
Compound Heterozygosis in AADC Deficiency and Its Complex Phenotype in Terms of AADC Protein Population


Abstract
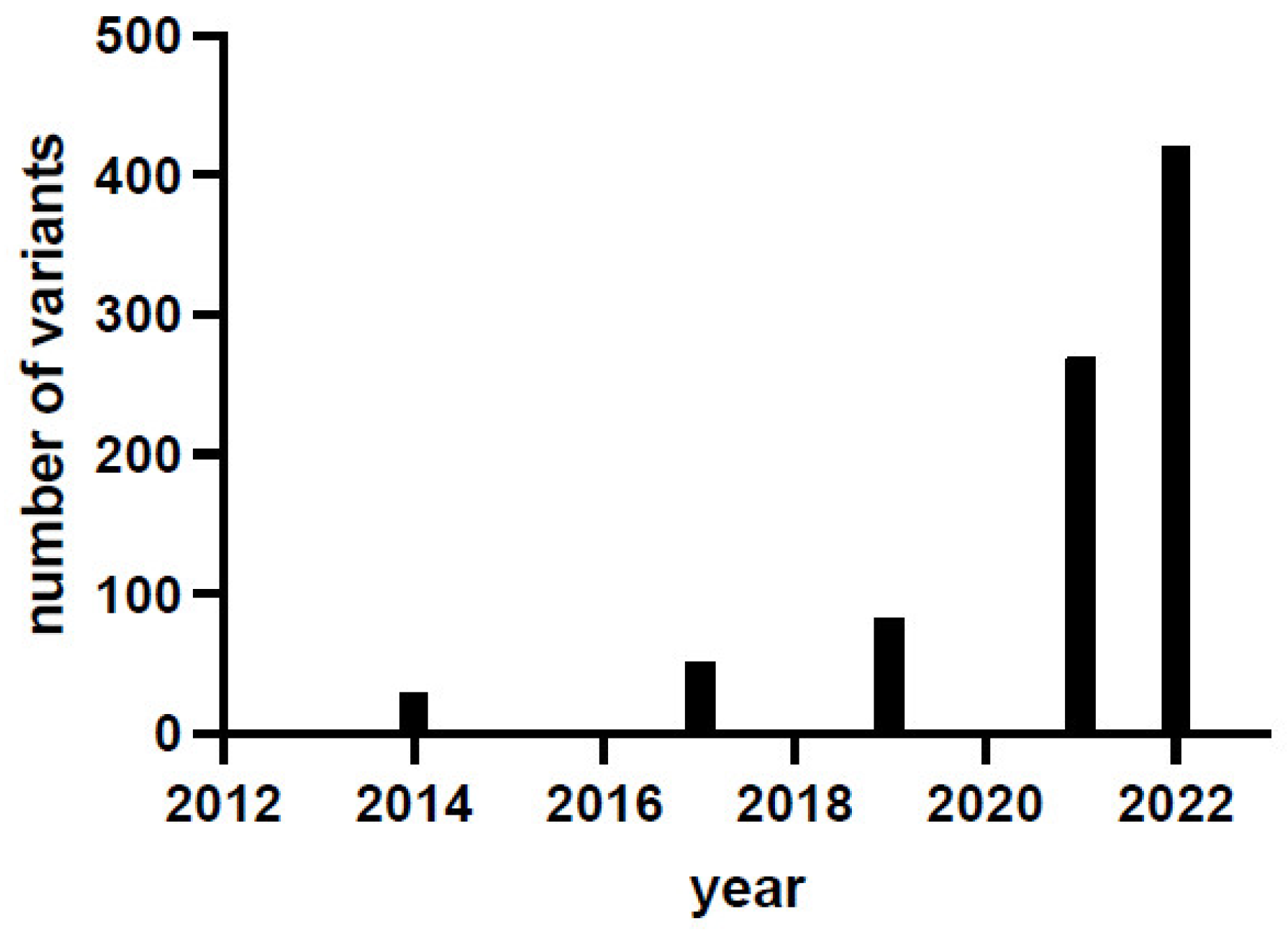
1. Introduction
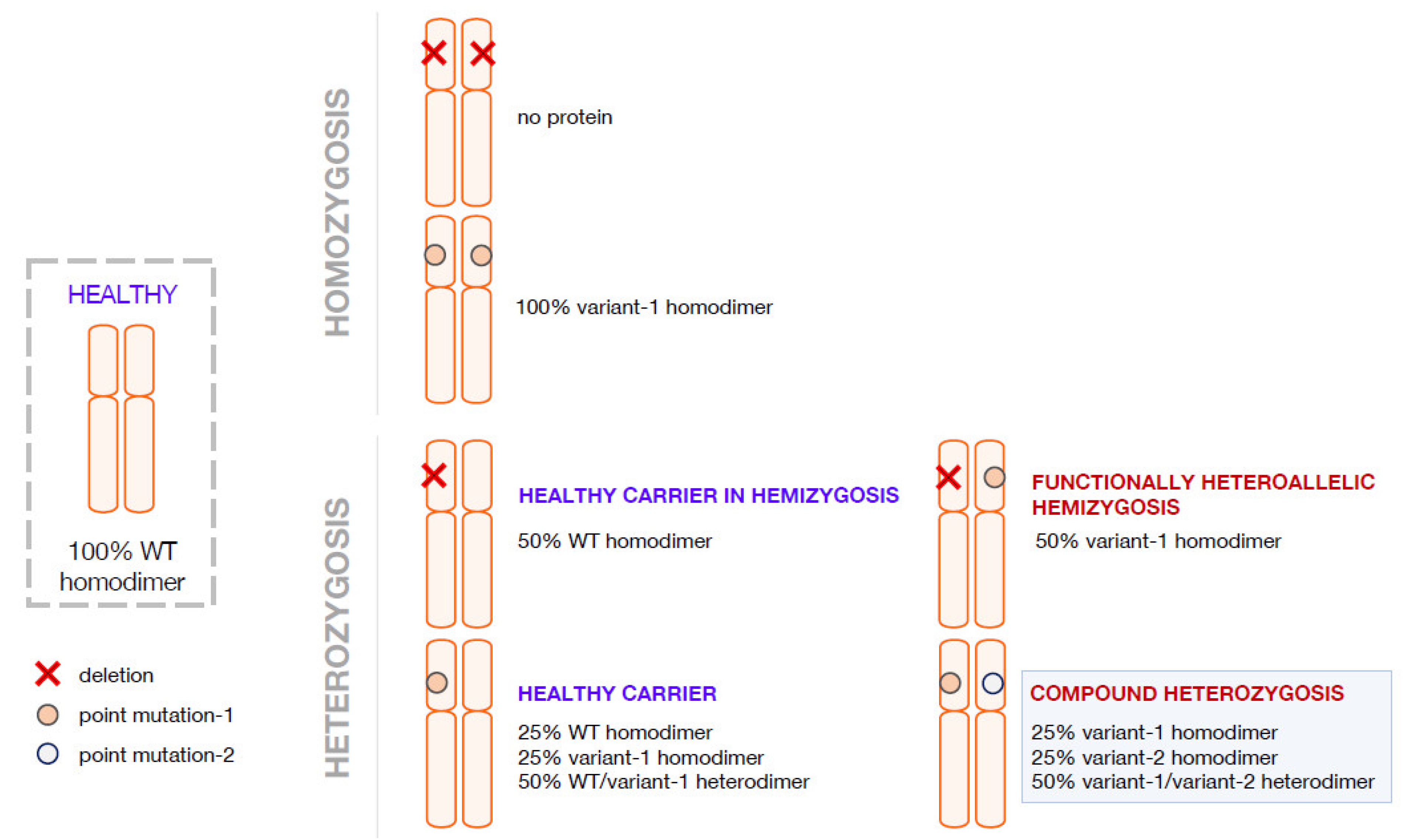
2. Compound Heterozygosis and the Interpretation of the Clinical Phenotype on the Basis of the Combination of Genetics, Computational Methods, and Functional Analyses
3. Compound Heterozygosis in AADC Deficiency
4. Pathogenic Heterozygosity in AADC Deficiency
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hyland, K.; Clayton, P.T. Aromatic amino acid decarboxylase deficiency in twins. J. Inherit. Metab. Dis. 1990, 13, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Wassenberg, T.; Molero-Luis, M.; Jeltsch, K.; Hoffmann, G.F.; Assmann, B.; Blau, N.; Garcia-Cazorla, A.; Artuch, R.; Pons, R.; Pearson, T.S.; et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J. Rare Dis. 2017, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, N.; Montioli, R.; Bertoldi, M.; Carducci, C.; Leuzzi, V.; Gemperle, C.; Berner, T.; Hyland, K.; Thöny, B.; Hoffmann, G.F.; et al. Aromatic amino acid decarboxylase deficiency: Molecular and metabolic basis and therapeutic outlook. Mol. Genet. Metab. 2019, 127, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.; Montioli, R.; Bisello, G.; Palazzi, L.; Mastrangelo, M.; Brennenstuhl, H.; de Laureto, P.P.; Opladen, T.; Leuzzi, V.; Bertoldi, M. Compound heterozygosis in AADC deficiency: A complex phenotype dissected through comparison among heterodimeric and homodimeric AADC proteins. Mol. Genet. Metab 2021, 134, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Hwu, W.L.; Muramatsu, S.; Tseng, S.H.; Tzen, K.Y.; Lee, N.C.; Chien, Y.H.; Snyder, R.O.; Byrne, B.J.; Tai, C.H.; Wu, R.M. Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci. Transl. Med. 2012, 4, 134ra161. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Tseng, S.H.; Tai, C.H.; Muramatsu, S.I.; Byrne, B.J.; Hwu, W.L. Efficacy and safety of AAV2 gene therapy in children with aromatic L-amino acid decarboxylase deficiency: An open-label, phase 1/2 trial. Lancet Child Adolesc. Health 2017, 1, 265–273. [Google Scholar] [CrossRef]
