
Progress and Current Status in Hajdu-Cheney Syndrome with Focus on Novel Genetic Research


Abstract
1. Background and Clinical Manifestations
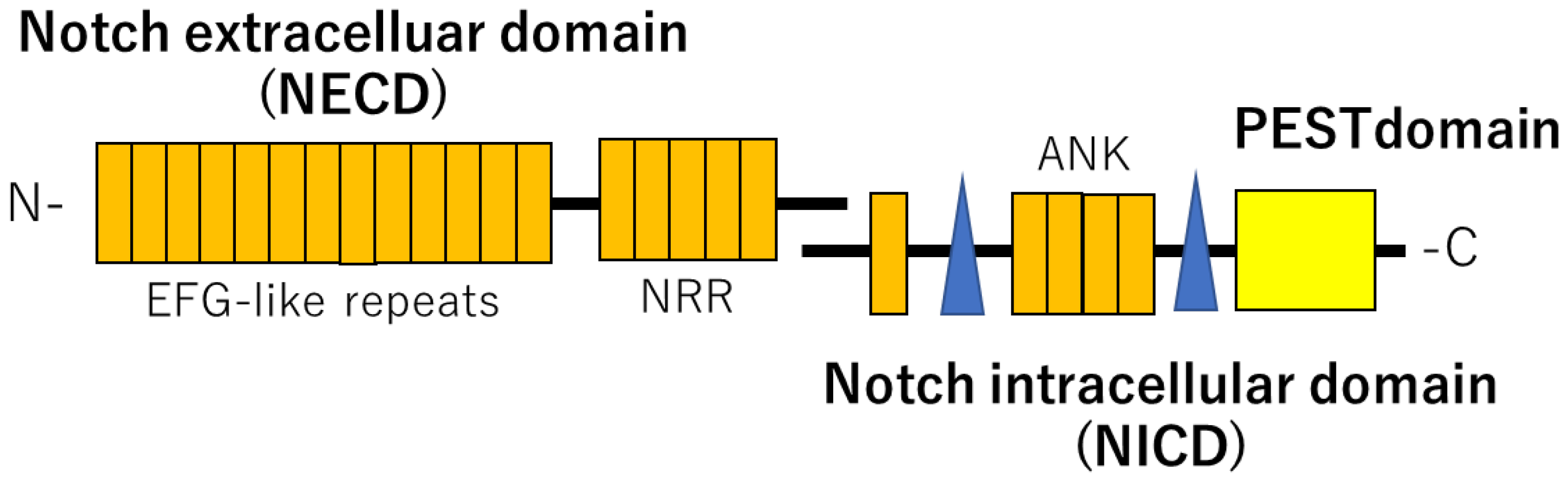
2. NOTCH Signaling
3. Gene Mutation in NOTCH2
4. Variants of the NOTCH2 Gene
5. Clinical Treatment
6. Recent Reports and Genetics of Mouse Models
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hajdu, N.; Kauntze, R. Cranio-skeletal dysplasia. Br. J. Radiol. 1948, 21, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Zanotti, S. Hajdu-Cheney syndrome: A review. Orphanet J. Rare Dis. 2014, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Vingerhoedt, E.; Bailleul-Forestier, I.; Fellus, P.; Schoenaers, J.; Frijns, J.P.; Carels, C. Syndrome of Hajdu-Cheney: Three cases. Cleft Palate-Craniofacial J. 2010, 47, 645–653. [Google Scholar] [CrossRef]
- Antoniades, K.; Kaklamanos, E.; Kavadia, S.; Hatzistilianou, M.; Antoniades, V. Hajdu-Cheney syndrome (acro-osteolysis): A case report of dental interest. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2003, 95, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Kaczoruk-Wieremczuk, M.; Adamska, P.; Adamski, L.J.; Wychowanski, P.; Jereczek-Fossa, B.A.; Starzynska, A. Oral surgery procedures in a patient with Hajdu-Cheney syndrome treated with denosumab—A rare case report. Int. J. Environ. Res. Public Health 2021, 18, 9099. [Google Scholar] [CrossRef]
- Brennan, A.M.; Pauli, R.M. Hajdu-Cheney syndrome: Evolution of phenotype and clinical problems. Am. J. Med. Genet. 2001, 100, 292–310. [Google Scholar] [CrossRef]
- Cortés-Martín, J.; Díaz-Rodríguez, L.; Piqueras-Sola, B.; Rodríguez-Blanque, R.; Bermejo-Fernández, A.; Sánchez-García, J.C. Hajdu–Cheney syndrome: A systematic review of the literature. Int. J. Environ. Res. Public Health 2020, 17, 6174. [Google Scholar] [CrossRef]
- Simpson, M.A.; Irving, M.D.; Asilmaz, E.; Gray, M.J.; Dafou, D.; Elmslie, F.V.; Mansour, S.; Holder, S.E.; Brain, C.E.; Burton, B.K.; et al. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011, 43, 303–305. [Google Scholar] [CrossRef]
- Isidor, B.; Lindenbaum, P.; Pichon, O.; Bézieau, S.; Dina, C.; Jacquemont, S.; Martin-Coignard, D.; Thauvin-Robinet, C.; Le Merrer, M.; Mandel, J.L.; et al. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat. Genet. 2011, 43, 306–308. [Google Scholar] [CrossRef]
- Gray, M.J.; Kim, C.A.; Bertola, D.R.; Arantes, P.R.; Stewart, H.; Simpson, M.A.; Irving, M.D.; Robertson, S.P. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur. J. Hum. Genet. 2012, 20, 122–124. [Google Scholar] [CrossRef]
- Han, M.S.; Ko, J.M.; Cho, T.J.; Park, W.Y.; Cheong, H.I. A novel NOTCH2 mutation identified in a Korean family with Hajdu-Cheney syndrome showing phenotypic diversity. Ann. Clin. Lab. Sci. 2015, 45, 110–114. [Google Scholar] [PubMed]
