

Molecular Mechanisms of Cellular Injury and Role of Toxic Heavy Metals in Chronic Kidney Disease

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
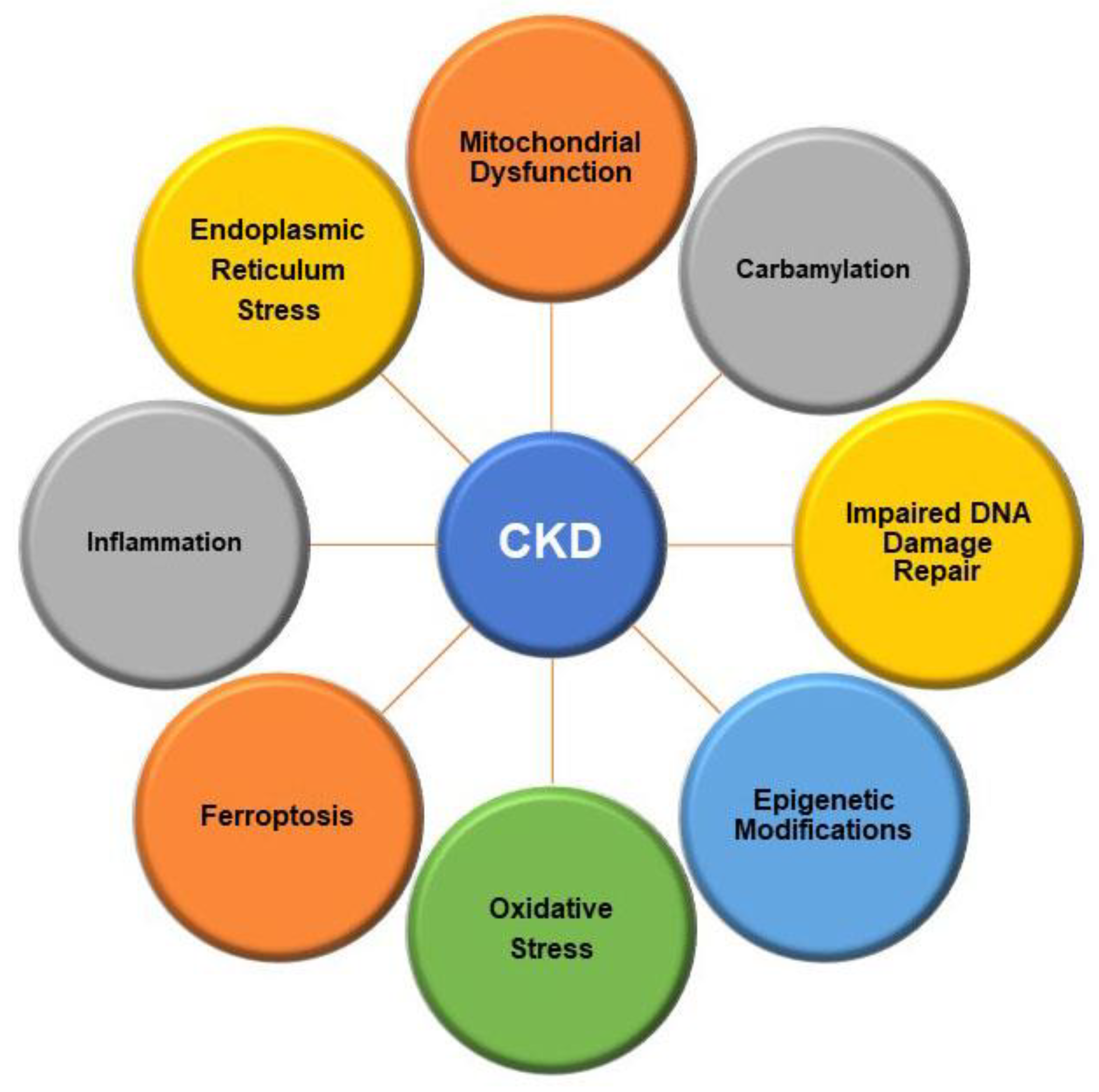
2. Pathophysiology of Chronic Kidney Disease
2.1. Structural Alterations within the Kidney
2.1.1. Podocytes
2.1.2. Capillary Network
2.1.3. Tubular Epithelial Cells
2.2. Intracellular Alterations
2.2.1. Mitochondrial Function
2.2.2. Oxidative Stress
2.2.3. Autophagy
2.2.4. Endoplasmic Reticulum Stress
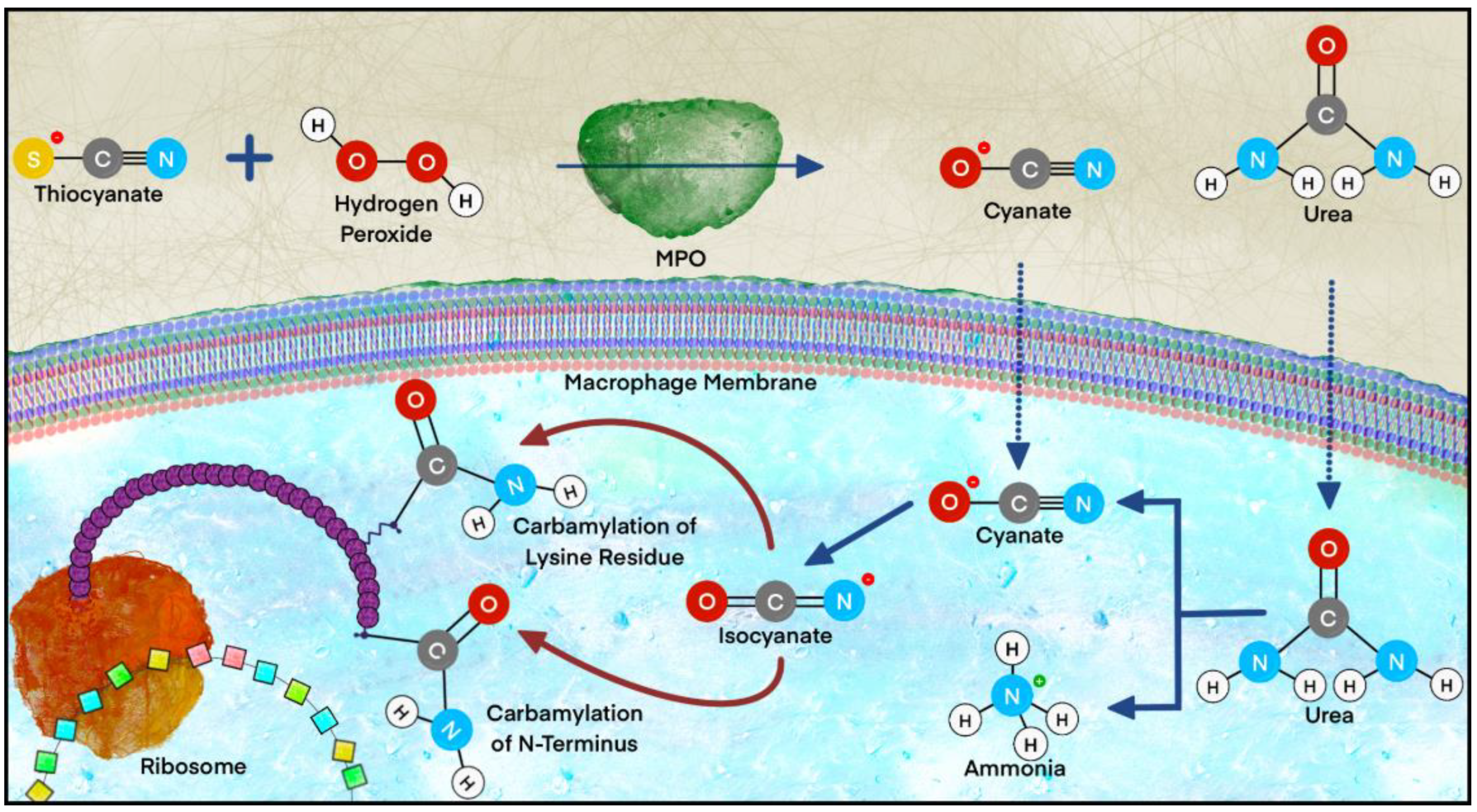
2.2.5. Carbamylation
2.2.6. Ferroptosis
2.2.7. DNA Damage and Repair
2.2.8. Epigenetic Modifications
2.2.9. Cellular Senescence
2.2.10. Inflammation
2.2.11. Fibrosis
3. Molecular Effects of Environmental Toxicants on CKD Progression
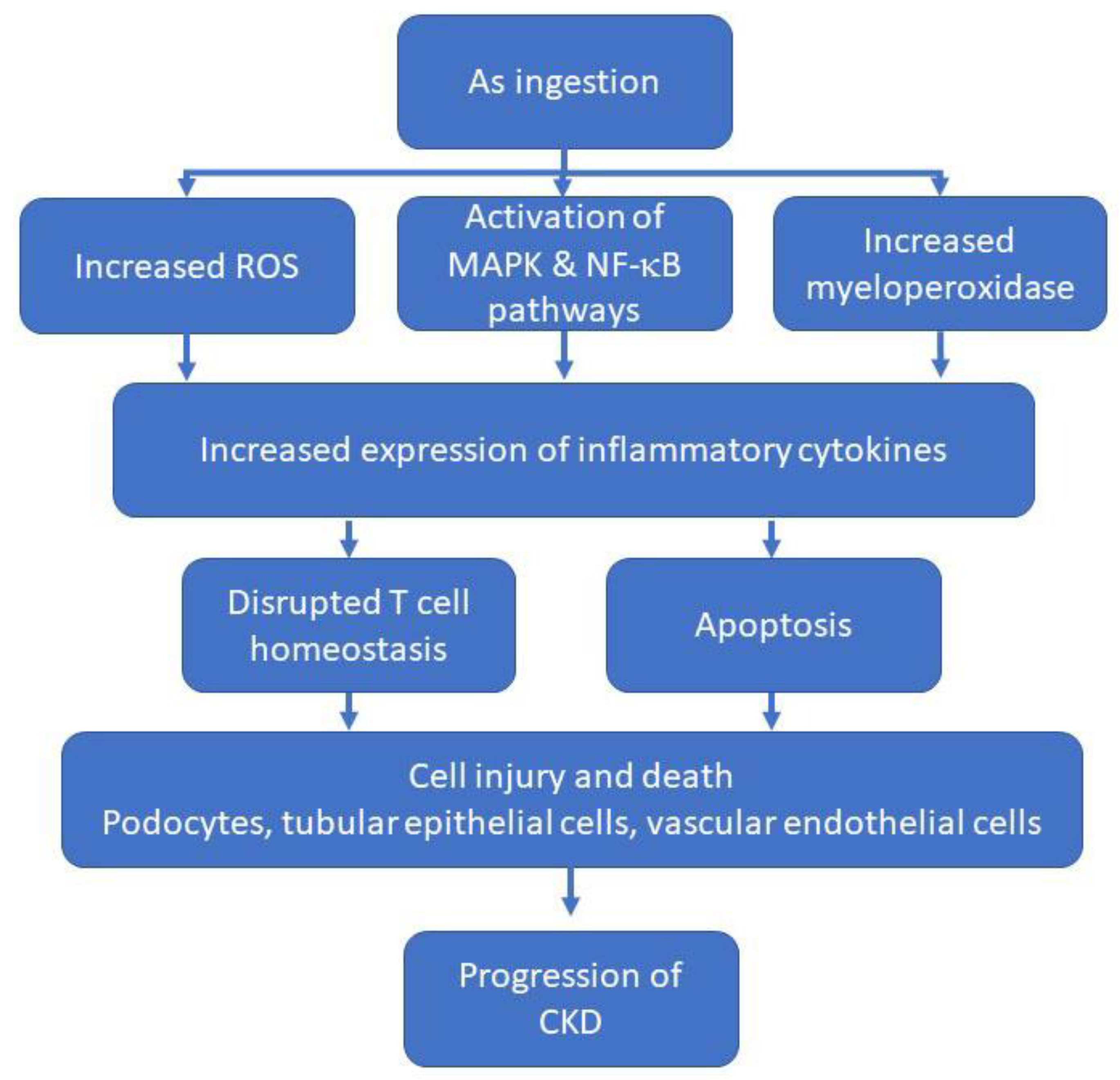
3.1. Arsenic
3.2. Cadmium
3.3. Mercury
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ATSDR. Toxicological Profile for Mercury; Public Health Service, Ed.; U.S. Department of Health and Human Services, Centers for Disease Control: Atlanta, GA, USA, 2008.
- Gaitonde, D.Y.; Cook, D.L.; Rivera, I.M. Chronic Kidney Disease: Detection and Evaluation. Am. Fam. Physician 2017, 96, 776–783. [Google Scholar] [PubMed]
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef]
- Levin, A.; Stevens, P.E. The authors reply. Kidney Int. 2013, 84, 623. [Google Scholar] [CrossRef]
- Li, L.; Astor, B.C.; Lewis, J.; Hu, B.; Appel, L.J.; Lipkowitz, M.S.; Toto, R.D.; Wang, X.; Wright, J.T., Jr.; Greene, T.H. Longitudinal progression trajectory of GFR among patients with CKD. Am. J. Kidney Dis. 2012, 59, 504–512. [Google Scholar] [CrossRef]
- Yan, M.T.; Chao, C.T.; Lin, S.H. Chronic Kidney Disease: Strategies to Retard Progression. Int. J. Mol. Sci. 2021, 22, 10084. [Google Scholar] [CrossRef]
- Ekrikpo, U.E.; Kengne, A.P.; Bello, A.K.; Effa, E.E.; Noubiap, J.J.; Salako, B.L.; Rayner, B.L.; Remuzzi, G.; Okpechi, I.G. Chronic kidney disease in the global adult HIV-infected population: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0195443. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Acute kidney injury and chronic kidney disease: From the laboratory to the clinic. Nephrol. Ther. 2016, 12 (Suppl. 1), S41–S48. [Google Scholar] [CrossRef]
- Charles, C.; Ferris, A.H. Chronic Kidney Disease. Prim. Care 2020, 47, 585–595. [Google Scholar] [CrossRef]
- Chevalier, R.L. Evolution, kidney development, and chronic kidney disease. Semin. Cell Dev. Biol. 2019, 91, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Hostetter, T.H.; Olson, J.L.; Rennke, H.G.; Venkatachalam, M.A. Hyperfiltration in remnant nephrons: A potentially adverse response to renal ablation. J. Am. Soc. Nephrol. 2001, 12, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, T.; Dai, H.; Heruth, D.P.; Alon, U.S.; Garola, R.E.; Zhou, J.; Duncan, R.S.; El-Meanawy, A.; McCarthy, E.T.; Sharma, R.; et al. Mechanotransduction signaling in podocytes from fluid flow shear stress. Am. J. Physiol. Ren. Physiol. 2018, 314, F22–F34. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef]