- Hwu, P.W.; Kiening, K.; Anselm, I.; Compton, D.R.; Nakajima, T.; Opladen, T.; Pearl, P.L.; Roubertie, A.; Roujeau, T.; Muramatsu, S.I. Gene therapy in the putamen for curing AADC deficiency and Parkinson’s disease. EMBO Mol. Med. 2021, 13, e14712. [Google Scholar] [CrossRef]
- Tai, C.H.; Lee, N.C.; Chien, Y.H.; Byrne, B.J.; Muramatsu, S.I.; Tseng, S.H.; Hwu, W.L. Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol. Ther. 2022, 30, 509–518. [Google Scholar] [CrossRef]
- Kojima, K.; Nakajima, T.; Taga, N.; Miyauchi, A.; Kato, M.; Matsumoto, A.; Ikeda, T.; Nakamura, K.; Kubota, T.; Mizukami, H.; et al. Gene therapy improves motor and mental function of aromatic l-amino acid decarboxylase deficiency. Brain 2019, 142, 322–333. [Google Scholar] [CrossRef]
- Onuki, Y.; Ono, S.; Nakajima, T.; Kojima, K.; Taga, N.; Ikeda, T.; Kuwajima, M.; Kurokawa, Y.; Kato, M.; Kawai, K.; et al. Dopaminergic restoration of prefrontal cortico-putaminal network in gene therapy for aromatic l-amino acid decarboxylase deficiency. Brain Commun. 2021, 3, fcab078. [Google Scholar] [CrossRef]
- Bankiewicz, K.S.; Pasterski, T.; Kreatsoulas, D.; Onikijuk, J.; Mozgiel, K.; Munjal, V.; Elder, J.B.; Lonser, R.R.; Zabek, M. Use of a novel ball-joint guide array for magnetic resonance imaging-guided cannula placement and convective delivery: Technical note. J. Neurosurg. 2020, 135, 651–657. [Google Scholar] [CrossRef]
- Pearson, T.S.; Gupta, N.; San Sebastian, W.; Imamura-Ching, J.; Viehoever, A.; Grijalvo-Perez, A.; Fay, A.J.; Seth, N.; Lundy, S.M.; Seo, Y.; et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency by MR-guided direct delivery of AAV2-AADC to midbrain dopaminergic neurons. Nat. Commun. 2021, 12, 4251. [Google Scholar] [CrossRef]
