
Antiviral Peptides as Anti-Influenza Agents

 ,
,  ,
, 
 , and
, and
Abstract
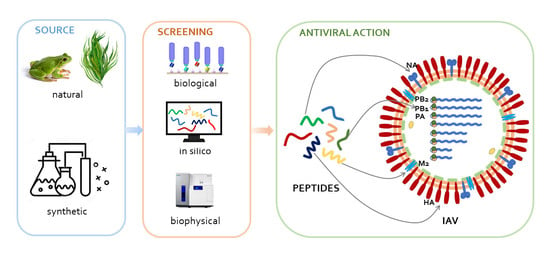
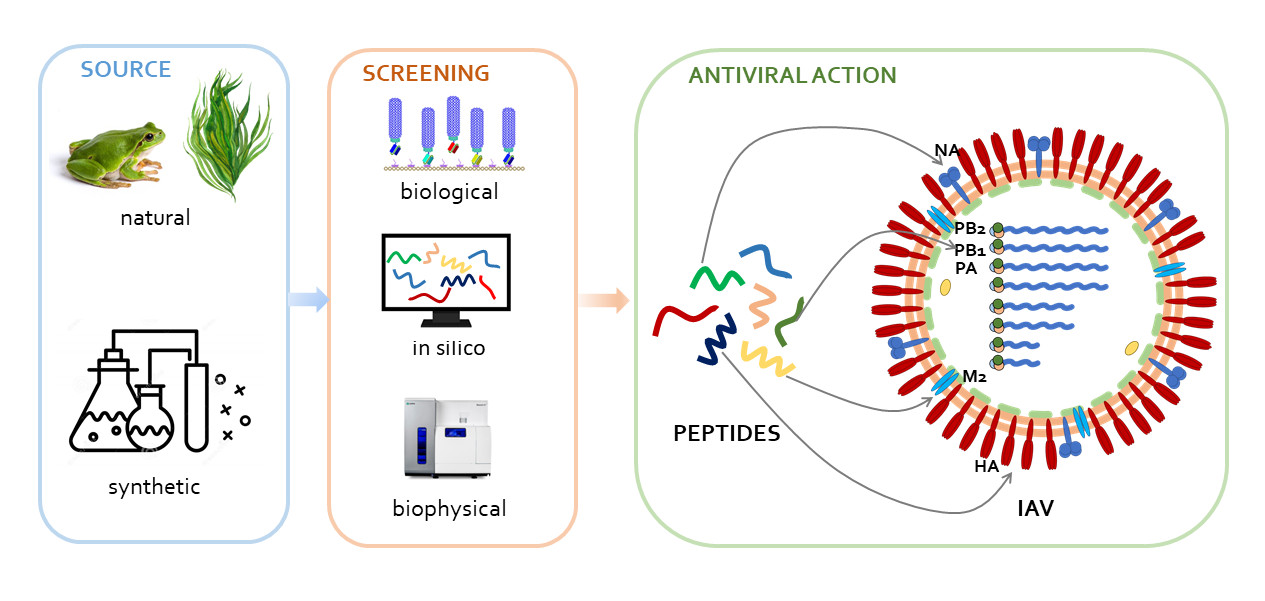
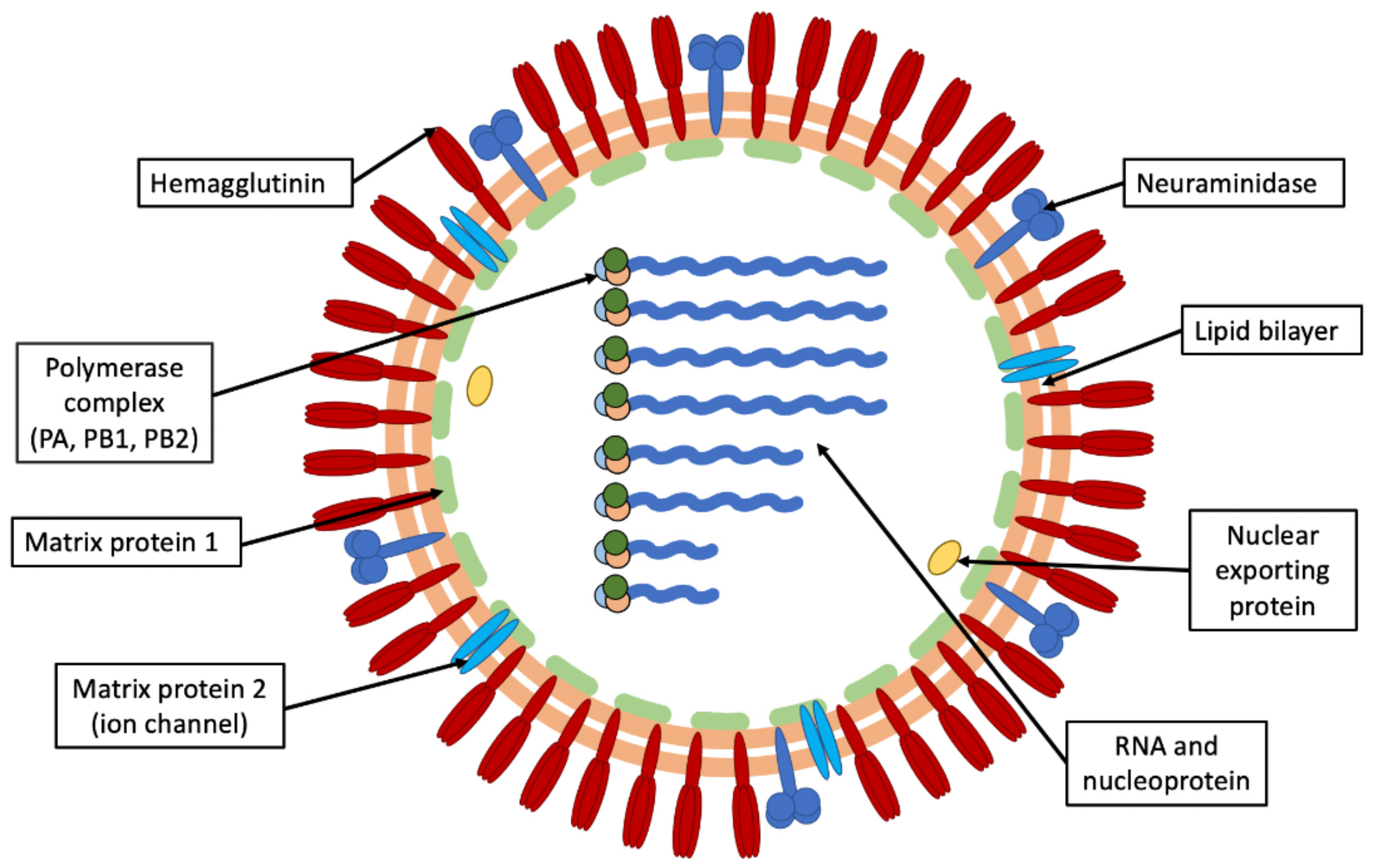
1. Introduction
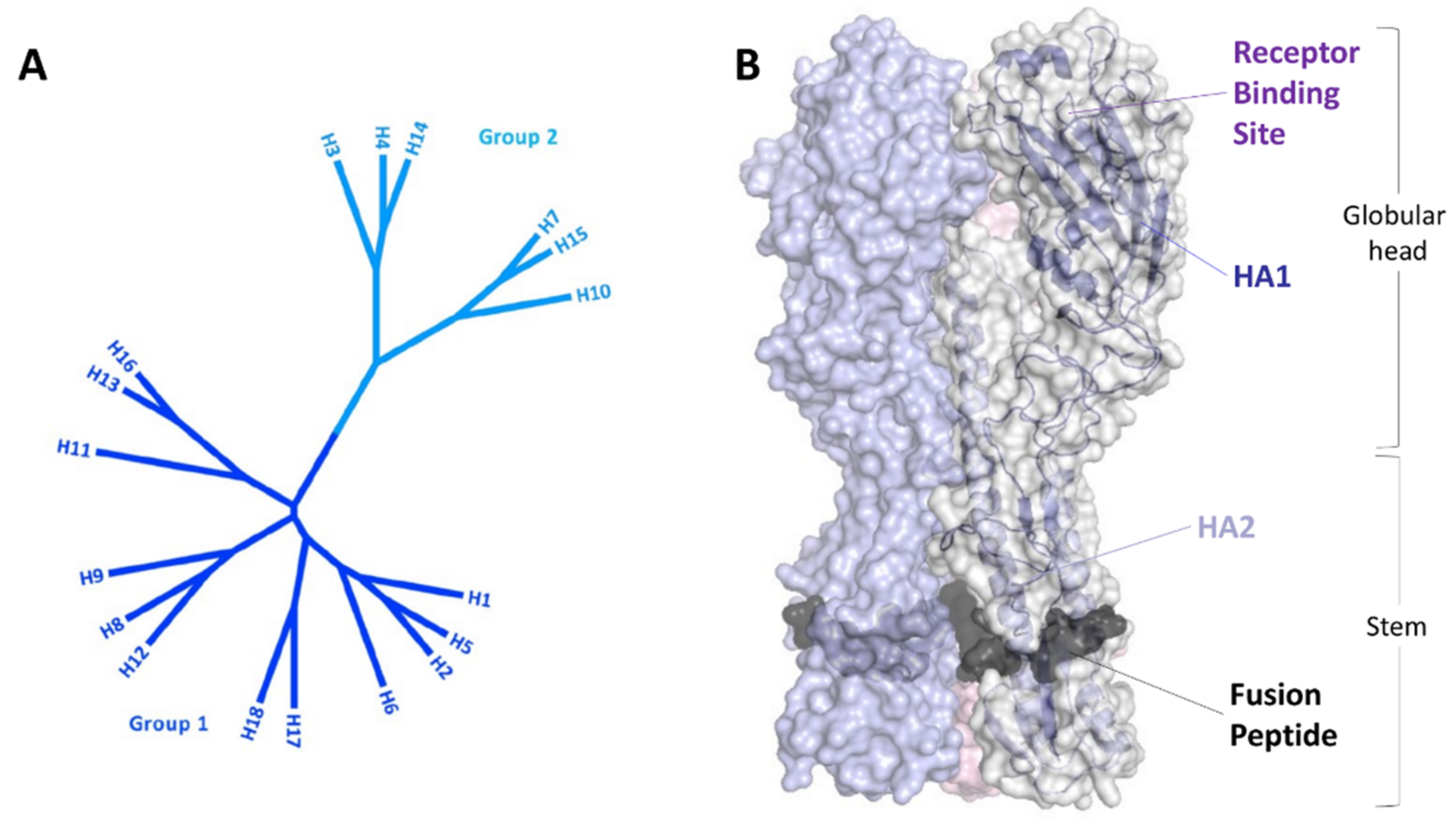
1.1. Influenza Virus
1.2. Treatment of Flu
2. Peptides as Drugs
3. Peptides Targeting HA
3.1. Peptides Interfering with the Sialic Acid Binding
3.2. Peptides Interfering with the Fusogenic Activity of HA
3.3. Entry Blockers Not Interacting with HA
4. Peptides Targeting NA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sequence | Strain | Assay | Activity (mM) | Ref |
|---|---|---|---|---|---|
| Peptide P | PGEKGPSGEAGTAGPPGTPGPQGL | A/Puerto Rico/8/1934 (H1N1) | Cytopathic Effect Reduction | 471 ± 12 g/mL. | [152] |
| P2 | errKPAQP | A/Puerto Rico/8/1934 (H1N1) | Cytopathic effect inhibition | 2.26 ± 0.40 | [153] |
| A/Vietnam/1203/2004 (H5N1) | 1.46 ± 0.12 | ||||
| IntPep | ELVDPVVAAGAVVTSSGIVFFS | Structural studies | [156] |
5. Peptides Targeting PB1
6. Other
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Uyeki, T.M. Influenza. Ann. Intern. Med. 2017, 167, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Schutten, M.; van Baalen, C.; Zoeteweij, P.; Fraaij, P. The influenza virus: Disease, diagnostics, and treatment. MLO Med. Lab. Obs. 2013, 45, 38–40. [Google Scholar] [PubMed]
- Giacchetta, I.; Primieri, C.; Cavalieri, R.; Domnich, A.; de Waure, C. The burden of seasonal influenza in Italy: A systematic review of influenza-related complications, hospitalizations, and mortality. Influenza Other Respir. Viruses 2022, 16, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef]
- Bedford, T.; Riley, S.; Barr, I.G.; Broor, S.; Chadha, M.; Cox, N.J.; Daniels, R.S.; Gunasekaran, C.P.; Hurt, A.C.; Kelso, A.; et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 2015, 523, 217–220. [Google Scholar] [CrossRef]
- Kilbourne, E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006, 12, 9–14. [Google Scholar] [CrossRef]
- Zambon, M.C. The pathogenesis of influenza in humans. Rev. Med. Virol. 2001, 11, 227–241. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The Mother of All Pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef]
- Cheng, V.C.C.; To, K.K.W.; Tse, H.; Hung, I.F.N.; Yuen, K.Y. Two years after pandemic influenza A/2009/H1N1: What have we learned? Clin. Microbiol. Rev. 2012, 25, 223–263. [Google Scholar] [CrossRef]
- Al Hajjar, S.; McIntosh, K. The first influenza pandemic of the 21st century. Ann. Saudi Med. 2010, 30, 1–10. [Google Scholar] [CrossRef]
- Baldo, V.; Bertoncello, C.; Cocchio, S.; Fonzo, M.; Pillon, P.; Buja, A.; Baldovin, T. The new pandemic influenza A/(H1N1) pdm09 virus: Is it really “new”? J. Prev. Med. Hyg. 2016, 57, E19–E22. [Google Scholar]
- Melidou, A.; Ködmön, C.; Nahapetyan, K.; Kraus, A.; Alm, E.; Adlhoch, C.; Mooks, P.; Dave, N.; Carvalho, C.; Meslé, M.M.; et al. Influenza returns with a season dominated by clade 3C.2a1b.2a.2 A(H3N2) viruses, WHO European Region, 2021/22. Eurosurveillance 2022, 27, 2200255. [Google Scholar] [CrossRef]
- Bagaria, J.; Jansen, T.; Marques, D.F.; Hooiveld, M.; McMenamin, J.; de Lusignan, S.; Vilcu, A.M.; Meijer, A.; Rodrigues, A.P.; Brytting, M.; et al. Rapidly adapting primary care sentinel surveillance across seven countries in Europe for COVID-19 in the first half of 2020: Strengths, challenges, and lessons learned. Eurosurveillance 2022, 27, 2100864. [Google Scholar] [CrossRef]
- Wu, X.; Cai, Y.; Huang, X.; Yu, X.; Zhao, L.; Wang, F.; Li, Q.; Gu, S.; Xu, T.; Li, Y.; et al. Co-infection with SARS-CoV-2 and influenza A virus in patient with pneumonia, China. Emerg. Infect. Dis. 2020, 26, 1324–1326. [Google Scholar] [CrossRef]
- D’Abramo, A.; Lepore, L.; Palazzolo, C.; Barreca, F.; Liuzzi, G.; Lalle, E.; Nicastri, E. Acute respiratory distress syndrome due to SARS-CoV-2 and influ- enza A co-infection in an Italian patient: Mini-review of the literature. Int. J. Infect. Dis. 2020, 97, 236–239. [Google Scholar] [CrossRef]
- Azekawa, S.; Namkoong, H.; Mitamura, K.; Kawaoka, Y.; Saito, F. Co-infection with SARS-CoV-2 and influenza A virus. IDCases 2020, 20, e00775. [Google Scholar] [CrossRef]
- Coutinho, A.; Riaz, A.; Makan, A.; Crawford, E.; Dev, D.; Srinivasan, K.; Ahmad, N.; Moudgil, H. Lessons of the month: Co-infection with SARS-CoV-2 and influenza B virus in a patient with community-acquired pneumonia. Clin. Med. 2020, 20, e262–e263. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Wehl, G.; Laible, M.; Rauchenzauner, M. Co-infection of SARS CoV-2 and influenza A in a pediatric patient in Germany. Klin. Padiatr. 2020, 232, 217–218. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, H.; Su, Z.; Li, W.; Yang, D.; Deng, F.; Chen, J. Co-infection of SARS-CoV-2 and influenza virus in early stage of the COVID-19 epi- demic in Wuhan, China. J. Infect. 2020, 81, e128–e129. [Google Scholar] [CrossRef]
- Covin, S.; Rutherford, G.W. Coinfection, severe acute respiratory syn- drome coronavirus 2 (SARS-CoV-2), and influenza: An evolving puzzle. Clin. Infect. Dis. 2021, 72, e993–e994. [Google Scholar] [CrossRef]
- Kim, E.H.; Nguyen, T.Q.; Casel, M.A.B.; Rollon, R.; Kim, S.M.; Kim, Y.I.; Yu, K.M.; Jang, S.G.; Yang, J.; Poo, H.; et al. Coinfection with SARS-CoV-2 and Influenza A Virus Increases Disease Severity and Impairs Neutralizing Antibody and CD4+ T Cell Responses. J. Virol. 2022, 96, e0187321. [Google Scholar] [CrossRef]
- Iacobucci, G. Covid and flu: What do the numbers tell us about morbidity and deaths? BMJ 2021, 375, n2514. [Google Scholar] [CrossRef]