- Blaine, J.; Dylewski, J. Regulation of the Actin Cytoskeleton in Podocytes. Cells 2020, 9, 1700. [Google Scholar] [CrossRef]
- Kim, J.J.; Wilbon, S.S.; Fornoni, A. Podocyte Lipotoxicity in CKD. Kidney360 2021, 2, 755–762. [Google Scholar] [CrossRef]
- Wanner, N.; Hartleben, B.; Herbach, N.; Goedel, M.; Stickel, N.; Zeiser, R.; Walz, G.; Moeller, M.J.; Grahammer, F.; Huber, T.B. Unraveling the role of podocyte turnover in glomerular aging and injury. J. Am. Soc. Nephrol. 2014, 25, 707–716. [Google Scholar] [CrossRef]
- Hodgin, J.B.; Bitzer, M.; Wickman, L.; Afshinnia, F.; Wang, S.Q.; O’Connor, C.; Yang, Y.; Meadowbrooke, C.; Chowdhury, M.; Kikuchi, M.; et al. Glomerular Aging and Focal Global Glomerulosclerosis: A Podometric Perspective. J. Am. Soc. Nephrol. 2015, 26, 3162–3178. [Google Scholar] [CrossRef]
- Wiggins, J.E.; Goyal, M.; Sanden, S.K.; Wharram, B.L.; Shedden, K.A.; Misek, D.E.; Kuick, R.D.; Wiggins, R.C. Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: Prevention by calorie restriction. J. Am. Soc. Nephrol. 2005, 16, 2953–2966. [Google Scholar] [CrossRef]
- Trimarchi, H. Mechanisms of Podocyte Detachment, Podocyturia, and Risk of Progression of Glomerulopathies. Kidney Dis. 2020, 6, 324–329. [Google Scholar] [CrossRef]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Hillebrand, U.; Amler, S.; Konig, M.; Larger, E.; Oberleithner, H.; Brand, E.; Pavenstadt, H.; Brand, M. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2009, 20, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Babickova, J.; Klinkhammer, B.M.; Buhl, E.M.; Djudjaj, S.; Hoss, M.; Heymann, F.; Tacke, F.; Floege, J.; Becker, J.U.; Boor, P. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017, 91, 70–85. [Google Scholar] [CrossRef]
- Ehling, J.; Babickova, J.; Gremse, F.; Klinkhammer, B.M.; Baetke, S.; Knuechel, R.; Kiessling, F.; Floege, J.; Lammers, T.; Boor, P. Quantitative Micro-Computed Tomography Imaging of Vascular Dysfunction in Progressive Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Urbieta-Caceres, V.H.; Syed, F.A.; Lin, J.; Zhu, X.Y.; Jordan, K.L.; Bell, C.C.; Bentley, M.D.; Lerman, A.; Khosla, S.; Lerman, L.O. Age-dependent renal cortical microvascular loss in female mice. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E979–E986. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.M.; Zhang, Z.J.; Wang, J.J.; Thomson, A.W.; Isenberg, J.S. CD47 regulates renal tubular epithelial cell self-renewal and proliferation following renal ischemia reperfusion. Kidney Int. 2016, 90, 334–347. [Google Scholar] [CrossRef]