- Hyland, K.; Reott, M. Prevalence of Aromatic l-Amino Acid Decarboxylase Deficiency in At-Risk Populations. Pediatr. Neurol. 2020, 106, 38–42. [Google Scholar] [CrossRef]
- Montioli, R.; Dindo, M.; Giorgetti, A.; Piccoli, S.; Cellini, B.; Voltattorni, C.B. A comprehensive picture of the mutations associated with aromatic amino acid decarboxylase deficiency: From molecular mechanisms to therapy implications. Hum. Mol. Genet. 2014, 23, 5429–5440. [Google Scholar] [CrossRef]
- Montioli, R.; Cellini, B.; Borri Voltattorni, C. Molecular insights into the pathogenicity of variants associated with the aromatic amino acid decarboxylase deficiency. J. Inherit. Metab. Dis. 2011, 34, 1213–1224. [Google Scholar] [CrossRef]
- Montioli, R.; Paiardini, A.; Kurian, M.A.; Dindo, M.; Rossignoli, G.; Heales, S.J.R.; Pope, S.; Voltattorni, C.B.; Bertoldi, M. The novel R347g pathogenic mutation of aromatic amino acid decarboxylase provides additional molecular insights into enzyme catalysis and deficiency. Biochim. Biophys. Acta 2016, 1864, 676–682. [Google Scholar] [CrossRef]
- Montioli, R.; Bisello, G.; Dindo, M.; Rossignoli, G.; Voltattorni, C.B.; Bertoldi, M. New variants of AADC deficiency expand the knowledge of enzymatic phenotypes. Arch. Biochem. Biophys. 2020, 682, 108263. [Google Scholar] [CrossRef]
- Montioli, R.; Oppici, E.; Cellini, B.; Roncador, A.; Dindo, M.; Voltattorni, C.B. S250F variant associated with aromatic amino acid decarboxylase deficiency: Molecular defects and intracellular rescue by pyridoxine. Hum. Mol. Genet. 2013, 22, 1615–1624. [Google Scholar] [CrossRef]
- Montioli, R.; Janson, G.; Paiardini, A.; Bertoldi, M.; Borri Voltattorni, C. Heterozygosis in aromatic amino acid decarboxylase deficiency: Evidence for a positive interallelic complementation between R347Q and R358H mutations. IUBMB Life 2018, 70, 215–223. [Google Scholar] [CrossRef]