- Stowe, J.; Tessier, E.; Zhao, H.; Guy, R.; Muller-Pebody, B.; Zambon, M.; Andrews, N.; Ramsay, M.; Lopez Bernal, J. Interactions between SARS-CoV-2 and influenza, and the impact of coinfection on disease severity: A test-negative design. Int. J. Epidemiol. 2021, 50, 1124–1133. [Google Scholar] [CrossRef]
- Lamb, R.A.; Krug, R.M. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2001; pp. 1487–1531. [Google Scholar]
- Wright, P.F.; Webster, R.G. Orthomyxoviruses. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2001; pp. 1533–1579. [Google Scholar]
- Jang, J.; Bae, S.E. Comparative Co-Evolution Analysis Between the HA and NA Genes of Influenza A Virus. Virology 2018, 9, 1178122X18788328. [Google Scholar] [CrossRef] [PubMed]
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins Publishers: Philadelphia, PA, USA, 2007; Volume 2. [Google Scholar]
- Rogers, G.N.; Paulson, J.C. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology 1983, 127, 361–373. [Google Scholar] [CrossRef]
- Rogers, G.N.; D’Souza, B.L. Receptor binding properties of human and animal H1 influenza virus isolates. Virology 1989, 173, 317–322. [Google Scholar] [CrossRef]
- Stevens, J.; Blixt, O.; Glaser, L.; Taubenberger, J.K.; Palese, P.; Paulson, J.C.; Wilson, I.A. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J. Mol. Biol. 2006, 355, 1143–1155. [Google Scholar] [CrossRef]
- Yamada, S.; Suzuki, Y.; Suzuki, T.; Le, M.Q.; Nidom, C.A.; Sakai-Tagawa, Y.; Muramoto, Y.; Ito, M.; Kiso, M.; Horimoto, T.; et al. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 2006, 444, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Sempere Borau, M.; Stertz, S. Entry of influenza A virus into host cells—recent progress and remaining challenges. Curr. Opin. Virol. 2021, 48, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Kuroda, K.; Kawasaki, K.; Yamashina, S.; Maeda, T.; Ohnishi, S. Infectious cell entry mechanism of influenza virus. J. Virol. 1982, 43, 284–293. [Google Scholar] [CrossRef] [PubMed]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.M.; de Haan, C.A.M. Dissection of the Influenza A Virus Endocytic Routes Reveals Macropinocytosis as an Alternative Entry Pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef] [PubMed]
- Akole, A.; Warner, J.M. Model of influenza virus acidification. PLoS ONE 2019, 14, 0214448. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef]
- Martin, K.; Helenius, A. Nuclear transport of influenza virus ribonucleoproteins: The viral matrix protein (M1) promotes export and inhibits import. Cell 1991, 67, 117–130. [Google Scholar] [CrossRef]
- Helenius, A. Unpacking the incoming influenza virus. Cell 1992, 69, 577–578. [Google Scholar] [CrossRef]
- Batishchev, O.V.; Shilova, L.A.; Kachala, M.V.; Tashkin, V.Y.; Sokolov, V.S.; Fedorova, N.V.; Baratova, L.A.; Knyazev, D.G.; Zimmerberg, J.; Chizmadzhev, Y.A. pH-Dependent Formation and Disintegration of the Influenza A Virus Protein Scaffold To Provide Tension for Membrane Fusion. J. Virol. 2015, 90, 575–585. [Google Scholar] [CrossRef]
- Lee, K.K. Architecture of a nascent viral fusion pore. EMBO J. 2010, 29, 1299–1311. [Google Scholar] [CrossRef]
- Chlanda, P.; Mekhedov, E.; Waters, H.; Schwartz, C.L.; Fischer, E.R.; Ryham, R.J.; Cohen, F.S.; Blank, P.S.; Zimmerberg, J. The hemifusion structure induced by influenza virus haemagglutinin is determined by physical properties of the target membranes. Nat. Microbiol. 2016, 1, 16050. [Google Scholar] [CrossRef]
- Lamb, R.A.; Choppin, P.W. The gene structure and replication of influenza virus. Annu. Rev. Biochem. 1983, 52, 467–506. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.; Fouchier, R.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis Primers 2018, 4, 3. [Google Scholar] [CrossRef]
- Janssens, Y.; Joye, J.; Waerlop, G.; Clement, F.; Leroux-Roels, G.; Leroux-Roels, I. The role of cell-mediated immunity against influenza and its implications for vaccine evaluation. Front. Immunol. 2022, 13, 959379. [Google Scholar] [CrossRef]
- Sparrow, E.; Wood, J.G.; Chadwick, C.; Newall, A.T.; Torvaldsen, S.; Moen, A.; Torelli, G. Global production capacity of seasonal and pandemic influenza vaccines in 2019. Vaccine 2021, 39, 512–520. [Google Scholar] [CrossRef]
- Uyeki, T.M.; Hui, D.S.; Zambon, M.; Wentworth, D.E.; Monto, A.S. Influenza. Lancet 2022, 400, 693–706. [Google Scholar] [CrossRef]
- Eshaghi, A.; Shalhoub, S.; Rosenfeld, P.; Li, A.; Higgins, R.R.; Stogios, P.J.; Savchenko, A.; Bastien, N.; Li, Y.; Rotstein, C.; et al. Multiple influenza A (H3N2) mutations conferring resistance to neuraminidase inhibitors in a bone marrow transplant recipient. Antimicrob. Agents Chemother. 2014, 58, 7188–7197. [Google Scholar] [CrossRef]
- Dobrovolny, H.M.; Beauchemin, C. Modelling the emergence of influenza drug resistance: The roles of surface proteins, the immune response and antiviral mechanisms. PLoS ONE 2017, 12, e0180582. [Google Scholar] [CrossRef]
- Liu, J.W.; Lin, S.H.; Wang, L.C.; Chiu, H.Y.; Lee, J.A. Comparison of Antiviral Agents for Seasonal Influenza Outcomes in Healthy Adults and Children: A Systematic Review and Network Meta-analysis. JAMA Netw. Open 2021, 4, e2119151. [Google Scholar] [CrossRef]
- Tejada, S.; Tejo, A.M.; Peña-López, Y.; Forero, C.G.; Corbella, X.; Rello, J. Neuraminidase inhibitors and single dose baloxavir are effective and safe in uncomplicated influenza: A meta-analysis of randomized controlled trials. Expert Rev. Clin. Pharmacol. 2021, 14, 901–918. [Google Scholar] [CrossRef]
- Roosenhoff, R.; Reed, V.; Kenwright, A.; Schutten, M.; Boucher, C.A.; Monto, A.; Clinch, B.; Kumar, D.; Whitley, R.; Nguyen-Van-Tam, J.S.; et al. Viral Kinetics and Resistance Development in Children Treated with Neuraminidase Inhibitors: The Influenza Resistance Information Study (IRIS). Clin. Infect. Dis. 2020, 71, 1186–1194. [Google Scholar] [CrossRef]