- Isidor, B.; Le Merrer, M.; Exner, G.U.; Pichon, O.; Thierry, G.; Guiochon-Mantel, A.; David, A.; Cormier-Daire, V.; Le Caignec, C. Serpentine fibula-polycystic kidney syndrome caused by truncating mutations in NOTCH2. Hum. Mutat. 2011, 32, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Schwartzentruber, J.A.; Caqueret, A.; Patry, L.; Marcadier, J.; Fryns, J.P.; Boycott, K.M.; Ste-Marie, L.G.; McKiernan, F.E.; Marik, I.; et al. Mutations in NOTCH2 in families with Hajdu-Cheney syndrome. Hum. Mutat. 2011, 32, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Gazave, E.; Lapébie, P.; Richards, G.S.; Brunet, F.; Ereskovsky, A.V.; Degnan, B.M.; Borchiellini, C.; Vervoort, M.; Renard, E. Origin and evolution of the Notch signalling pathway: An overview from eukaryotic genomes. BMC Evol. Biol. 2009, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Helmi, S.A.; Rohani, L.; Zaher, A.R.; El Hawary, Y.M.; Rancourt, D.E. Enhanced osteogenic differentiation of pluripotent stem cells via γ-secretase inhibition. Int. J. Mol. Sci. 2021, 22, 5215. [Google Scholar] [CrossRef]
- Fukushima, H.; Nakao, A.; Okamoto, F.; Shin, M.; Kajiya, H.; Sakano, S.; Bigas, A.; Jimi, E.; Okabe, K. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Mol. Cell Biol. 2008, 28, 6402–6412. [Google Scholar] [CrossRef]
- Yu, J.; Canalis, E. The Hajdu Cheney mutation sensitizes mice to the osteolytic actions of tumor necrosis factor α. J. Biol. Chem. 2019, 294, 14203–14214. [Google Scholar] [CrossRef]
- Onodera, S.; Nakamura, Y.; Azuma, T. Gorlin Syndrome: Recent advances in genetic testing and molecular and cellular biol ogical research. Int. J. Mol. Sci. 2020, 21, 7559. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Adami, G.; Rossini, M.; Gatti, D.; Orsolini, G.; Idolazzi, L.; Viapiana, O.; Scarpa, A.; Canalis, E. Hajdu Cheney Syndrome; report of a novel NOTCH2 mutation and treatment with denosumab. Bone 2016, 92, 150–156. [Google Scholar] [CrossRef]
- Nancollas, G.; Tang, R.; Phipps, R.; Henneman, Z.; Gulde, S.; Wu, W.; Mangood, A.; Russell, R.G.G.; Ebetino, F. Novel insights into actions of bisphosphonates on bone: Differences in interactions with hydroxyapatite. Bone 2006, 38, 617–627. [Google Scholar] [CrossRef]
- Abed, H.H.; Al-Sahafi, E.N. The role of dental care providers in the management of patients prescribed bisphosphonates: Brief clinical guidance. Gen. Dent. 2018, 66, 18–24. [Google Scholar]
- Polyzos, S.A.; Makras, P.; Tournis, S.; Anastasilakis, A.D. Off-label uses of denosumab in metabolic bone diseases. Bone 2019, 129, 115048. [Google Scholar] [CrossRef]
- Kumaki, D.; Nakamura, Y.; Suzuki, T.; Kato, H. Efficacy of denosumab for osteoporosis in two patients with adult-onset Still’s disease—Denosumab efficacy in osteoporotic Still’s disease patients. J. Clin. Med. 2018, 7, 63. [Google Scholar] [CrossRef]
- Assili, Z.; Dolivet, G.; Salleron, J.; Griffaton-Tallandier, C.; Egloff-Juras, C.; Phulpin, B. A Comparison of the clinical and radiological extent of denosumab (Xgeva®) related osteonecrosis of the jaw: A retrospective study. J. Clin. Med. 2021, 10, 2390. [Google Scholar] [CrossRef]
