

Increased Co-Occurrence of Pathogenic Variants in Hereditary Breast and Ovarian Cancer and Lynch Syndromes: A Consequence of Multigene Panel Genetic Testing?

 , , , , and
, , , , and
Abstract
1. Introduction
2. Patients and Methods
3. Mutation Analysis
4. Results
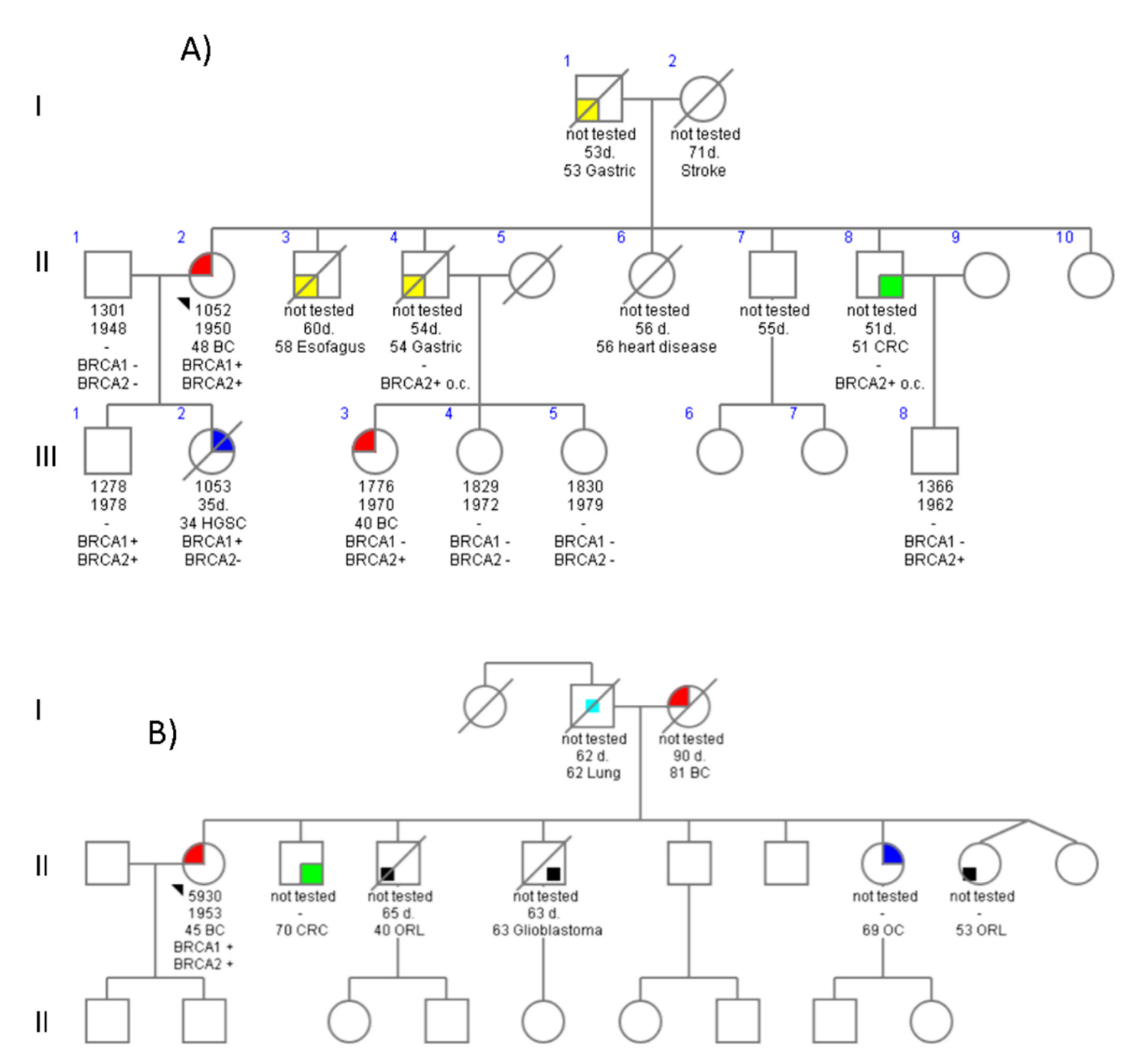
4.1. Family 678
4.2. Family 776
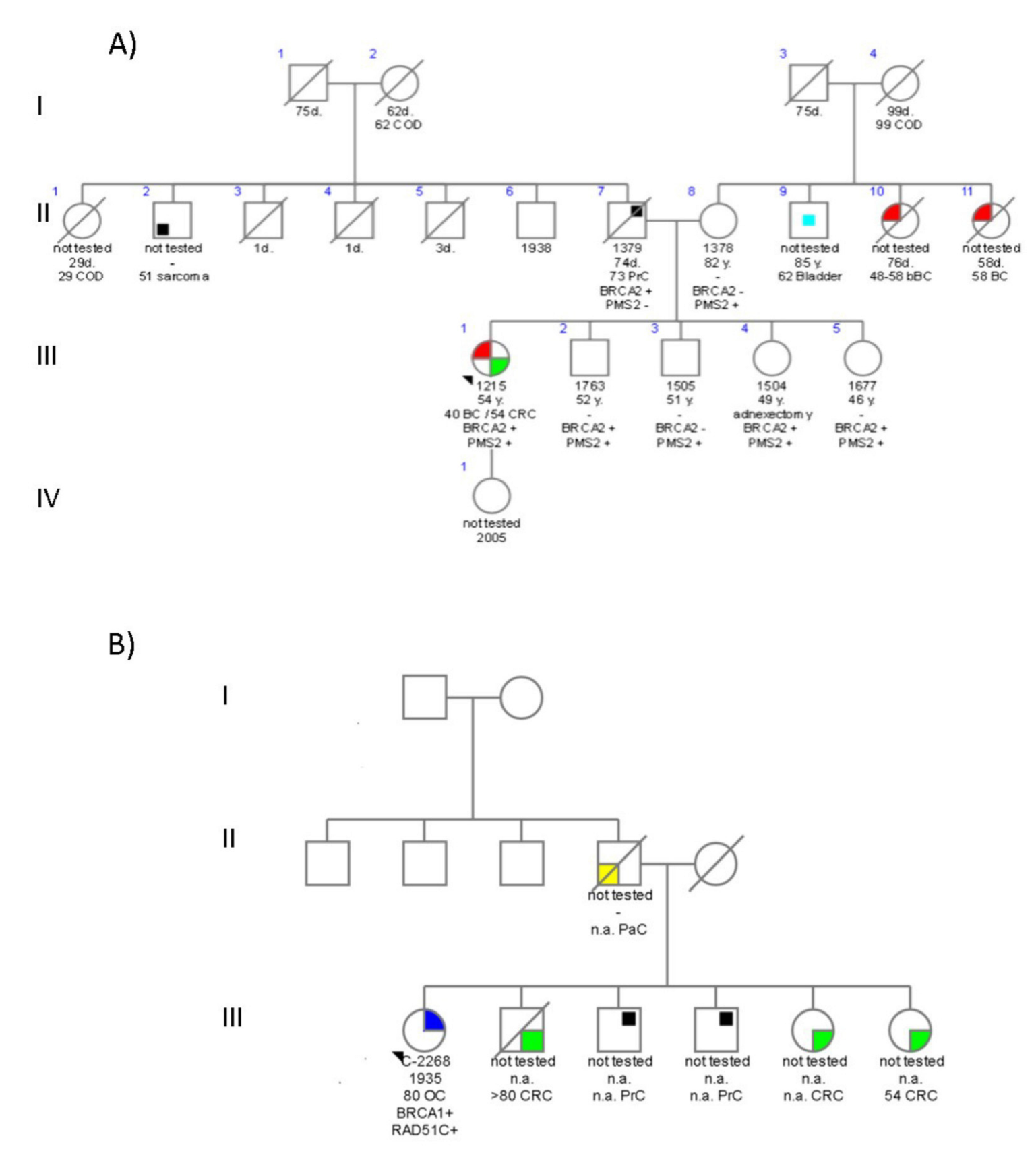
4.3. Family 3699
4.4. Family C1423
5. Discussion
5.1. BRCA DH
5.2. BRCA and Non-BRCA DH
5.3. Impact of DH on Phenotype
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mavaddat, N.; Antoniou, A.C.; Easton, D.F.; Garcia-Closas, M. Genetic Susceptibility to Breast Cancer. Mol. Oncol. 2010, 4, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch Syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Le Page, C.; Rahimi, K.; Rodrigues, M.; Heinzelmann-Schwarz, V.; Recio, N.; Tommasi, S.; Bataillon, G.; Portelance, L.; Golmard, L.; Meunier, L.; et al. Clinicopathological Features of Women with Epithelial Ovarian Cancer and Double Heterozygosity for BRCA1 and BRCA2: A Systematic Review and Case Report Analysis. Gynecol. Oncol. 2020, 156, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.T.; Caliendo, G.; D’Elia, G.; Resse, M.; Casamassimi, A.; Minucci, P.B.; Dello Ioio, C.; Cioffi, M.; Molinari, A.M. Five Italian Families with Two Mutations in BRCA Genes. Genes 2020, 11, 1451. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, C.K.; Kim, D.L.; Esteban-Cardeñosa, E.M.; Avila Cobos, F.; Lastra, E.; Abella, L.E.; de la Cruz, V.; Lobatón, C.D.; Claes, K.B.; Durán, M.; et al. Germline Genetic Findings Which May Impact Therapeutic Decisions in Families with a Presumed Predisposition for Hereditary Breast and Ovarian Cancer. Cancers 2020, 12, 2151. [Google Scholar] [CrossRef]