- Memoli, M.J.; Athota, R.; Reed, S.; Czajkowski, L.; Bristol, T.; Proudfoot, K.; Hagey, R.; Voell, J.; Fiorentino, C.; Ademposi, A.; et al. The natural history of influenza infection in the severely immunocompromised vs nonimmunocompromised hosts. Clin. Infect. Dis. 2014, 58, 214–224. [Google Scholar] [CrossRef]
- Omoto, S.; Speranzini, V.; Hashimoto, T.; Noshi, T.; Yamaguchi, H.; Kawai, M.; Kawaguchi, K.; Uehara, T.; Shishido, T.; Naito, A.; et al. Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci. Rep. 2018, 8, 9633. [Google Scholar] [CrossRef]
- Lüscher-Mattli, M. Influenza chemotherapy: A review of the present state of art and of new drugs in development. Arch. Virol. 2000, 145, 2233–2248. [Google Scholar] [CrossRef]
- Anovadiya, A.P.; Barvaliya, M.J.; Shah, R.A.; Ghori, V.M.; Sanmukhani, J.J.; Patel, T.K.; Tripathi, C.B. Adverse drug reaction profile of oseltamivir in Indian population: A prospective observational study. Indian J. Pharmacol. 2011, 43, 258–261. [Google Scholar] [CrossRef]
- Antipov, E.A.; Pokryshevskaya, E.B. The effects of adverse drug reactions on patients’ satisfaction: Evidence from publicly available data on Tamiflu (oseltamivir). Int. J. Med. Inform. 2019, 125, 30–36. [Google Scholar] [CrossRef]
- Dufrasne, F. Baloxavir Marboxil: An Original New Drug against Influenza. Pharmaceuticals 2021, 15, 28. [Google Scholar] [CrossRef]
- Berkhout, B.; Sanders, R.W. Molecular strategies to design an escape-proof antiviral therapy. Antiviral. Res. 2011, 92, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Król, E.; Rychłowska, M.; Szewczyk, B. Antivirals—Current trends in fighting influenza. Acta. Biochim. Pol. 2014, 61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Zaoral, M.; Kolc, J.; Sorm, F. Amino acids and peptides. LXXI. Synthesis of 1-deamino-8-D-gamma-aminobutyrine vasopressin, 1-deamino-8-D-lysine vasopressin, and 1-deamino-8-D-arginine vasopressin. Collect. Czech. Chem. Commun. 1967, 32, 1250–1257. [Google Scholar] [CrossRef]
- Melin, P.; Trojnar, J.; Johansson, B.; Vilhardt, H.; Akerlund, M. Synthetic antagonists of the myometrial response to vasopressin and oxytocin. J. Endocrinol. 1986, 111, 125–131. [Google Scholar] [CrossRef]
- Kyncl, J.; Rudinger, J. Excretion of antidiuretic activity in the urine of cats and rats after administration of the synthetic hormonogen, N alpha-glycyl-glycyl-glycyl-[8-lysine]-vasopressin (triglycylvasopressin). J. Endocrinol. 1970, 48, 157–165. [Google Scholar] [CrossRef]
- Infoholic Research LLP. Global Human Insulin Market 2018–2024; Research and Market., Region: Global; Infoholic Research LLP: Karnataka, India, 2018; pp. 1–77. [Google Scholar]
- Janecka, A.; Zubrzycka, M.; Janecki, T. Somatostatin analogs. J. Pept. Res. 2001, 58, 91–107. [Google Scholar] [CrossRef]
- Machado, E.S.; Passoni, L.F.; Sidi, L.C.; Andrade, H.B.; De Menezes, J.A. Successful desensitization of enfuvirtide after a first attempt failure. AIDS 2006, 20, 2130–2131. [Google Scholar] [CrossRef]
- Bourinet, E.; Zamponi, G.W. Block of voltage-gated calcium channels by peptide toxins. Neuropharmacology 2017, 127, 109–115. [Google Scholar] [CrossRef]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef]
- Telenti, A.; Arvin, A.; Corey, L.; Corti, D.; Diamond, M.S.; García-Sastre, A.; Garry, R.F.; Holmes, E.C.; Pang, P.S.; Virgin, H.W. After the pandemic: Perspectives on the future trajectory of COVID-19. Nature 2021, 596, 495–504. [Google Scholar] [CrossRef]
- Da Zhu, J.; Meng, W.; Wang, X.J.; Wang, H.C. R Broad-spectrum antiviral agents. Front. Microbiol. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Vilas Boas, L.C.P.; Campos, M.L.; Berland, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef]
- Musiime, V.; Kaudha, E.; Kayiwa, J.; Mirembe, G.; Odera, M.; Kizito, H.; Nankya, I.; Ssali, F.; Kityo, C.; Colebunders, R.; et al. Antiretroviral drug resistance profiles and response to second-line therapy among HIV type 1-infected Ugandan children. AIDS Res. Hum. Retroviruses 2013, 29, 449–455. [Google Scholar] [CrossRef]
- Deming, P.; McNicholl, I.R. Coinfection with human immunodeficiency virus and hepatitis C virus: Challenges and therapeutic advances. Insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy 2011, 31, 357–368. [Google Scholar] [CrossRef]
- Bontempia, E.; Vergalli, S.; Squazzoni, F. Understanding COVID-19 diffusion requires an interdisciplinary, multi-dimensional approach. Environ. Res. 2020, 188, 109814. [Google Scholar] [CrossRef]
- Marston, B.J.; Dokubo, E.K.; van Steelandt, A. Ebola response impact on public health programs, West Africa, 2014–2017. Emerg. Infect. Dis. 2017, 23, S25–S32. [Google Scholar] [CrossRef]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.G.; Honório, N.A.; Kuper, H.; Carvalho, M.S. The zika virus epidemic in brazil: From discovery to future implications. Int. J. Environ. Res. Public Health 2018, 15, 96. [Google Scholar] [CrossRef]
- Chernysh, S.; Kim, S.I.; Bekker, G.; Pleskach, V.A.; Filatova, N.A.; Anikin, V.B.; Platonov, V.G.; Bulet, P. Antiviral and antitumor peptides from insects. Proc. Natl. Acad. Sci. USA 2002, 99, 12628–12632. [Google Scholar] [CrossRef]
- Skwarecki, A.S.; Nowak, M.G.; Milewska, M.J. Amino Acid and Peptide-Based Antiviral Agents. ChemMedChem 2021, 16, 3106–3135. [Google Scholar] [CrossRef]
- Agarwal, G.; Gabrani, R. Antiviral Peptides: Identification and Validation. Int. J. Pept. Res. Ther. 2021, 27, 149–168. [Google Scholar] [CrossRef]