- Montioli, R.; Battini, R.; Paiardini, A.; Tolve, M.; Bertoldi, M.; Carducci, C.; Leuzzi, V.; Borri Voltattorni, C. A novel compound heterozygous genotype associated with aromatic amino acid decarboxylase deficiency: Clinical aspects and biochemical studies. Mol. Genet. Metab. 2019, 127, 132–137. [Google Scholar] [CrossRef]
- McInnes, R.R.; Shih, V.; Chilton, S. Interallelic complementation in an inborn error of metabolism: Genetic heterogeneity in argininosuccinate lyase deficiency. Proc. Natl. Acad. Sci. USA 1984, 81, 4480–4484. [Google Scholar] [CrossRef]
- Janata, J.; Kogekar, N.; Fenton, W.A. Expression and kinetic characterization of methylmalonyl-CoA mutase from patients with the mut-phenotype: Evidence for naturally occurring interallelic complementation. Hum. Mol. Genet. 1997, 6, 1457–1464. [Google Scholar] [CrossRef]
- Ledley, F.D.; Rosenblatt, D.S. Mutations in mut methylmalonic acidemia: Clinical and enzymatic correlations. Hum. Mutat. 1997, 9, 1–6. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Shen, N.; Heintz, C.; Thiel, C.; Okun, J.G.; Hoffmann, G.F.; Blau, N. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol. Genet. Metab. 2016, 117, 328–335. [Google Scholar] [CrossRef]
- Heintz, C.; Cotton, R.G.; Blau, N. Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase, and importance of genotypes for pharmacological therapy of phenylketonuria. Hum. Mutat. 2013, 34, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Leandro, J.; Nascimento, C.; de Almeida, I.T.; Leandro, P. Co-expression of different subunits of human phenylalanine hydroxylase: Evidence of negative interallelic complementation. Biochim. Biophys. Acta 2006, 1762, 544–550. [Google Scholar] [CrossRef][Green Version]
- Leandro, J.; Leandro, P.; Flatmark, T. Heterotetrameric forms of human phenylalanine hydroxylase: Co-expression of wild-type and mutant forms in a bicistronic system. Biochim. Biophys. Acta 2011, 1812, 602–612. [Google Scholar] [CrossRef]