- McGuigan, A.; Whitworth, J.; Andreou, A.; Hearn, T.; Genomics England Research Consortium; Tischkowitz, M.; Maher, E.R. Multilocus Inherited Neoplasia Allele Syndrome (MINAS): An Update. Eur. J. Hum. Genet. 2022, 30, 265–270. [Google Scholar] [CrossRef]
- Whitworth, J.; Skytte, A.-B.; Sunde, L.; Lim, D.H.; Arends, M.J.; Happerfield, L.; Frayling, I.M.; van Minkelen, R.; Woodward, E.R.; Tischkowitz, M.D.; et al. Multilocus Inherited Neoplasia Alleles Syndrome: A Case Series and Review. JAMA Oncol. 2016, 2, 373. [Google Scholar] [CrossRef]
- Llort, G.; Chirivella, I.; Morales, R.; Serrano, R.; Sanchez, A.B.; Teulé, A.; Lastra, E.; Brunet, J.; Balmaña, J.; Graña, B. SEOM Clinical Guidelines in Hereditary Breast and Ovarian Cancer. Clin. Transl. Oncol. 2015, 17, 956–961. [Google Scholar] [CrossRef]
- Guillén-Ponce, C.; Lastra, E.; Lorenzo-Lorenzo, I.; Martín Gómez, T.; Morales Chamorro, R.; Sánchez-Heras, A.B.; Serrano, R.; Soriano Rodríguez, M.C.; Soto, J.L.; Robles, L. SEOM Clinical Guideline on Hereditary Colorectal Cancer (2019). Clin. Transl. Oncol. 2020, 22, 201–212. [Google Scholar] [CrossRef]
- Velázquez, C.; Lastra, E.; Avila Cobos, F.; Abella, L.; de la Cruz, V.; Hernando, B.A.; Hernández, L.; Martínez, N.; Infante, M.; Durán, M. A Comprehensive Custom Panel Evaluation for Routine Hereditary Cancer Testing: Improving the Yield of Germline Mutation Detection. J. Transl. Med. 2020, 18, 232. [Google Scholar] [CrossRef]