- Sivolella, S.; Lumachi, F.; Stellini, E.; Favero, L. Denosumab and anti-angiogenetic drug-related osteonecrosis of the jaw: An uncommon but potentially severe disease. Anticancer Res. 2013, 33, 1793–1797. [Google Scholar]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef]
- Khosla, S.; Bilezikian, J.P.; Dempster, D.W.; Lewiecki, E.M.; Miller, P.D.; Neer, R.M.; Recker, R.R.; Shane, E.; Shoback, D.; Potts, J.T. Benefits and risks of bisphosphonate Ttherapy for osteoporosis. J. Clin. Endocrinol. Metab. 2012, 97, 2272–2282. [Google Scholar] [CrossRef]
- Jensen, P.R.; Andersen, T.L.; Chavassieux, P.; Roux, J.P.; Delaisse, J.M. Bisphosphonates impair the onset of bone formation at remodeling sites. Bone 2021, 145, 115850. [Google Scholar] [CrossRef]
- Bazopoulou-Kyrkanidou, E.; Vrahopoulos, T.P.; Eliades, G.; Vastardis, H.; Tosios, K.; Vrotsos, I.A. Periodontitis associated with Hajdu-Cheney syndrome. J. Periodontol. 2007, 78, 1831–1838. [Google Scholar] [CrossRef]
- Shen, J.; Bronson, R.T.; Chen, D.F.; Xia, W.; Selkoe, D.J.; Tonegawa, S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 1997, 89, 629–639. [Google Scholar] [CrossRef]
- Wong, P.C.; Zheng, H.; Chen, H.; Becher, M.W.; Sirinathsinghji, D.J.; Trumbauer, M.E.; Chen, H.Y.; Price, D.L.; Van der Ploeg, L.H.; Sisodia, S.S. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 1997, 387, 288–292. [Google Scholar] [CrossRef]
- Dunwoodie, S.L.; Clements, M.; Sparrow, D.B.; Sa, X.; Conlon, R.A.; Beddington, R.S. Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development 2002, 129, 1795–1806. [Google Scholar] [CrossRef]
- Canalis, E. Clinical and experimental aspects of notch receptor signaling: Hajdu-Cheney syndrome and related disorders. Metabolism 2018, 80, 48–56. [Google Scholar] [CrossRef]
- Pittaway, J.F.; Harrison, C.; Rhee, Y.; Holder-Espinasse, M.; Fryer, A.E.; Cundy, T.; Drake, W.M.; Irving, M.D. Bisphosphonate therapy for spinal osteoporosis in Hajdu-Cheney syndrome—New data and literature review. Orphanet J. Rare Dis. 2018, 13, 47. [Google Scholar] [CrossRef]
- Engin, F.; Yao, Z.; Yang, T.; Zhou, G.; Bertin, T.; Jiang, M.M.; Chen, Y.; Wang, L.; Zheng, H.; Sutton, R.E.; et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 2008, 14, 299–305. [Google Scholar] [CrossRef]
- Zanotti, S.; Yu, J.; Sanjay, A.; Schilling, L.; Schoenherr, C.; Economides, A.N.; Canalis, E. Sustained Notch2 signaling in osteoblasts, but not in osteoclasts, is linked to osteopenia in a mouse model of Hajdu-Cheney syndrome. J. Biol. Chem. 2017, 292, 12232–12244. [Google Scholar] [CrossRef]
- Drake, W.M.; Hiorns, M.P.; Kendler, D.L. Hajdu-Cheney syndrome: Response to therapy with bisphosphonates in two patients. J. Bone Miner. Res. 2003, 18, 131–133. [Google Scholar] [CrossRef]
- Canalis, E.; Schilling, L.; Yee, S.P.; Lee, S.K.; Zanotti, S. Hajdu Cheney mouse mutants exhibit osteopenia, increased osteoclastogenesis, and bone resorption. J. Biol. Chem. 2016, 291, 1538–1551. [Google Scholar] [CrossRef]
- Canalis, E.; Grossman, T.M.; Carrer, M.; Schilling, L.; Yu, J. Antisense oligonucleotides targeting Notch2 ameliorate the osteopenic phenotype in a mouse model of Hajdu-Cheney syndrome. J. Biol. Chem. 2020, 295, 3952–3964. [Google Scholar] [CrossRef]