- Monroe, M.K.; Wang, H.; Anderson, C.F.; Jia, H.; Flexner, C.; Cui, H. Leveraging the therapeutic, biological, and self-assembling potential of peptides for the treatment of viral infections. J. Control. Release 2022, 348, 1028–1049. [Google Scholar] [CrossRef]
- Al-Azzam, S.; Ding, Y.; Liu, J.; Pandya, P.; Ting, J.P.; Afshar, S. Peptides to combat viral infectious diseases. Peptides 2020, 134, 170402. [Google Scholar] [CrossRef]
- Skalickova, S.; Heger, Z.; Krejcova, L.; Pekarik, V.; Bastl, K.; Janda, J.; Kostolansky, F.; Vareckova, E.; Zitka, O.; Adam, V.; et al. Perspective of use of antiviral peptides against influenza virus. Viruses 2015, 7, 5428–5442. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Sauter, N.K.; Hanson, J.E.; Glick, G.D.; Brown, J.H.; Crowther, R.L.; Park, S.J.; Skehel, J.J.; Wiley, D.C. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: Analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry 1992, 31, 9609–9621. [Google Scholar] [CrossRef]
- Skehel, J.J.; Bizebard, T.; Bullough, P.A.; Hughson, F.M.; Knossow, M.; Steinhauer, D.A.; Wharton, S.A.; Wiley, D.C. Membrane fusion by influenza hemagglutinin. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 573–580. [Google Scholar] [CrossRef]
- Jones, J.C.; Turpin, E.A.; Bultmann, H.; Brandt, C.R.; Schultz-Cherry, S. Inhibition of Influenza Virus Infection by a Novel Antiviral Peptide That Targets Viral Attachment to Cells. J. Virol. 2006, 80, 11960–11967. [Google Scholar] [CrossRef]
- Jones, J.C.; Settles, E.W.; Brandt, C.R.; Schultz-Cherry, S. Identification of the minimal active sequence of an anti-influenza virus peptide. Antimicrob. Agents Chemother. 2011, 55, 1810–1813. [Google Scholar] [CrossRef]
- Jones, J.C.; Settles, E.W.; Brandt, C.R.; Schultz-Cherry, S. Virus aggregating peptide enhances the cell-mediated response to influenza virus vaccine. Vaccine 2011, 29, 7696–7703. [Google Scholar] [CrossRef]
- Lu, R.; Müller, P.; Downard, K.M. Molecular basis of influenza hemagglutinin inhibition with an entry-blocker peptide by computational docking and mass spectrometry. Antivir. Chem. Chemother. 2015, 24, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Barrera, K.L.; Soria-Guerra, R.E.; López-Martínez, R.; Huerta, L.; Salinas-Jazmín, N.; Cabello-Gutiérrez, C.; Alpuche-Solís, Á.G. The Entry Blocker Peptide Produced in Chlamydomonas reinhardtii Inhibits Influenza Viral Replication in vitro. Front. Plant. Sci. 2021, 12, 641420. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Sumi, M.; Kubota, H.; Taki, T.; Okahata, Y.; Sato, T. Inhibition of influenza virus infections by sialylgalactose-binding peptides selected from a phage library. J. Med. Chem. 2009, 52, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Onishi, A.; Saito, T.; Shimada, A.; Inoue, H.; Taki, T.; Nagata, K.; Okahata, Y.; Sato, T. Sialic Acid-Mimic Peptides As Hemagglutinin Inhibitors for Anti-Influenza Therapy. J. Med. Chem. 2010, 53, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Hüttl, C.; Hettrich, C.; Miller, R.; Paulke, B.R.; Henklein, P.; Rawel, H.; Bier, F.F. Self-assembled peptide amphiphiles function as multivalent binder with increased hemagglutinin affinity. BMC Biotechnol. 2013, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Hatano, K.; Matsubara, T.; Muramatsu, Y.; Ezure, M.; Koyama, T.; Matsuoka, K.; Kuriyama, R.; Kori, H.; Sato, T. Synthesis and Influenza Virus Inhibitory Activities of Carbosilane Dendrimers Peripherally Functionalized with Hemagglutinin-Binding Peptide. J. Med. Chem. 2014, 57, 8332–8339. [Google Scholar] [CrossRef]
- Matsubara, T.; Shibata, R.; Sato, T. Binding of hemagglutinin and influenza virus to a peptide-conjugated lipid membrane. Front. Microbiol. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Matsubara, T.; Onishi, A.; Yamaguchi, D.; Sato, T. Heptapeptide ligands against receptor-binding sites of influenza hemagglutinin toward anti-influenza therapy. Bioorg. Med. Chem. 2016, 24, 1106–1114. [Google Scholar] [CrossRef]
- Mammari, N.; Krier, Y.; Albert, Q.; Devocelle, M.; Varbanov, M. Plant-derived antimicrobial peptides as potential antiviral agents in systemic viral infections. Pharmaceuticals 2021, 14, 774. [Google Scholar] [CrossRef]
- Sencanski, M.; Radosevic, D.; Perovic, V.; Gemovic, B.; Stanojevic, M.; Veljkovic, N.; Glisic, S. Natural Products as Promising Therapeutics for Treatment of Influenza Disease. Curr. Pharm. Des. 2015, 21, 5573–5588. [Google Scholar] [CrossRef]
- Sato, Y.; Hirayama, M.; Morimoto, K.; Yamamoto, N.; Okuyama, S.; Hori, K. High mannose-binding lectin with preference for the cluster of α1-2-mannose from the green alga Boodlea coacta is a potent entry inhibitor of HIV-1 and influenza viruses. J. Biol. Chem. 2011, 286, 19446–19458. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, Z.; Zhou, D.; Chen, Y.; Hong, W.; Cao, L.; Yang, J.; Zhang, Y.; Shi, W.; Cao, Z.; et al. Virucidal activity of a scorpion venom peptide variant mucroporin-M1 against measles, SARS-CoV and influenza H5N1 viruses. Peptides 2011, 32, 1518–1525. [Google Scholar] [CrossRef]
- Superti, F. Lactoferrin from Bovine Milk: A Protective Companion for Life. Nutrients 2020, 12, 2562. [Google Scholar] [CrossRef]
- Pietrantoni, A.; Dofrelli, E.; Tinari, A.; Ammendolia, M.G.; Puzelli, S.; Fabiani, C.; Donatelli, I.; Superti, F. Bovine lactoferrin inhibits Influenza A virus induced programmed cell death in vitro. Biometals 2010, 23, 465–475. [Google Scholar] [CrossRef]
- Pietrantoni, A.; Ammendolia, M.G.; Superti, F. Bovine lactoferrin: Involvement of metal saturation and carbohydrates in the inhibition of influenza virus infection. Biochem. Cell Biol. 2012, 90, 442–448. [Google Scholar] [CrossRef]