- Velasco, E.; Infante, M.; Durán, M.; Pérez-Cabornero, L.; Sanz, D.J.; Esteban-Cardeñosa, E.; Miner, C. Heteroduplex Analysis by Capillary Array Electrophoresis for Rapid Mutation Detection in Large Multiexon Genes. Nat. Protoc. 2007, 2, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Laish, I.; Friedman, E.; Levi-Reznick, G.; Kedar, I.; Katz, L.; Levi, Z.; Halpern, N.; Parnasa, S.; Abu-Shatya, A.; Half, E.; et al. Double Heterozygotes of BRCA1/BRCA2 and Mismatch Repair Gene Pathogenic Variants: Case Series and Clinical Implications. Breast Cancer Res. Treat. 2021, 188, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Mitra, N.; Wan, F.; Chen, S.; Andrulis, I.L.; Apostolou, P.; Arnold, N.; Arun, B.K.; Barrowdale, D.; et al. Inheritance of Deleterious Mutations at Both BRCA1 and BRCA2 in an International Sample of 32,295 Women. Breast Cancer Res. 2016, 18, 112. [Google Scholar] [CrossRef] [PubMed]
- Infante, M.; Durán, M.; Acedo, A.; Pérez-Cabornero, L.; Sanz, D.; García-González, M.; Beristain, E.; Esteban-Cardeñosa, E.; De La Hoya, M.; Teulé, A.; et al. BRCA1 5272-1G>A and BRCA2 5374delTATG Are Founder Mutations of High Relevance for Genetic Counselling in Breast/Ovarian Cancer Families of Spanish Origin. Clin. Genet. 2010, 77, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.-H.; et al. Mutational Spectrum in a Worldwide Study of 29,700 Families with BRCA1 or BRCA2 Mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, N.C.; Oosthuizen, J.; Theron, M.; Chong, G.; Foulkes, W.D. The Contribution of Large Genomic Rearrangements in BRCA1 and BRCA2 to South African Familial Breast Cancer. BMC Cancer 2020, 20, 391. [Google Scholar] [CrossRef]
- Pilato, B.; De Summa, S.; Danza, K.; Lambo, R.; Paradiso, A.; Tommasi, S. Maternal and Paternal Lineage Double Heterozygosity Alteration in Familial Breast Cancer: A First Case Report. Breast Cancer Res. Treat. 2010, 124, 875–878. [Google Scholar] [CrossRef]