- Himmelreich, N.; Shen, N.; Okun, J.G.; Thiel, C.; Hoffmann, G.F.; Blau, N. Relationship between genotype, phenylalanine hydroxylase expression and in vitro activity and metabolic phenotype in phenylketonuria. Mol. Genet. Metab. 2018, 125, 86–95. [Google Scholar] [CrossRef]
- Garbade, S.F.; Shen, N.; Himmelreich, N.; Haas, D.; Trefz, F.K.; Hoffmann, G.F.; Burgard, P.; Blau, N. Allelic phenotype values: A model for genotype-based phenotype prediction in phenylketonuria. Genet. Med. 2019, 21, 580–590. [Google Scholar] [CrossRef]
- Wang, R.; Shen, N.; Ye, J.; Han, L.; Qiu, W.; Zhang, H.; Liang, L.; Sun, Y.; Fan, Y.; Wang, L.; et al. Mutation spectrum of hyperphenylalaninemia candidate genes and the genotype-phenotype correlation in the Chinese population. Clin. Chim. Acta 2018, 481, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Brennenstuhl, H.; Didiasova, M.; Assmann, B.; Bertoldi, M.; Molla, G.; Jung-Klawitter, S.; Kuseyri Hübschmann, O.; Schröter, J.; Opladen, T.; Tikkanen, R. Succinic Semialdehyde Dehydrogenase Deficiency: In Vitro and In Silico Characterization of a Novel Pathogenic Missense Variant and Analysis of the Mutational Spectrum of. Int. J. Mol. Sci. 2020, 21, 8578. [Google Scholar] [CrossRef] [PubMed]
- Hübschmann, O.K.; Juliá-Palacios, N.A.; Olivella, M.; Guder, P.; Zafeiriou, D.I.; Horvath, G.; Kulhánek, J.; Pearson, T.S.; Kuster, A.; Cortès-Saladelafont, E.; et al. Integrative Approach to Predict Severity in Nonketotic Hyperglycinemia. Ann. Neurol. 2022, 92, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.A.; Coughlin, C.R.; Scharer, G.H.; Szerlong, H.J.; Bjoraker, K.J.; Spector, E.B.; Creadon-Swindell, G.; Mahieu, V.; Matthijs, G.; Hennermann, J.B.; et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann. Neurol. 2015, 78, 606–618. [Google Scholar] [CrossRef]
- Farris, J.; Calhoun, B.; Alam, M.S.; Lee, S.; Haldar, K. Large scale analyses of genotype-phenotype relationships of glycine decarboxylase mutations and neurological disease severity. PLoS Comput. Biol. 2020, 16, e1007871. [Google Scholar] [CrossRef]