- Ammendolia, M.G.; Agamennone, M.; Pietrantoni, A.; Lannutti, F.; Siciliano, R.A.; de Giulio, B.; Amici, C.; Superti, F. Bovine lactoferrin-derived peptides as novel broad-spectrum inhibitors of influenza virus. Pathog. Glob. Health 2012, 106, 12–19. [Google Scholar] [CrossRef]
- Superti, F.; Agamennone, M.; Ammendolia, M.G.; Pietrantoni, A.; Lannutti, F. European Patent n. EP2780365B1. Available online: https://worldwide.espacenet.com/patent/search/family/045491710/publication/EP2780365B1?q=EP2780365B1 (accessed on 30 August 2022).
- Scala, M.C.; Sala, M.; Pietrantoni, A.; Spensiero, A.; Di Micco, S.; Agamennone, M.; Bertamino, A.; Novellino, E.; Bifulco, G.; Gomez-Monterrey, I.M.; et al. Lactoferrin-derived Peptides Active towards Influenza: Identification of Three Potent Tetrapeptide Inhibitors. Sci. Rep. 2017, 7, 10593. [Google Scholar] [CrossRef]
- Scala, M.C.; Agamennone, M.; Pietrantoni, A.; Di Sarno, V.; Bertamino, A.; Superti, F.; Campiglia, P.; Sala, M. Discovery of a novel tetrapeptide against influenza a virus: Rational design, synthesis, bioactivity evaluation and computational studies. Pharmaceuticals 2021, 14, 959. [Google Scholar] [CrossRef]
- Memczak, H.; Lauster, D.; Kar, P.; Lella, S.D.; Volkmer, R.; Knecht, V.; Herrmann, A.; Ehrentreich-Förster, E.; Bier, F.F.; Stöcklein, W.F.M. Anti-Hemagglutinin Antibody Derived Lead Peptides for Inhibitors of Influenza Virus Binding. PLoS ONE 2016, 11, e0159074. [Google Scholar] [CrossRef]
- Lauster, D.; Pawolski, D.; Storm, J.; Ludwig, K.; Volkmer, R.; Memczak, H.; Herrmann, A.; Bhatia, S. Potential of acylated peptides to target the influenza A virus. Beilstein J. Org. Chem. 2015, 11, 589–595. [Google Scholar] [CrossRef]
- Lauster, D. Multivalent Peptide Nanoparticle Conjugates for Influenza-Virus Inhibition. Angew. Chem. Int. Ed. Engl. 2017, 56, 5931–5936. [Google Scholar] [CrossRef]
- Rajik, M.; Jahanshiri, F.; Omar, A.; Ideris, A.; Hassan, S.; Yusoff, K. Identification and characterisation of a novel anti-viral peptide against avian influenza virus H9N2. Virol. J. 2009, 6, 74. [Google Scholar] [CrossRef]
- Rajik, M.; Omar, A.R.; Ideris, A.; Hassan, S.S.; Yusoff, K. A novel peptide inhibits the influenza virus replication by preventing the viral attachment to the host cells. Int. J. Biol. Sci. 2009, 5, 543–548. [Google Scholar] [CrossRef]
- Arbi, M.; Larbi, I.; Nsiri, J.; Behi, I.E.; Rejeb, A.; Miled, K.; Ghram, A.; Houimel, M. Inhibition of avian influenza virus H9N2 infection by antiviral hexapeptides that target viral attachment to epithelial cells. Virus Res. 2022, 313, 198745. [Google Scholar] [CrossRef]
- Saito, M.; Itoh, Y.; Yasui, F.; Munakata, T.; Yamane, D.; Ozawa, M.; Ito, R.; Katoh, T.; Ishigaki, H.; Nakayama, M.; et al. Macrocyclic peptides exhibit antiviral effects against influenza virus HA and prevent pneumonia in animal models. Nat. Commun. 2021, 12, 2654. [Google Scholar] [CrossRef]
- Omi, J.; Watanabe-Takahashi, M.; Igai, K.; Shimizu, E.; Tseng, C.Y.; Miyasaka, T.; Waku, T.; Hama, S.; Nakanishi, R.; Goto, Y.; et al. The inducible amphisome isolates viral hemagglutinin and defends against influenza A virus infection. Nat. Commun. 2020, 11, 162. [Google Scholar] [CrossRef]
- Zhao, R.; Yu, X.; Liu, H.; Zhai, L.; Xiong, S.; Su, T.; Liu, G. Study on the degeneracy of antisense peptides using affinity chromatography. J. Chromatogr. A 2001, 913, 421–428. [Google Scholar] [CrossRef]
- Zhao, R.; Fang, C.; Yu, X.; Liu, Y.; Luo, J.; Shangguan, D.; Xiong, S.; Su, T.; Liu, G. Screening of inhibitors for influenza A virus using high-performance affinity chromatography and combinatorial peptide libraries. J. Chromatogr. A 2005, 1064, 59–66. [Google Scholar] [CrossRef]
- Wu, W.; Lin, D.; Shen, X.; Li, F.; Fang, Y.; Li, K.; Xun, T.; Yang, G.; Yang, J.; Liu, S.; et al. New influenza A Virus Entry Inhibitors Derived from the Viral Fusion Peptides. PLoS ONE 2015, 10, e0138426. [Google Scholar] [CrossRef]
- López-Mart\’\inez, R.; Ram\’\irez-Salinas, G.L.; Correa-Basurto, J.; Barrón, B.L. Inhibition of Influenza A Virus Infection In Vitro by Peptides Designed In Silico. PLoS ONE 2013, 8, e76876. [Google Scholar] [CrossRef]
- Lee, K.K.; Pessi, A.; Gui, L.; Santoprete, A.; Talekar, A.; Moscona, A.; Porotto, M. Capturing a Fusion Intermediate of Influenza Hemagglutinin with a Cholesterol-conjugated Peptide, a New Antiviral Strategy for Influenza Virus. J. Biol. Chem. 2011, 286, 42141–42149. [Google Scholar] [CrossRef] [PubMed]
- Figueira, T.N.; Augusto, M.T.; Rybkina, K.; Stelitano, D.; Noval, M.G.; Harder, O.E.; Veiga, A.S.; Huey, D.; Alabi, C.A.; Biswas, S.; et al. Effective in Vivo Targeting of Influenza Virus through a Cell-Penetrating/Fusion Inhibitor Tandem Peptide Anchored to the Plasma Membrane. Bioconjug. Chem. 2018, 29, 3362–3376. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhao, L.; Xia, S.; Zhang, T.; Cao, R.; Liang, G.; Li, Y.; Meng, G.; Wang, W.; Shi, W.; et al. De Novo Design of α-Helical Lipopeptides Targeting Viral Fusion Proteins: A Promising Strategy for Relatively Broad-Spectrum Antiviral Drug Discovery. J. Med. Chem. 2018, 61, 8734–8745. [Google Scholar] [CrossRef] [PubMed]
- Fleishman, S.J.; Whitehead, T.A.; Ekiert, D.C.; Dreyfus, C.; Corn, J.E.; Strauch, E.; Wilson, I.A.; Baker, D. Computational Design of Proteins Targeting the Conserved Stem Region of Influenza Hemagglutinin. Science 2011, 979, 816–822. [Google Scholar] [CrossRef]
- Koday, M.T.; Nelson, J.; Chevalier, A.; Koday, M.; Kalinoski, H.; Stewart, L.; Carter, L.; Nieusma, T.; Lee, P.S.; Ward, A.B.; et al. A Computationally Designed Hemagglutinin Stem-Binding Protein Provides In Vivo Protection from Influenza Independent of a Host Immune Response. PLoS Pathog. 2016, 12, 1–23. [Google Scholar] [CrossRef]