- Sambiasi, D.; Lambo, R.; Pilato, B.; Tommasi, S.; Trojano, G.; Kardhashi, A.; Digennaro, M.; Trojano, V.; Simone, G.; Paradiso, A. BRCA1/2 and Clinical Outcome in a Monoinstitutional Cohort of Women with Hereditary Breast Cancer. Oncol. Rep. 2014, 31, 365–369. [Google Scholar] [CrossRef][Green Version]
- de Juan, I.; Palanca, S.; Domenech, A.; Feliubadaló, L.; Segura, Á.; Osorio, A.; Chirivella, I.; de la Hoya, M.; Sánchez, A.B.; Infante, M.; et al. BRCA1 and BRCA2 Mutations in Males with Familial Breast and Ovarian Cancer Syndrome. Results of a Spanish Multicenter Study. Fam. Cancer 2015, 14, 505–513. [Google Scholar] [CrossRef]
- Mampel, A.; Sottile, M.L.; Denita-Juárez, S.P.; Vargas, A.L.; Vargas-Roig, L.M. Double Heterozygous Pathogenic Variants in the BRCA1 and BRCA2 Genes in a Patient with Bilateral Metachronous Breast Cancer. Cancer Genet. 2022, 261, 14–17. [Google Scholar] [CrossRef]
- van der Klift, H.M.; Mensenkamp, A.R.; Drost, M.; Bik, E.C.; Vos, Y.J.; Gille, H.J.J.P.; Redeker, B.E.J.W.; Tiersma, Y.; Zonneveld, J.B.M.; García, E.G.; et al. Comprehensive Mutation Analysis of PMS2 in a Large Cohort of Probands Suspected of Lynch Syndrome or Constitutional Mismatch Repair Deficiency Syndrome. Hum. Mutat. 2016, 37, 1162–1179. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer Risks by Gene, Age, and Gender in 6350 Carriers of Pathogenic Mismatch Repair Variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- NCCN Guidelines CRC. Available online: https://www.nccn.org/guidelines/guidelines-detail (accessed on 9 June 2022).