- Canalis, E.; Zanotti, S. Hajdu-Cheney syndrome, a disease associated with NOTCH2 mutations. Curr. Osteoporos. Rep. 2016, 14, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, S.; Yu, J.; Bridgewater, D.; Wolf, J.M.; Canalis, E. Mice harboring a Hajdu Cheney syndrome mutation are sensitized to osteoarthritis. Bone 2018, 114, 198205. [Google Scholar] [CrossRef] [PubMed]
- Vollersen, N.; Hermans-Borgmeyer, I.; Cornils, K.; Fehse, B.; Rolvien, T.; Triviai, I.; Jeschke, A.; Oheim, R.; Amling, M.; Schinke, T.; et al. High bone turnover in mice carrying a pathogenic NOTCH2 mutation causing Hajdu-Cheney syndrome. J. Bone Miner. Res. 2018, 33, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Yorgan, T.; Vollersen, N.; Riedel, C.; Jeschke, A.; Peters, S.; Busse, B.; Amling, M.; Schinke, T. Osteoblast-specific Notch2 inactivation causes increased trabecular bone mass at specific sites of the appendicular skeleton. Bone 2016, 87, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, H.; Shimizu, K.; Watahiki, A.; Hoshikawa, S.; Kosho, T.; Oba, D.; Sakano, S.; Arakaki, M.; Yamada, A.; Nagashima, K.; et al. NOTCH2 Hajdu-Cheney mutations escape SCF FBW7-dependent proteolysis to promote osteoporosis. Mol. Cell. 2017, 68, 645–658.e5. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Craniofacial Abnormality | Dental Abnormality | Skeletal Abnormality | Cardiac Diseases | Others |
|---|---|---|---|---|
| Micrognathism | Highly arched palates | Acroosteolysis | Cardiovascular abnormalities | Polycystic kidneys |
| Facial dysmorphism | Caries | Fibular deformities, severe osteoporosis | Valvular insufficiency | Neurological disorders |
| Open sutures, wormian bones | Severe periodontal disease | Severe osteoporosis | ||
| Platybasia and basilar invagination | Premature tooth loss | Fractures | ||
| Abnormal redness of gingiva | Joint hyperlaxity | |||
| Abnormal of tooth eruption | Compression fractures and deformities | |||
| Short stature, developmental delay |
| Human Genome Variation Society (HGVS) | Variant Type | Nucleotide Change | Protein (Amino Acid Definition) | Molecular Consequence (Functional Effect) | dbSNP |
|---|---|---|---|---|---|
| M_024408.4(NOTCH2):c.1668C>A(p.Cys556Ter) | SNV | c.1668C>A | p.Cys556Ter | nonsense | - |
| NM_024408.4(NOTCH2):c.2235_2236del (p.Cys745_Asp746delinsTer) | Microsatelite | c.2235_2236del | p.Cys745_Asp746delinsTer | nonsense | - |
| NM_024408.4(NOTCH2):c.3415del(p.Leu1139fs) | Deletion | c.3415del | p.Leu1139fs | frameshift | - |
| NM_024408.4(NOTCH2):c.4174C>T(p.Gln1392Ter) | SNV | c.4174C>T | p.Gln1392Ter | nonsense | rs1649449471 |
| NM_024408.4(NOTCH2):c.5123_5132delinsAGA(p.Gln1392Ter) | Indel | c.5123_5132delinsAGA | p.Gln1392Ter | nonsense | rs1649314295 |
| NM_024408.4(NOTCH2):c.5345del(p.Asp1782fs) | Deletion | c.5345del | p.Asp1782fs | frameshift | rs1553193977 |
| NM_024408.4(NOTCH2):c.6272del(p.Phe2091fs) | Deletion | c.6272del | p.Phe2091fs | frameshift | rs1557802353 |