- Kadam, R.U.; Juraszek, J.; Brandenburg, B.; Buyck, C.; Schepens, W.B.G.; Kesteleyn, B.; Stoops, B.; Vreeken, R.J.; Vermond, J.; Goutier, W.; et al. Potent peptidic fusion inhibitors of influenza virus. Science 2017, 358, 496–502. [Google Scholar] [CrossRef]
- Nicol, M.Q.; Ligertwood, Y.; Bacon, M.N.; Dutia, B.M.; Nash, A.A. A novel family of peptides with potent activity against influenza a viruses. J. Gen. Virol. 2012, 93, 980–986. [Google Scholar] [CrossRef]
- Holthausen, D.J.; Lee, S.H.; Kumar, V.T.; Bouvier, N.M.; Krammer, F.; Ellebedy, A.H.; Wrammert, J.; Lowen, A.C.; George, S.; Pillai, M.R.; et al. An Amphibian Host Defense Peptide Is Virucidal for Human H1 Hemagglutinin-Bearing Influenza Viruses. Immunity 2017, 46, 587–595. [Google Scholar] [CrossRef]
- Wu, W.; Wang, J.; Lin, D.; Chen, L.; Xie, X.; Shen, X.; Yang, Q.; Wu, Q.; Yang, J.; He, J.; et al. Super short membrane-active lipopeptides inhibiting the entry of influenza A virus. Biochim. Biophys. Acta. Biomembr. 2015, 1848, 2344–2350. [Google Scholar] [CrossRef]
- Lin, D.; Li, F.; Wu, Q.; Xie, X.; Wu, W.; Wu, J.; Chen, Q.; Liu, S.; He, J. A “building block” approach to the new influenza A virus entry inhibitors with reduced cellular toxicities. Sci. Rep. 2016, 6, 22790. [Google Scholar] [CrossRef]
- Chen, Q.; Guo, Y. Influenza Viral Hemagglutinin Peptide Inhibits Influenza Viral Entry by Shielding the Host Receptor. ACS Infect. Dis. 2016, 2, 187–193. [Google Scholar] [CrossRef]
- Leikina, E.; Delanoe-Ayari, H.; Melikov, K.; Cho, M.-S.; Chen, A.; Waring, A.J.; Wang, W.; Xie, Y.; Loo, J.A.; Lehrer, R.I.; et al. Carbohydrate-binding molecules inhibit viral fusion and entry by crosslinking membrane glycoproteins. Nat. Immunol. 2005, 6, 995–1001. [Google Scholar] [CrossRef]
- Salvatore, M.; García-Sastre, A.; Ruchala, P.; Lehrer, R.I.; Chang, T.; Klotman, M.E. A-Defensin Inhibits Influenza Virus Replication By Cell-Mediated Mechanism(S). J. Infect. Dis. 2007, 196, 835–843. [Google Scholar] [CrossRef]
- Doss, M.; White, M.R.; Tecle, T.; Gantz, D.; Crouch, E.C.; Jung, G.; Ruchala, P.; Waring, A.J.; Lehrer, R.I.; Hartshorn, K.L. Interactions of α-, β-, and θ-Defensins with Influenza A Virus and Surfactant Protein D. J. Immunol. 2009, 182, 7878–7887. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Y.; Kuang, Y.; Wang, B.; Li, W.; Gong, T.; Jiang, Z.; Yang, D.; Li, M. Expression of mouse beta-defensin-3 in MDCK cells and its anti-influenza-virus activity. Arch. Virol. 2009, 154, 639–647. [Google Scholar] [CrossRef]
- Gong, T.; Jiang, Y.; Wang, Y.; Yang, D.; Li, W.; Zhang, Q.; Feng, W.; Wang, B.; Jiang, Z.; Li, M. Recombinant mouse beta-defensin 2 inhibits infection by influenza A virus by blocking its entry. Arch. Virol. 2010, 155, 491–498. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, J.; Zhang, K.; Chu, H.; Liu, D.; Poon, V.K.M.; Chan, C.C.S.; Leung, H.C.; Fai, N.; Lin, Y.P.; et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 2016, 6, 22008. [Google Scholar] [CrossRef]
- Zhao, H.; To, K.K.W.; Sze, K.H.; Yung, T.T.M.; Bian, M.; Lam, H.; Yeung, M.L.; Li, C.; Chu, H.; Yuen, K.Y. A broad-spectrum virus- and host-targeting peptide against respiratory viruses including influenza virus and SARS-CoV-2. Nat. Commun. 2020, 11, 4252. [Google Scholar] [CrossRef]
- Barlow, P.G.; Svoboda, P.; Mackellar, A.; Nash, A.A.; York, I.A.; Pohl, J.; Davidson, D.J.; Donis, R.O. Antiviral Activity and Increased Host Defense against Influenza Infection Elicited by the Human Cathelicidin LL-37. PLoS ONE 2011, 6, e25333. [Google Scholar] [CrossRef]
- Tripathi, S.; Tecle, T.; Verma, A.; Crouch, E.; White, M.; Hartshorn, K.L. The human cathelicidin LL-37 inhibits influenza a viruses through a mechanism distinct from that of surfactant protein d or defensins. J. Gen. Virol. 2013, 94, 40–49. [Google Scholar] [CrossRef]
- He, G.; Massarella, J.; Ward, P. Clinical pharmacokinetics of the prodrug oseltamivir and its active metabolite Ro 64-0802. Clin. Pharmacokinet. 1999, 37, 471–484. [Google Scholar] [CrossRef]
- Greengard, O.; Poltoratskaia, N.; Leikina, E.; Zimmerberg, J.; Moscona, A. The anti-influenza virus ahent 4-GU-DANA (Zanamivir) inhibits cell fusion mediated by human parainfluenza virus and influenza virus HA. J. Virol. 2000, 74, 11108–11114. [Google Scholar] [CrossRef]
- Hayden, F.G.; Pavia, A.T. Antiviral management of seasonal and pandemic influenza. J. Infect. Dis. 2006, 194, 119–126. [Google Scholar] [CrossRef]
- Yen, H.L.; Mckimm-Breschkin, J.L.; Choy, K.T.; Wong, D.D.Y.; Cheung, P.P.H.; Zhou, J.; Ng, I.H.; Zhu, H.; Webby, R.J.; Guan, Y.; et al. Resistance to neuraminidase inhibitors conferred by an R292K mutation in a human influenza virus H7N9 isolate can be masked by a mixed R/K viral population. MBio 2013, 4, e00396-13. [Google Scholar] [CrossRef] [PubMed]
- Amri, N.; Parikesit, A.A.; Tambunan, U.S.F. In silico design of cyclic peptides as influenza virus, a subtype H1N1 neuraminidase inhibitor. Afr. J. Biotechnol. 2013, 11, 11474–11491. [Google Scholar]
- Upadhyay, A.; Chompoo, J.; Taira, N.; Fukuta, M.; Gima, S.; Tawata, S. Solid-phase synthesis of mimosine ntetrapeptides and their inhibitory activities on neuraminidase and tyrosinase. J. Agric. Food Chem. 2011, 59, 12858–12863. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, Y.; Yuan, N.; Zeng, M.; Zhao, Y.; Yu, R.; Liu, Z.; Wu, H.; Dong, S. A Novel Natural Influenza A H1N1 Virus Neuraminidase Inhibitory Peptide Derived from Cod Skin Hydrolysates and Its Antiviral Mechanism. Mar. Drugs. 2018, 16, 377. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Feng, S.; Xu, Y.; Huang, X.; Zhang, J.; Chen, J.; An, X.; Zhang, Y.; Ning, X. Discovery and characterization of a novel peptide inhibitor against influenza neuraminidase. RSC Med. Chem. 2020, 11, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Arpel, A.; Gamper, C.; Spenlé, C.; Fernandez, A.; Jacob, L.; Baumlin, N.; Laquerriere, P.; Orend, G.; Crémel, G.; Bagnard, D. Inhibition of primary breast tumor growth and metastasis using a neuropilin-1 transmembrane domain interfering peptide. Oncotarget 2016, 7, 54723–54732. [Google Scholar] [CrossRef]