- Montioli, R.; Roncador, A.; Oppici, E.; Mandrile, G.; Giachino, D.F.; Cellini, B.; Borri Voltattorni, C. S81L and G170R mutations causing Primary Hyperoxaluria type I in homozygosis and heterozygosis: An example of positive interallelic complementation. Hum. Mol. Genet. 2014, 23, 5998–6007. [Google Scholar] [CrossRef]
- Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.N.; Malashkevich, V.N. Structural insight into Parkinson’s disease treatment from drug-inhibited DOPA decarboxylase. Nat. Struct. Biol. 2001, 8, 963–967. [Google Scholar] [CrossRef]
- Paiardini, A.; Giardina, G.; Rossignoli, G.; Voltattorni, C.B.; Bertoldi, M. New Insights Emerging from Recent Investigations on Human Group II Pyridoxal 5′-Phosphate Decarboxylases. Curr. Med. Chem. 2017, 24, 226–244. [Google Scholar] [CrossRef]
- Portaro, S.; Gugliandolo, A.; Scionti, D.; Cammaroto, S.; Morabito, R.; Leonardi, S.; Fraggetta, F.; Bramanti, P.; Mazzon, E. When dysphoria is not a primary mental state: A case report of the role of the aromatic L-aminoacid decarboxylase. Medicine 2018, 97, e10953. [Google Scholar] [CrossRef]
- Leuzzi, V.; Mastrangelo, M.; Polizzi, A.; Artiola, C.; van Kuilenburg, A.B.; Carducci, C.; Ruggieri, M.; Barone, R.; Tavazzi, B.; Abeling, N.G.; et al. Report of two never treated adult sisters with aromatic L-amino Acid decarboxylase deficiency: A portrait of the natural history of the disease or an expanding phenotype? JIMD Rep. 2015, 15, 39–45. [Google Scholar] [CrossRef]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Li, M.H. Natural History of Aromatic L-Amino Acid Decarboxylase Deficiency in Taiwan. JIMD Rep. 2018, 40, 1–6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
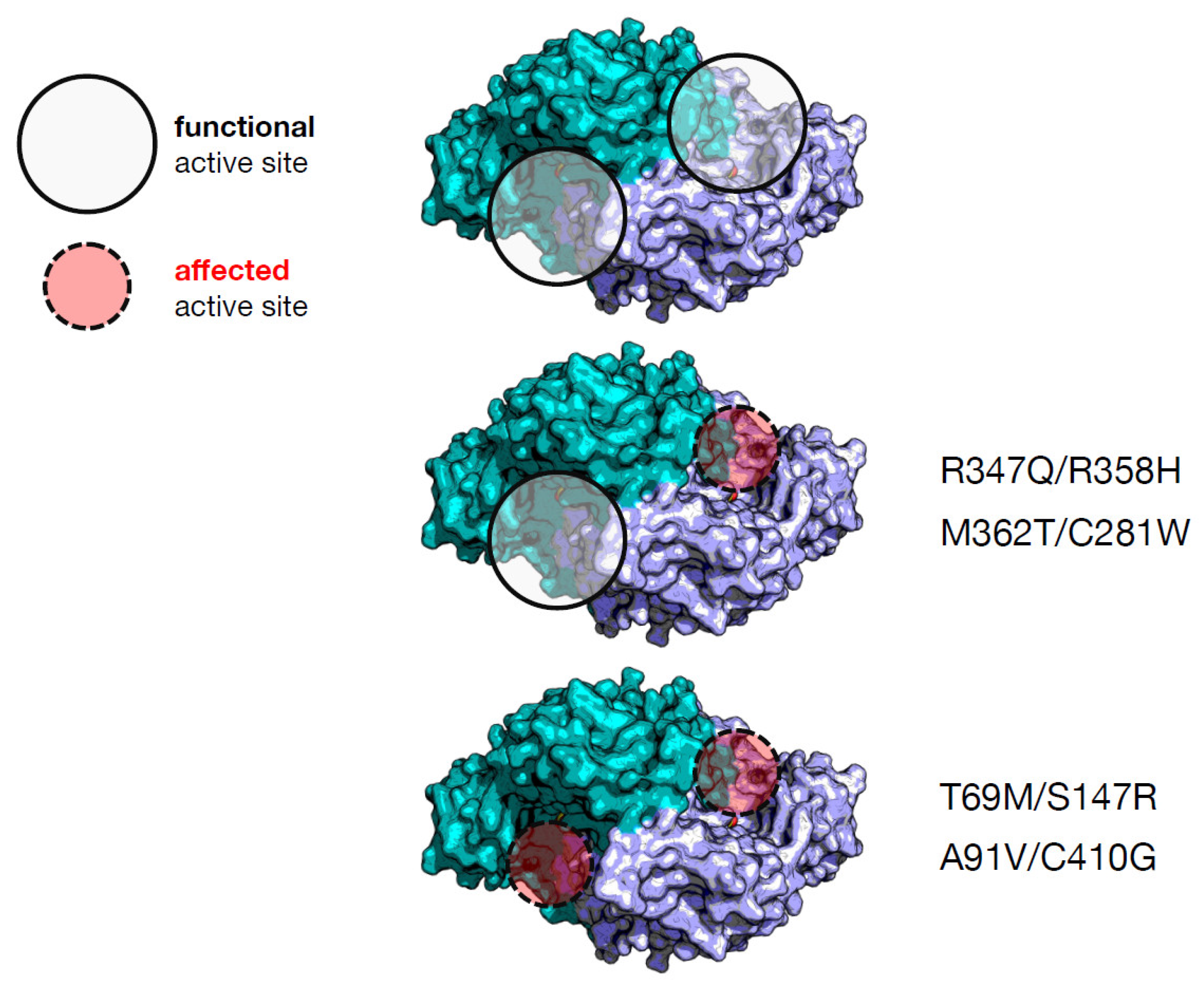
| Heterodimer | Individual Effect on Activity | Interallelic Complementation in Activity | Number of Active Sites Affected | Catalytic Constant kcat (s−1) d | PLP Binding Affinity (nM) d | Clinical Phenotype Output |