- Kida, Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression. Int. J. Mol. Sci. 2020, 21, 8255. [Google Scholar] [CrossRef]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef]
- Uesugi, N.; Shimazu, Y.; Kikuchi, K.; Nagata, M. Age-Related Renal Microvascular Changes: Evaluation by Three-Dimensional Digital Imaging of the Human Renal Microcirculation Using Virtual Microscopy. Int. J. Mol. Sci. 2016, 17, 1831. [Google Scholar] [CrossRef]
- Berkenkamp, B.; Susnik, N.; Baisantry, A.; Kuznetsova, I.; Jacobi, C.; Sorensen-Zender, I.; Broecker, V.; Haller, H.; Melk, A.; Schmitt, R. In vivo and in vitro analysis of age-associated changes and somatic cellular senescence in renal epithelial cells. PLoS ONE 2014, 9, e88071. [Google Scholar] [CrossRef]
- Forbes, M.S.; Thornhill, B.A.; Park, M.H.; Chevalier, R.L. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am. J. Pathol. 2007, 170, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Noiri, E.; Satoh, H.; Taguchi, J.; Brodsky, S.V.; Nakao, A.; Ogawa, Y.; Nishijima, S.; Yokomizo, T.; Tokunaga, K.; Fujita, T. Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension 2002, 40, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, J.M. Mitochondrial biogenesis in kidney disease. J. Am. Soc. Nephrol. 2011, 22, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Bohman, R.; Navas, P.; Norman, J.T.; Bradley, T.; Fine, L.G. Hypertrophy of renal mitochondria. J. Am. Soc. Nephrol. 1990, 1, 822–827. [Google Scholar] [CrossRef]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Eirin, A.; Lerman, A.; Lerman, L.O. The Emerging Role of Mitochondrial Targeting in Kidney Disease. In Handbook of Experimental Pharmacology; Springer: New York, NY, USA, 2017; Volume 240, pp. 229–250. [Google Scholar]
- Layton, A.T.; Edwards, A.; Vallon, V. Adaptive changes in GFR, tubular morphology, and transport in subtotal nephrectomized kidneys: Modeling and analysis. Am. J. Physiol. Ren. Physiol. 2017, 313, F199–F209. [Google Scholar] [CrossRef]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. Punctum on two different transcription factors regulated by PGC-1alpha: Nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim. Biophys. Acta 2013, 1830, 4137–4146. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Parikh, S.M. PGC1alpha in the kidney. Am. J. Physiol. Ren. Physiol. 2018, 314, F1–F8. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef]