- Espenschied, C.R.; LaDuca, H.; Li, S.; McFarland, R.; Gau, C.-L.; Hampel, H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.E.; Jackson, S.A.; Susswein, L.R.; Zeinomar, N.; Ma, X.; Marshall, M.L.; Stettner, A.R.; Milewski, B.; Xu, Z.; Solomon, B.D.; et al. MSH6 and PMS2 Germ-Line Pathogenic Variants Implicated in Lynch Syndrome Are Associated with Breast Cancer. Genet. Med. 2018, 20, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Blanco, A.; Gutiérrez-Enríquez, S.; Santamariña, M.; Montalban, G.; Bonache, S.; Balmaña, J.; Carracedo, A.; Diez, O.; Vega, A. RAD51C Germline Mutations Found in Spanish Site-Specific Breast Cancer and Breast-Ovarian Cancer Families. Breast Cancer Res. Treat. 2014, 147, 133–143. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Carter, N.J.; Marshall, M.L.; Susswein, L.R.; Zorn, K.K.; Hiraki, S.; Arvai, K.J.; Torene, R.I.; McGill, A.K.; Yackowski, L.; Murphy, P.D.; et al. Germline Pathogenic Variants Identified in Women with Ovarian Tumors. Gynecol. Oncol. 2018, 151, 481–488. [Google Scholar] [CrossRef]
- Nurmi, A.; Muranen, T.A.; Pelttari, L.M.; Kiiski, J.I.; Heikkinen, T.; Lehto, S.; Kallioniemi, A.; Schleutker, J.; Bützow, R.; Blomqvist, C.; et al. Recurrent Moderate-Risk Mutations in Finnish Breast and Ovarian Cancer Patients. Int. J. Cancer 2019, 145, 2692–2700. [Google Scholar] [CrossRef]
- Ahlborn, L.B.; Steffensen, A.Y.; Jønson, L.; Djursby, M.; Nielsen, F.C.; Gerdes, A.-M.; Hansen, T.V.O. Identification of a Breast Cancer Family Double Heterozygote for RAD51C and BRCA2 Gene Mutations. Fam. Cancer 2015, 14, 129–133. [Google Scholar] [CrossRef]
- Fostira, F.; Kostantopoulou, I.; Apostolou, P.; Papamentzelopoulou, M.S.; Papadimitriou, C.; Faliakou, E.; Christodoulou, C.; Boukovinas, I.; Razis, E.; Tryfonopoulos, D.; et al. One in Three Highly Selected Greek Patients with Breast Cancer Carries a Loss-of-Function Variant in a Cancer Susceptibility Gene. J. Med. Genet. 2020, 57, 53–61. [Google Scholar] [CrossRef]
- Cummings, S.; Roman, S.S.; Saam, J.; Bernhisel, R.; Brown, K.; Lancaster, J.M.; Usha, L. Age of Ovarian Cancer Diagnosis among BRIP1, RAD51C, and RAD51D Mutation Carriers Identified through Multi-Gene Panel Testing. J. Ovarian Res. 2021, 14, 61. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Family | Proband Phenotype and Age at dx | Gene | Pathogenic Variant DNA Change (Protein Change) | Evidence for P/LP Classification (n° Submissions) | First Degree Cancer Relatives Type and Age at dx | |||