- Albrecht, C.; Kuznetsov, A.S.; Appert-Collin, A.; Dhaideh, Z.; Callewaert, M.; Bershatsky, Y.V.; Urban, A.S.; Bocharov, E.V.; Bagnard, D.; Baud, S.; et al. Transmembrane Peptides as a New Strategy to Inhibit Neuraminidase-1 Activation. Front. Cell Dev. Biol. 2020, 8, 611121. [Google Scholar] [CrossRef]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [CrossRef]
- Tiley, L.S.; Hagen, M.; Mathews, J.T.; Krystal, M. Sequence- specific binding of the influenza virus RNA polymerase to sequences located at the 5′ends of the viral RNAs. J. Virol. 1994, 68, 5108–5116. [Google Scholar] [CrossRef]
- Herz, C.; Stavnezer, E.; Krug, R.M.; Gurney, T. Influenza virus, an RNA virus, synthesizes its messenger RNA in the nucleus of infected cells. Cell 1981, 26, 391–400. [Google Scholar] [CrossRef]
- Obayashi, E.; Yoshida, H.; Kawai, F.; Shibayama, N.; Kawaguchi, A.; Nagata, K.; Tame, J.R.; Park, S.Y. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 2008, 454, 1127–1131. [Google Scholar] [CrossRef]
- González, S.; Ortín, J. Characterization of influenza virus PB1 protein binding to viral RNA: Two separate regions of the protein contribute to the interaction domain. J. Virol. 1999, 73, 631–637. [Google Scholar] [CrossRef]
- Sanz-Ezquerro, J.J.; Zürcher, T.; de la Luna, S.; Ortín, J.; Nieto, A. The amino-terminal one-third of the influenza virus PA protein is responsible for the induction of proteolysis. J. Virol. 1996, 70, 1905–1911. [Google Scholar] [CrossRef]
- Ulmanen, I.; Broni, B.A.; Krug, R.M. The role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc. Natl. Acad. Sci. USA 1981, 78, 7355–7359. [Google Scholar] [CrossRef]
- Hagen, M.; Chung, T.D.; Butcher, J.A.; Krystal, M. Recombinant influenza virus polymerase: Requirement of both 5’ and 3’ viral ends for endonuclease activity. J. Virol. 1994, 68, 1509–1515. [Google Scholar] [CrossRef]
- D’Agostino, I.; Giacchello, I.; Nannetti, G.; Fallacara, A.L.; Deodato, D.; Musumeci, F.; Grossi, G.; Palu, G.; Cau, Y.; Trist, I.M.; et al. Synthesis and biological evaluation of a library of hybrid derivatives as inhibitors of influenza virus PA-PB1 interaction. Eur. J. Med. Chem. 2018, 157, 743–758. [Google Scholar] [CrossRef]
- Hejdánek, J.; Radilová, K.; Pachl, P.; Hodek, J.; Machara, A.; Weber, J.; Řezáčová, P.; Konvalinka, J.; Kožíšek, M. Structural characterization of the interaction between the C-terminal domain of the influenza polymerase PA subunit and an optimized small peptide inhibitor. Antivir. Res. 2021, 185, 104971. [Google Scholar] [CrossRef]
- Mohammadi-Barzelighi, H.; Nasr-Esfahani, B.; Bakhshi, B.; Daraei, B.; Moghim, S.; Fazeli, H. Analysis of antibacterial and antibiofilm activity of purified recombinant Azurin from Pseudomonas aeruginosa. Iran. J. Microbiol. 2019, 11, 166–176. [Google Scholar] [CrossRef]
- Chakrabarty, A.M. Bacterial azurin in potential cancer therapy. Cell Cycle 2016, 15, 1665. [Google Scholar] [CrossRef]
- Chaudhari, A.; Fialho, A.M.; Ratner, D.; Gupta, P.; Hong, C.S.; Kahali, S.; Yamada, T.; Haldar, K.; Cho, W.; Chauhan, V.S. Azurin, Plasmodium falciparum malaria and HIV/AIDS: Inhibition of parasitic and viral growth by Azurin. Cell Cycle 2006, 5, 1642–1648. [Google Scholar] [CrossRef]
- Sasidharan, S.; Gosu, V.; Shin, D.; Nath, S.; Tripathi, T.; Saudagar, P. Therapeutic p28 peptide targets essential H1N1 influenza virus proteins: Insights from docking and molecular dynamics simulations. Mol. Divers. 2021, 25, 1929–1943. [Google Scholar] [CrossRef]
- Arivajiagane, A.; Ravi Varadharajulu, N.; Seerangan, K.; Rattinam, R. In silico structure-based design of enhanced peptide inhibitors targeting RNA polymerase PAN-PB1Cinteraction. Comput. Biol. Chem. 2019, 78, 273–281. [Google Scholar] [CrossRef]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef]
- Ma, K.; Wang, Y.J.; Wang, J.F. Influenza virus assembly and budding in lipid rafts. Prog. Biochem. Biophys. 2015, 42, 495–500. [Google Scholar] [CrossRef]
- Krammer, F. Emerging influenza viruses and the prospect of a universal influenza virus vaccine. Biotechnol. J. 2015, 10, 690–701. [Google Scholar] [CrossRef]
- Krammer, F.; Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef]
- Petrie, J.G.; Malosh, R.E.; Cheng, C.K.; Ohmit, S.E.; Martin, E.T.; Johnson, E.; Truscon, R.; Eichelberger, M.C.; Gubareva, L.V.; Fry, A.M.; et al. The Household Influenza Vaccine Effectiveness Study: Lack of Antibody Response and Protection Following Receipt of 2014-2015 Influenza Vaccine. Clin. Infect. Dis. 2017, 65, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Antonny, B. Amphipathic helices and membrane curvature. FEBS Lett. 2010, 584, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Martyna, A.; Bahsoun, B.; Badham, M.D.; Srinivasan, S.; Howard, M.J.; Rossman, J.S. Membrane remodeling by the M2 amphipathic helix drives influenza virus membrane scission. Sci. Rep. 2017, 7, 44695. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kong, B.; Moon, S.; Yu, S.H.; Chung, J.; Ban, C.; Chung, W.J.; Kim, S.G.; Kweon, D.H. Envelope-deforming antiviral peptide derived from influenza virus M2 protein. Biochem. Biophys. Res. Commun. 2019, 517, 507–512. [Google Scholar] [CrossRef]