- Namba, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Yamamoto, T.; Matsuda, J.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Iwatani, H.; et al. Autophagic clearance of mitochondria in the kidney copes with metabolic acidosis. J. Am. Soc. Nephrol. 2014, 25, 2254–2266. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; He, L.; Liu, J.; Dong, Z. Mitophagy: Basic Mechanism and Potential Role in Kidney Diseases. Kidney Dis. 2015, 1, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sun, Z.; Varghese, Z.; Guo, Y.; Moorhead, J.F.; Unwin, R.J.; Ruan, X.Z. Nonesterified free fatty acids enhance the inflammatory response in renal tubules by inducing extracellular ATP release. Am. J. Physiol. Ren. Physiol. 2020, 319, F292–F303. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Gyuraszova, M.; Gurecka, R.; Babickova, J.; Tothova, L. Oxidative Stress in the Pathophysiology of Kidney Disease: Implications for Noninvasive Monitoring and Identification of Biomarkers. Oxidative Med. Cell. Longev. 2020, 2020, 5478708. [Google Scholar] [CrossRef]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, R.E.; Mohammed-Ali, Z.; Lu, C.; Yousof, T.; Tat, V.; Nademi, S.; MacDonald, M.E.; Austin, R.C.; Dickhout, J.G. TDAG51 induces renal interstitial fibrosis through modulation of TGF-beta receptor 1 in chronic kidney disease. Cell Death Dis. 2021, 12, 921. [Google Scholar] [CrossRef] [PubMed]
- Bignon, Y.; Rinaldi, A.; Nadour, Z.; Poindessous, V.; Nemazanyy, I.; Lenoir, O.; Fohlen, B.; Weill-Raynal, P.; Hertig, A.; Karras, A.; et al. Cell stress response impairs de novo NAD+ biosynthesis in the kidney. JCI Insight 2022, 7, e153019. [Google Scholar] [CrossRef]
- Gorisse, L.; Jaisson, S.; Pietrement, C.; Gillery, P. Carbamylated Proteins in Renal Disease: Aggravating Factors or Just Biomarkers? Int. J. Mol. Sci. 2022, 23, 574. [Google Scholar] [CrossRef]
- Zhang, X.; Li, X. Abnormal Iron and Lipid Metabolism Mediated Ferroptosis in Kidney Diseases and Its Therapeutic Potential. Metabolites 2022, 12, 58. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef]
- Pan, M.R.; Li, K.; Lin, S.Y.; Hung, W.C. Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci. 2016, 17, 685. [Google Scholar] [CrossRef]
- Kishi, S.; Brooks, C.R.; Taguchi, K.; Ichimura, T.; Mori, Y.; Akinfolarin, A.; Gupta, N.; Galichon, P.; Elias, B.C.; Suzuki, T.; et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J. Clin. Investig. 2019, 129, 4797–4816. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Castellano, G.; Franzin, R.; Sallustio, F.; Stasi, A.; Banelli, B.; Romani, M.; De Palma, G.; Lucarelli, G.; Divella, C.; Battaglia, M.; et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/betacatenin signaling after ischemia/reperfusion injury. Aging 2019, 11, 4382–4406. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.Y.; Tin, A.; Schlosser, P.; Ko, Y.A.; Qiu, C.; Yao, C.; Joehanes, R.; Grams, M.E.; Liang, L.; Gluck, C.A.; et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 2017, 8, 1286. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Marchant, V.; Rodrigues-Diez, R.; Dolade, N.; Suarez-Alvarez, B.; Kerr, B.; Valdivielso, J.M.; Ruiz-Ortega, M.; Rayego-Mateos, S. Epigenetic Modification Mechanisms Involved in Inflammation and Fibrosis in Renal Pathology. Mediat. Inflamm. 2018, 2018, 2931049. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Karimi, M.; Johansson, S.; Axelsson, J.; Suliman, M.; Lindholm, B.; Heimburger, O.; Barany, P.; Alvestrand, A.; Nordfors, L.; et al. Impact of inflammation on epigenetic DNA methylation—A novel risk factor for cardiovascular disease? J. Intern. Med. 2007, 261, 488–499. [Google Scholar] [CrossRef]
- Zhang, H.; Cai, X.; Yi, B.; Huang, J.; Wang, J.; Sun, J. Correlation of CTGF gene promoter methylation with CTGF expression in type 2 diabetes mellitus with or without nephropathy. Mol. Med. Rep. 2014, 9, 2138–2144. [Google Scholar] [CrossRef]
- Franzin, R.; Stasi, A.; Ranieri, E.; Netti, G.S.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Castellano, G. Targeting Premature Renal Aging: From Molecular Mechanisms of Cellular Senescence to Senolytic Trials. Front. Pharmacol. 2021, 12, 630419. [Google Scholar] [CrossRef]
- Sanchez-Navarro, A.; Perez-Villalva, R.; Murillo-de-Ozores, A.R.; Martinez-Rojas, M.A.; Rodriguez-Aguilera, J.R.; Gonzalez, N.; Castaneda-Bueno, M.; Gamba, G.; Recillas-Targa, F.; Bobadilla, N.A. Vegfa promoter gene hypermethylation at HIF1alpha binding site is an early contributor to CKD progression after renal ischemia. Sci. Rep. 2021, 11, 8769. [Google Scholar] [CrossRef]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Wing, M.R.; Ramezani, A.; Gill, H.S.; Devaney, J.M.; Raj, D.S. Epigenetics of progression of chronic kidney disease: Fact or fantasy? Semin. Nephrol. 2013, 33, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Kim, J.G.; Kim, H.J.; Kwon, H.K.; Cho, I.J.; Choi, D.W.; Lee, W.H.; Kim, W.D.; Hwang, S.J.; Choi, S.; et al. Discovery of an integrative network of microRNAs and transcriptomics changes for acute kidney injury. Kidney Int. 2014, 86, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ma, S.X.; Shang, Y.Q.; Zhang, H.Q.; Su, W. microRNAs in chronic kidney disease. Clin. Chim. Acta 2019, 491, 59–65. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.J.; Cai, G.Y.; Chen, X.M. Cellular senescence, senescence-associated secretory phenotype, and chronic kidney disease. Oncotarget 2017, 8, 64520–64533. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Melk, A. New insights on molecular mechanisms of renal aging. Am. J. Transplant. 2012, 12, 2892–2900. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Falke, L.L.; Nguyen, T.Q.; Goldschmeding, R. Cellular senescence in the aging and diseased kidney. J. Cell Commun. Signal 2018, 12, 69–82. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, S.; Zhang, Q.; Jin, B.; Lv, D.; Li, W.; Zhao, M.; Jiang, C.; Dai, C.; Liu, Z. Integrin beta3 Induction Promotes Tubular Cell Senescence and Kidney Fibrosis. Front. Cell Dev. Biol. 2021, 9, 733831. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Crowley, S.D. The varying roles of macrophages in kidney injury and repair. Curr. Opin. Nephrol. Hypertens. 2020, 29, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Hostetter, T.H. Hyperfiltration and glomerulosclerosis. Semin. Nephrol. 2003, 23, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Taal, M.W.; Omer, S.A.; Nadim, M.K.; Mackenzie, H.S. Cellular and molecular mediators in common pathway mechanisms of chronic renal disease progression. Curr. Opin. Nephrol. Hypertens. 2000, 9, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.E.; Kelly, D.J.; Cox, A.J.; Zhang, Y.; Gow, R.M.; Gilbert, R.E. Mast cell infiltration and chemokine expression in progressive renal disease. Kidney Int. 2003, 64, 906–913. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Paton, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. Macrophage Phenotype in Kidney Injury and Repair. Kidney Dis. 2015, 1, 138–146. [Google Scholar] [CrossRef]
- DeWolf, S.E.; Kasimsetty, S.G.; Hawkes, A.A.; Stocks, L.M.; Kurian, S.M.; McKay, D.B. DAMPs Released From Injured Renal Tubular Epithelial Cells Activate Innate Immune Signals in Healthy Renal Tubular Epithelial Cells. Transplantation 2022, 106, 1589–1599. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, X.; Yang, S.; Peng, Y.; Hou, F.; Zhou, Q. High salt aggravates renal inflammation via promoting pro-inflammatory macrophage in 5/6-nephrectomized rat. Life Sci. 2021, 274, 119109. [Google Scholar] [CrossRef]
- Montford, J.R.; Bauer, C.; Rahkola, J.; Reisz, J.A.; Floyd, D.; Hopp, K.; Soranno, D.E.; Klawitter, J.; Weiser-Evans, M.C.M.; Nemenoff, R.; et al. 15-Lipoxygenase worsens renal fibrosis, inflammation, and metabolism in a murine model of ureteral obstruction. Am. J. Physiol. Ren. Physiol. 2022, 322, F105–F119. [Google Scholar] [CrossRef]
- Huang, C.C.; Chou, C.A.; Chen, W.Y.; Yang, J.L.; Lee, W.C.; Chen, J.B.; Lee, C.T.; Li, L.C. Empagliflozin Ameliorates Free Fatty Acid Induced-Lipotoxicity in Renal Proximal Tubular Cells via the PPARgamma/CD36 Pathway in Obese Mice. Int. J. Mol. Sci. 2021, 22, 12408. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.Y.; Jing, H.Y.; Ma, Y.R. Interleukin-33/Suppression of Tumorigenicity 2 in Renal Fibrosis: Emerging Roles in Prognosis and Treatment. Front. Physiol. 2021, 12, 792897. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.; Gavela, E.; Vizcaino, B.; Huarte, E.; Carrero, J.J. Optimizing Diet to Slow CKD Progression. Front. Med. 2021, 8, 654250. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Duffield, J.S.; Humphreys, B.D. The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 2012, 27, 183–193. [Google Scholar] [CrossRef]
