 2019, 142, 2276–2287. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Bar-Or, A.; Lassmann, H.; Uccelli, A.; Hartung, H.-P.; Montalban, X.; Sørensen, P.S.; Hohlfeld, R.; Hauser, S.L. Expert Panel of the 27th Annual Meeting of the European Charcot Foundation Role of B Cells in Multiple Sclerosis and Related Disorders. Ann. Neurol. 2021, 89, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Dobson, R.; Ramagopalan, S.; Davis, A.; Giovannoni, G. Cerebrospinal Fluid Oligoclonal Bands in Multiple Sclerosis and Clinically Isolated Syndromes: A Meta-Analysis of Prevalence, Prognosis and Effect of Latitude. J. Neurol. Neurosurg. Psychiatry 2013, 84, 909–914. [Google Scholar] [CrossRef]
- Arrambide, G.; Tintore, M.; Espejo, C.; Auger, C.; Castillo, M.; Río, J.; Castilló, J.; Vidal-Jordana, A.; Galán, I.; Nos, C.; et al. The Value of Oligoclonal Bands in the Multiple Sclerosis Diagnostic Criteria. Brain J. Neurol. 2018, 141, 1075–1084. [Google Scholar] [CrossRef]
- Pfuhl, C.; Grittner, U.; Gieß, R.M.; Scheel, M.; Behrens, J.R.; Rasche, L.; Pache, F.C.; Wenzel, R.; Brandt, A.U.; Bellmann-Strobl, J.; et al. Intrathecal IgM Production Is a Strong Risk Factor for Early Conversion to Multiple Sclerosis. Neurology 2019, 93, e1439–e1451. [Google Scholar] [CrossRef]
- Capuano, R.; Zubizarreta, I.; Alba-Arbalat, S.; Sepulveda, M.; Sola-Valls, N.; Pulido-Valdeolivas, I.; Andorra, M.; Martinez-Heras, E.; Solana, E.; Lopez-Soley, E.; et al. Oligoclonal IgM Bands in the Cerebrospinal Fluid of Patients with Relapsing MS to Inform Long-Term MS Disability. Mult. Scler. Houndmills Basingstoke Engl. 2021, 27, 1706–1716. [Google Scholar] [CrossRef]
- Konen, F.F.; Schwenkenbecher, P.; Jendretzky, K.F.; Gingele, S.; Sühs, K.-W.; Tumani, H.; Süße, M.; Skripuletz, T. The Increasing Role of Kappa Free Light Chains in the Diagnosis of Multiple Sclerosis. Cells 2021, 10, 3056. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biomarkers in EAE | Biomarkers in MS | References | |
|---|---|---|---|
| Inflammation | |||
| CXCL13 | CXCL13 expression is upregulated in the CNS of the EAE model | Highly upregulated in the active MS lesions and CSF of RRMS patients during relapses | [13,14] |
| OPN | OPN expression is upregulated in EAE lesions | -OPN expression is upregulated in MS lesions compared to the healthy brain -Elevated CSF levels in MS patients -Higher plasma levels in SPMS patients compared to healthy and RRMS patients | [24,26,27] |
| IL-17 | IL17 expression is increased in EAE CNS and EAE-derived lymphocytes | IL17 levels are increased in MS lesions and MS patients-derived blood and CSF lymphocytes | [39,41,42] |
| BBB breakdown | |||
| MMP9 | Increased levels coincide with disease severity | MMP9 CSF and serum levels correlate with EDSS score and Gadolinium enhancements | [55,56,57,62,64] |
| ADAMTS13 | ADAMTS13 plasma activity is decreased in EAE mice | ADAMTS13 plasma level is lower in MS patients compared to healthy subjects | [70,71] |
| Astrogliosis | |||
| GFAP | GFAP is upregulated in EAE lesions during the peak of the disease | GFAP levels are highly expressed in the brain/CSF/plasma of RRMS patients | [82,85,86] |
| Myelin/axonal damage | |||
| MOG | MOG antibodies titers correlate with demyelination activity in the EAE model | MOG circulating-free DNA (cfDNA) is found in the serum of RRMS patients | [104,114,115] |
| NF | High NfH serum levels correlate with acute axonal injury at the peak of EAE disease | NfL levels increase in the CSF of RRMS patients and predict the conversion from CIS to MS | [128,131,133] |
| Repair | |||
| BDNF | BDNF expression levels decrease in the spinal cord of EAE mice during the peak of the disease | BDNF levels are decreased in the plasma of MS patients | [150,152,153,154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Birmpili, D.; Charmarke Askar, I.; Bigaut, K.; Bagnard, D. The Translatability of Multiple Sclerosis Animal Models for Biomarkers Discovery and Their Clinical Use. Int. J. Mol. Sci. 2022, 23, 11532. https://doi.org/10.3390/ijms231911532
Birmpili D, Charmarke Askar I, Bigaut K, Bagnard D. The Translatability of Multiple Sclerosis Animal Models for Biomarkers Discovery and Their Clinical Use. International Journal of Molecular Sciences. 2022; 23(19):11532. https://doi.org/10.3390/ijms231911532
Chicago/Turabian StyleBirmpili, Dafni, Imane Charmarke Askar, Kévin Bigaut, and Dominique Bagnard. 2022. "The Translatability of Multiple Sclerosis Animal Models for Biomarkers Discovery and Their Clinical Use" International Journal of Molecular Sciences 23, no. 19: 11532. https://doi.org/10.3390/ijms231911532
APA StyleBirmpili, D., Charmarke Askar, I., Bigaut, K., & Bagnard, D. (2022). The Translatability of Multiple Sclerosis Animal Models for Biomarkers Discovery and Their Clinical Use. International Journal of Molecular Sciences, 23(19), 11532. https://doi.org/10.3390/ijms231911532