|---|---|---|---|---|---|---|---|---|
| ClinVar | LOVD | Our Series | Carriers | Unknown Carriers | ||||
| 678 | BC 48y CDI RRHH- | BRCA1 | c.34C>T (p.Gln13*) | P (18) | P (22) | 1 | OC 34y HGSC | Esophagus, Gastric |
| BRCA2 | c.1587delTinsCA (p.Glu532Argfs*3) | P (2) | P (3) | 5 | BC 40y RRHH+/HER2- Gastric 54y; CRC 51y | |||
| 776 | BC 40y IDC RRHH+/HER2- CRC 54y | BRCA2 | c.5146_5149delTATG (p.Tyr1716Lysfs*8) | P (10) | P (8) | 26 | PrC 78y | Sarcoma |
| PMS2 | c.903G>T (p.Lys301Asn) | LP (7) | LP/P (9) | 1 | 2 BC, Bladder | |||
| 3699 | BC 45y IDC RRHH- | BRCA1 | c.-232_*1383{0} (p.Met1ValfsX13) | - | P (31) | 1 | OC, CRC, BC, 2 ORL, Lung, glioblastoma | |
| BRCA2 | c.5796_5797delTA (p.His1932Glnfs*12) | P (14) | P (20) | 1 | ||||
| C1423 | OC 81y HGSC | BRCA1 | c.4165_4166delAG (p.Gln1388_Ser1389ins*) | P (14) | P (43) | 6 | PaC, PrC, 3 CRC | |
| RAD51C | c.709C>T (p.Arg237*) | P (14) | P (3) | 1 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Infante, M.; Arranz-Ledo, M.; Lastra, E.; Abella, L.E.; Ferreira, R.; Orozco, M.; Hernández, L.; Martínez, N.; Durán, M. Increased Co-Occurrence of Pathogenic Variants in Hereditary Breast and Ovarian Cancer and Lynch Syndromes: A Consequence of Multigene Panel Genetic Testing? Int. J. Mol. Sci. 2022, 23, 11499. https://doi.org/10.3390/ijms231911499
Infante M, Arranz-Ledo M, Lastra E, Abella LE, Ferreira R, Orozco M, Hernández L, Martínez N, Durán M. Increased Co-Occurrence of Pathogenic Variants in Hereditary Breast and Ovarian Cancer and Lynch Syndromes: A Consequence of Multigene Panel Genetic Testing? International Journal of Molecular Sciences. 2022; 23(19):11499. https://doi.org/10.3390/ijms231911499
Chicago/Turabian StyleInfante, Mar, Mónica Arranz-Ledo, Enrique Lastra, Luis Enrique Abella, Raquel Ferreira, Marta Orozco, Lara Hernández, Noemí Martínez, and Mercedes Durán. 2022. "Increased Co-Occurrence of Pathogenic Variants in Hereditary Breast and Ovarian Cancer and Lynch Syndromes: A Consequence of Multigene Panel Genetic Testing?" International Journal of Molecular Sciences 23, no. 19: 11499. https://doi.org/10.3390/ijms231911499
APA StyleInfante, M., Arranz-Ledo, M., Lastra, E., Abella, L. E., Ferreira, R., Orozco, M., Hernández, L., Martínez, N., & Durán, M. (2022). Increased Co-Occurrence of Pathogenic Variants in Hereditary Breast and Ovarian Cancer and Lynch Syndromes: A Consequence of Multigene Panel Genetic Testing? International Journal of Molecular Sciences, 23(19), 11499. https://doi.org/10.3390/ijms231911499