- Strutz, F.; Zeisberg, M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 2992–2998. [Google Scholar] [CrossRef]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef]
- Lovisa, S.; Zeisberg, M.; Kalluri, R. Partial Epithelial-to-Mesenchymal Transition and Other New Mechanisms of Kidney Fibrosis. Trends Endocrinol. Metab. TEM 2016, 27, 681–695. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, H.; Jiang, H.; Fan, Y.; Zhang, J.; Ma, X.; Yang, X.; Sun, Y.; Zhao, X. The ILEI/LIFR complex induces EMT via the Akt and ERK pathways in renal interstitial fibrosis. J. Transl. Med. 2022, 20, 54. [Google Scholar] [CrossRef]
- Ostendorf, T.; Boor, P.; van Roeyen, C.R.; Floege, J. Platelet-derived growth factors (PDGFs) in glomerular and tubulointerstitial fibrosis. Kidney Int. Suppl. (2011) 2014, 4, 65–69. [Google Scholar] [CrossRef]
- Buhl, E.M.; Djudjaj, S.; Klinkhammer, B.M.; Ermert, K.; Puelles, V.G.; Lindenmeyer, M.T.; Cohen, C.D.; He, C.; Borkham-Kamphorst, E.; Weiskirchen, R.; et al. Dysregulated mesenchymal PDGFR-beta drives kidney fibrosis. EMBO Mol. Med. 2020, 12, e11021. [Google Scholar] [CrossRef] [PubMed]
- WHO Ten Chemicals of Major Public Health Concern. Available online: http://www.who.int/ipcs/assessment/public_health/chemicals_phc/en/ (accessed on 23 July 2018).
- ATSDR. Public Health Statement: Arsenic; Center for Disease Control and Prevention: Atlanta, GA, USA, 2007.
- Nriagu, J.O.; Bhattacharya, P.; Mukherjee, A.B.; Bundschuh, J.; Zevenhoven, R.; Leoppert, R.H. Arsenic in soil and graoundwater: An overview. In Trace Metals and Other Contaminants in the Environment; Bhattacharya, P., Mukherjee, A.B., Bundschuh, J., Zevenhoven, R., Loeppert, R.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 9, pp. 3–30. [Google Scholar]
- Shankar, S.; Shanker, U.; Shikha. Arsenic contamination of groundwater: A review of sources, prevalence, health risks, and strategies for mitigation. Sci. World J. 2014, 2014, 304524. [Google Scholar] [CrossRef] [PubMed]
- van Halem, D.; Bakker, S.A.; Amy, G.L.; van Dijk, J.C. Arsenic in drinking water: A worldwide quality concern for water supply companies. Drink. Water Eng. Sci. 2009, 2, 29–34. [Google Scholar] [CrossRef]
- Hong, Y.S.; Song, K.H.; Chung, J.Y. Health effects of chronic arsenic exposure. J. Prev. Med. Public Health 2014, 47, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Howard, G. Arsenic, Drinking-Water and Health Risks Substitution in Arsenic Mitigation: A Discussion Paper; World Health Organization: Geneva, Switzerland, 2003.