|---|---|---|---|---|---|---|
| T69M/S147R a | Mild/severe | negative | 2 | 0.27 (4%) e | 310 (3.9-fold) | severe |
| M362T/C281W a | Mild/mild | negative | 1 | 1.8 (29%) | 1232 (15.4-fold) | mild |
| A91V/C410G b | Severe/mild | negative | 2 | 0.38 (6%) | ≈000 (≈12.5-fold) | mild |
| R347Q/R358H c | Severe/severe | positive | 1 | 0.45 (7%) | 360 (4.5-fold) | severe |
| Homodimer | Expression a | Catalytic Constant kcat (s−1) b per Dimer | PLP Binding Affinity (nM) b | Homozygous Patient Phenotype |
|---|---|---|---|---|
| T69M | good | 3.5 | 100 | mild |
| S147R | quite good | 0.0090 | 846 c | n.p. |
| M362T | good | 4.6 | 271 | n.p. |
| C281W | poor | n.d. | n.d. | n.p. |
| A91V | quite good | 0.0083 | 782 | n.p. |
| C410G | good | 4.4 | 1070 | n.p. |
| R347Q | good | 0.087 | 54 | severe |
| R358H | good | 0.030 | 1300 | n.p. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisello, G.; Bertoldi, M. Compound Heterozygosis in AADC Deficiency and Its Complex Phenotype in Terms of AADC Protein Population. Int. J. Mol. Sci. 2022, 23, 11238. https://doi.org/10.3390/ijms231911238
Bisello G, Bertoldi M. Compound Heterozygosis in AADC Deficiency and Its Complex Phenotype in Terms of AADC Protein Population. International Journal of Molecular Sciences. 2022; 23(19):11238. https://doi.org/10.3390/ijms231911238
Chicago/Turabian StyleBisello, Giovanni, and Mariarita Bertoldi. 2022. "Compound Heterozygosis in AADC Deficiency and Its Complex Phenotype in Terms of AADC Protein Population" International Journal of Molecular Sciences 23, no. 19: 11238. https://doi.org/10.3390/ijms231911238
APA StyleBisello, G., & Bertoldi, M. (2022). Compound Heterozygosis in AADC Deficiency and Its Complex Phenotype in Terms of AADC Protein Population. International Journal of Molecular Sciences, 23(19), 11238. https://doi.org/10.3390/ijms231911238