| RNA Segments | Genes | Proteins |
|---|---|---|
| 1 | PB2 | Basic polymerase 2 * |
| 2 | PB1 | Basic polymerase 1 * |
| PB1-F2 | PB1-F2 protein * | |
| 3 | PA | Acidic polymerase |
| 4 | HA | Hemagglutinin |
| 5 | NP | Nucleoprotein |
| 6 | NA | Neuraminidase |
| 7 | M1 | Matrix 1 protein |
| M2 | Matrix 2 protein | |
| 8 | NS1 | Non-structural protein 1 |
| NS2/NEP | Nuclear export protein |
| ID | Sequence | Strain | Assay | Activity (μM) | Ref |
|---|---|---|---|---|---|
| EB | RRKK AAVA LLPA VLLA LLAP | A/Puerto Rico/8 (H1N1) | Plaque reduction | 7.0 ± 2.1 | [92] |
| B10NP | RRKK ______L_A VLLA LLA | A/Puerto Rico/8 (H1N1) | Plaque reduction | 5.0 ± 4.0 | [92] |
| B7NP | RRKK __VA LL _A VLLA LLA | A/Puerto Rico/8 (H1N1) | Plaque reduction | 0.3 ± 0.2 | [92] |
| EB extract | RRKK AAVA LLPA VLLA LLAP DDDD KHHH HHH | A(H1N1) pdm | Viral inhibition replication | 0.00202 ± 0.001027 | [96] |
| C18-s2 | C17H35CO-ARLPRTMVHPKPAQP-NH2 | A/Puerto Rico/8 (H1N1) | Plaque reduction | 11 | [98] |
| C18-s2(1–5) | C17H35CO-ARLPR-NH2 | A/Puerto Rico/8 (H1N1) | Plaque reduction | 1.9 | [98] |
| 4 | Dumbbell(1)6-ARLPR | A/Puerto Rico/8 (H1N1) | Plaque reduction | 0.72 | [100] |
| 7-1 peptide | C17H35CO-LVRPLAL | A/Aichi/2/68 (H3N2) | Plaque reduction | 6.4 | [102] |
| P1 | SKHSSLDCVLRP | A/Parma/24/09 (H1N1) | Neutralization | 3.1 ± 0.12 | [110] |
| P2 | AGDDQGLDKCVPNSKEK | A/Parma/24/09 (H1N1) | Neutralization | 3.4 ± 0.14 | [110] |
| P3 | NGESTADWAKN | A/Parma/24/09 (H1N1) | Neutralization | 0.05 ± 0.0014 | [110] |
| 14 | VLRP | A/Parma/24/09 (H1N1) | Neutralization | 1 ± 0.05 | [112] |
| 15 | SLDC | A/Parma/24/09 (H1N1) | Neutralization | 4.6 ± 0.05 | [112] |
| 17 | SKHS | A/Parma/24/09 (H1N1) | Neutralization | 0.048 ± 0.0012 | [112] |
| 4 | SAHS | A/Parma/24/09 (H1N1) | Neutralization | 0.0004 ± 0.00003 | [113] |
| PeB | ARDFYDYDVFYYAMD | A/Aichi/2/68 X31 (H3N2) | Infection inhibition | 32 ± 5 | [114] |
| PeBGF | ARDFYGYDVFFYAMD | A/Aichi/2/68 X31 (H3N2) | Infection inhibition | 25 ± 6 | [114] |
| C18-PeBGFa | C17H35CO-ARDFYGYDVFFYAMD | A/Aichi/2/68 X31 (H3N2) | Infection inhibition | 5.9 | [115] |
| 4b | dPG340PeB9 Ligand | A/Aichi/2/68 X31 (H3N2) | Infection inhibition | 0.3 ± 0.1 | [116] |
| 4b | dPG340PeB9 Nanoparticle | A/Aichi/2/68 X31 (H3N2) | Infection inhibition | 0.0006 ± 0.0003 | [116] |
| L-P1 | NDFRSKT | A/chicken/Iran/16/2000 (H9N2) | In ovo antiviral activity | 48 | [117] |
| C-P1 | CNDFRSKTC | A/chicken/Iran/16/2000 (H9N2) | In ovo antiviral activity | 71 | [118] |
| P1 | LSRMPK | A/chicken/Tunisia/12/2010 (H9N2) | In ovo antiviral activity | 870 | [119] |
| P2 | FAPRWR | A/chicken/Tunisia/12/2010 (H9N2) | In ovo antiviral activity | 620 | [119] |
| iHA-100 | Ac-WTGDFFSSHYTVPRC | H5 HA | Surface Plasmon Resonance | 0.0015 | [120] |
| PVF-tet | (MA-RRPVNHF-AU)4-3Lys | A/Puerto Rico/8 (H1N1) | Infection inhibition | 1.4 | [121] |
| ID | Sequence | Strain | Assay | Activity (μM) | Ref |
|---|---|---|---|---|---|
| FHRKKGRGKHK | A/Hufang/7/1999 (H1N1) | Neutralization | 1 log unit inhibitory activity | [123] | |
| HA-FP-1 | GLFGAIAGFIKNGWKGMIKG | A/Puerto Rico/8/34 (H1N1) | Cytopathic Effect inhibition | 9.61 (µg/mL) | [124] |
| A/Aichi/2/68 (H3N2) | 5.90 (µg/mL) | ||||
| C3LB-HA | Residues 270–285 of the HA1 C-ter | A/Puerto Rico/916/34 (H1N1) | Cytopathic Effect inhibition | 27.21 | [125] |
| A (H1N1)pdm2009 | 26.45 | ||||
| P155–185-Chol | GTYDHDVYRDEALNNRFQIKGVELKSGYKDWGSGSG-C(PEG4-Chol)NH2 | A/Hong Kong/8/68 (H3N2) | Plaque reduction | 0.4 | [126] |
| IIQ | AcIEEIQKKIEEIQKKIEEIQKKIEEIQKKIEEIQKKβAKC16 | A/Puerto Rico/8/34 (H1N1) | Cytopathic Effect reduction | 1.73 | [128] |
| HB36.6 | 97 aa sequence | A/California/2009 (H1N1) | Cytopathic Effect reduction | 0.18 µg/mL | [130] |
| A/Puerto Rico/1934 (H1N1) | 0.58 µg/mL | ||||
| A/New Caledonia/1999 (H1N1) | 1.26 µg/mL | ||||
| A/Hong Kong/2003 (H5N1) | 12 µg/mL | ||||
| P7 |  | A/California/07/2009 (H1N1) A/New Caledonia/20/1999 (H1N1) A/Vietnam/1203/2004 (H5N1) | AlphaLisa competition | 0.03–0.07 | [131] |
| FP4 | RRKKWLVFFVIFYFFR | A/Winsconsin/33 (H1N1) | Plaque reduction | 0.00004 | [132] |
| Urumin | IPLRGAFINGRWDSQCHRFSNGAIACA | A/Puerto Rico/8/34 (H1N1) | Plaque reduction | 3.4 | [133] |
| C12-OOWO | A/Puerto Rico/8/34 (H1N1) | Virus titer reduction | 8.48 ± 0.74 (mg/L) | [134] | |
| C12-KKWK | A/Puerto Rico/8/34 (H1N1) | Virus titer reduction | 7.30 ± 1.57 (mg/L) | [134] | |
| C20-Jp-Hp | C20-ARLPRKKWK | A/Puerto Rico/8/34 (H1N1) | Cytopathic Effect inhibition | 0.53 ± 0.25 | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agamennone, M.; Fantacuzzi, M.; Vivenzio, G.; Scala, M.C.; Campiglia, P.; Superti, F.; Sala, M. Antiviral Peptides as Anti-Influenza Agents. Int. J. Mol. Sci. 2022, 23, 11433. https://doi.org/10.3390/ijms231911433
Agamennone M, Fantacuzzi M, Vivenzio G, Scala MC, Campiglia P, Superti F, Sala M. Antiviral Peptides as Anti-Influenza Agents. International Journal of Molecular Sciences. 2022; 23(19):11433. https://doi.org/10.3390/ijms231911433
Chicago/Turabian StyleAgamennone, Mariangela, Marialuigia Fantacuzzi, Giovanni Vivenzio, Maria Carmina Scala, Pietro Campiglia, Fabiana Superti, and Marina Sala. 2022. "Antiviral Peptides as Anti-Influenza Agents" International Journal of Molecular Sciences 23, no. 19: 11433. https://doi.org/10.3390/ijms231911433
APA StyleAgamennone, M., Fantacuzzi, M., Vivenzio, G., Scala, M. C., Campiglia, P., Superti, F., & Sala, M. (2022). Antiviral Peptides as Anti-Influenza Agents. International Journal of Molecular Sciences, 23(19), 11433. https://doi.org/10.3390/ijms231911433