| NM_024408.4(NOTCH2):c.6386del(p.Ser2129fs) | Deletion | c.6386del | p.Ser2129fs | frameshift | - |
| NM_024408.4(NOTCH2):c.6403_6404del(p.Leu2135fs) | Microsatelite | c.6403_6404del | p.Leu2135fs | frameshift | rs1649067817 |
| NM_024408.4(NOTCH2):c.6424_6427del(p.Ser2142fs) | Deletion | c.6424_6427del | p.Ser2142fs | frameshift | rs1064793515 |
| NM_024408.4(NOTCH2):c.6426_6427insTT(p.Leu2135fs) | Insertion | c.6426_6427insTT | p.Leu2135fs | frameshift | rs1649066485 |
| NM_024408.4(NOTCH2):c.6449_6450del(p.Pro2150fs) | Deletion | c.6449_6450del | p.Pro2150fs | frameshift | rs1553193574 |
| NM_024408.4(NOTCH2):c.6503del(p.Pro2168fs) | Deletion | c.6503del | p.Pro2168fs | frameshift | rs1557802165 |
| NM_024408.4(NOTCH2):c.6622C>T(p.Gln2208Ter) | SNV | c.6622C>T | p.Gln2208Ter | nonsense | rs387906746 |
| NM_024408.4(NOTCH2):c.6832dup(p.Thr2278fs) | Duplication | c.6832dup | p.Thr2278fs | frameshift | - |
| NM_024408.4(NOTCH2):c.6853C>T(p.Gln2285Ter) | SNV | c.6853C>T | p.Gln2285Ter | nonsense | rs1553193507 |
| NM_024408.4(NOTCH2):c.6877del(p.His2293fs) | Deletion | c.6877del | p.His2293fs | frameshift | rs1649047546 |
| NM_024408.4(NOTCH2):c.6895G>T(p.Glu2299Ter) | SNV | c.6895G>T | p.Glu2299Ter | nonsense | rs387906748 |
| NM_024408.4(NOTCH2):c.6909del(p.Ile2304fs) | Deletion | c.6909del | p.Ile2304fs | frameshift | rs771237928 |
| NM_024408.4(NOTCH2):c.6909dup(p.Ile2304fs) | Duplication | c.6909dup | p.Ile2304fs | frameshift | rs771237928 |
| NM_024408.4(NOTCH2):c.6949C>T(p.Gln2317Ter) | SNV | c.6949C>T | p.Gln2317Ter | nonsense | rs387906747 |
| NM_024408.4(NOTCH2):c.7078C>T(p.Gln2360Ter) | SNV | c.7078C>T | p.Gln2360Ter | nonsense | rs1553193485 |
| NM_024408.4(NOTCH2):c.7090del(p.Gln2364fs) | Deletion | c.7090del | p.Gln2364fs | frameshift | rs1649037695 |
| NM_024408.4(NOTCH2):c.7119T>G(p.Tyr2373Ter) | SNV | c.7119T>G | p.Tyr2373Ter | nonsense | rs1557801639 |
| NM_024408.4(NOTCH2):c.7165C>T(p.Gln2389Ter) | SNV | c.7165C>T | p.Gln2389Ter | nonsense | rs387906749 |
| NOTCH2, 1-BP DEL, 6460T | Deletion | 6460T | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aida, N.; Ohno, T.; Azuma, T. Progress and Current Status in Hajdu-Cheney Syndrome with Focus on Novel Genetic Research. Int. J. Mol. Sci. 2022, 23, 11374. https://doi.org/10.3390/ijms231911374
Aida N, Ohno T, Azuma T. Progress and Current Status in Hajdu-Cheney Syndrome with Focus on Novel Genetic Research. International Journal of Molecular Sciences. 2022; 23(19):11374. https://doi.org/10.3390/ijms231911374
Chicago/Turabian StyleAida, Natsuko, Tatsukuni Ohno, and Toshifumi Azuma. 2022. "Progress and Current Status in Hajdu-Cheney Syndrome with Focus on Novel Genetic Research" International Journal of Molecular Sciences 23, no. 19: 11374. https://doi.org/10.3390/ijms231911374
APA StyleAida, N., Ohno, T., & Azuma, T. (2022). Progress and Current Status in Hajdu-Cheney Syndrome with Focus on Novel Genetic Research. International Journal of Molecular Sciences, 23(19), 11374. https://doi.org/10.3390/ijms231911374