- Ratnaike, R.N. Acute and chronic arsenic toxicity. Postgrad. Med. J. 2003, 79, 391–396. [Google Scholar] [CrossRef]
- Martinez, V.D.; Vucic, E.A.; Becker-Santos, D.D.; Gil, L.; Lam, W.L. Arsenic exposure and the induction of human cancers. J. Toxicol. 2011, 2011, 431287. [Google Scholar] [CrossRef]
- Hafey, M.J.; Aleksunes, L.M.; Bridges, C.C.; Brouwer, K.R.; Chien, H.C.; Leslie, E.M.; Hu, S.; Li, Y.; Shen, J.; Sparreboom, A.; et al. Transporters and Toxicity: Insights from the International Transporter Consortium Workshop 4. Clin. Pharmacol. Ther. 2022, 112, 527–539. [Google Scholar] [CrossRef]
- Jalili, C.; Kazemi, M.; Cheng, H.; Mohammadi, H.; Babaei, A.; Taheri, E.; Moradi, S. Associations between exposure to heavy metals and the risk of chronic kidney disease: A systematic review and meta-analysis. Crit. Rev. Toxicol. 2021, 51, 165–182. [Google Scholar] [CrossRef]
- Robles-Osorio, M.L.; Sabath-Silva, E.; Sabath, E. Arsenic-mediated nephrotoxicity. Ren. Fail. 2015, 37, 542–547. [Google Scholar] [CrossRef]
- Roggenbeck, B.A.; Banerjee, M.; Leslie, E.M. Cellular arsenic transport pathways in mammals. J. Environ. Sci. (China) 2016, 49, 38–58. [Google Scholar] [CrossRef]
- Jin, W.; Xue, Y.; Xue, Y.; Han, X.; Song, Q.; Zhang, J.; Li, Z.; Cheng, J.; Guan, S.; Sun, S.; et al. Tannic acid ameliorates arsenic trioxide-induced nephrotoxicity, contribution of NF-kappaB and Nrf2 pathways. Biomed. Pharmacother. 2020, 126, 110047. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Xu, G.; Li, J.; Yan, N.; Li, X.; Liu, X.; Li, B. Arsenic Induces Continuous Inflammation and Regulates Th1/Th2/Th17/Treg Balance in Liver and Kidney In Vivo. Mediat. Inflamm. 2022, 2022, 8414047. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Gu, Y.; Yan, N.; Li, Y.; Sun, L.; Li, B. Curcumin functions as an anti-inflammatory and antioxidant agent on arsenic-induced hepatic and kidney injury by inhibiting MAPKs/NF-kappaB and activating Nrf2 pathways. Environ. Toxicol. 2021, 36, 2161–2173. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, H.; Shao, Y.; Liu, J.; Li, J.; Xing, M. Copper or/and arsenic induce oxidative stress-cascaded, nuclear factor kappa B-dependent inflammation and immune imbalance, trigging heat shock response in the kidney of chicken. Oncotarget 2017, 8, 98103–98116. [Google Scholar] [CrossRef][Green Version]
- Bermejo, S.; Bolufer, M.; Riveiro-Barciela, M.; Soler, M.J. Immunotherapy and the Spectrum of Kidney Disease: Should We Individualize the Treatment? Front. Med. (Lausanne) 2022, 9, 906565. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.P.; Tsai, K.S.; Chen, Y.W.; Huang, C.F.; Yang, R.S.; Liu, S.H. Arsenic induces apoptosis in myoblasts through a reactive oxygen species-induced endoplasmic reticulum stress and mitochondrial dysfunction pathway. Arch. Toxicol. 2012, 86, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- ATSDR. Toxicological Profile for Cadmium; U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control: Atlanta, GA, USA, 2008.
- WHO. Cadmium—Environmental Health Criteria 134; WHO: Geneva, Switzerland, 1992.
- MartzEmerson, M. FAQs about Cadmium in Fertilizer: Fertilizer Laws and Limits; Pacific Northwest Pollution Prevention Resource Center: Seattle, WA, USA, 2017; pp. 1–10. [Google Scholar]
- Ashraf, M.W. Levels of heavy metals in popular cigarette brands and exposure to these metals via smoking. Sci. World J. 2012, 2012, 729430. [Google Scholar] [CrossRef]
- Adams, S.V.; Newcomb, P.A. Cadmium blood and urine concentrations as measures of exposure: NHANES 1999-2010. J. Expo. Sci. Environ. Epidemiol. 2014, 24, 163–170. [Google Scholar] [CrossRef]
- Souza, V.; Bucio, L.; Gutierrez-Ruiz, M.C. Cadmium uptake by a human hepatic cell line (WRL-68 cells). Toxicology 1997, 120, 215–220. [Google Scholar] [CrossRef]
- Drobna, Z.; Styblo, M.; Thomas, D.J. An Overview of Arsenic Metabolism and Toxicity. Curr. Protoc. Toxicol. 2009, 42, 4–31. [Google Scholar]
- Khairul, I.; Wang, Q.Q.; Jiang, Y.H.; Wang, C.; Naranmandura, H. Metabolism, toxicity and anticancer activities of arsenic compounds. Oncotarget 2017, 8, 23905–23926. [Google Scholar] [CrossRef]
- Fujishiro, H.; Yano, Y.; Takada, Y.; Tanihara, M.; Himeno, S. Roles of ZIP8, ZIP14, and DMT1 in transport of cadmium and manganese in mouse kidney proximal tubule cells. Met. Integr. Biomet. Sci. 2012, 4, 700–708. [Google Scholar] [CrossRef]
- Jarup, L.; Hellstrom, L.; Alfven, T.; Carlsson, M.D.; Grubb, A.; Persson, B.; Pettersson, C.; Spang, G.; Schutz, A.; Elinder, C.G. Low level exposure to cadmium and early kidney damage: The OSCAR study. Occup. Environ. Med. 2000, 57, 668–672. [Google Scholar] [CrossRef]
- Ginsberg, G.L. Cadmium risk assessment in relation to background risk of chronic kidney disease. J. Toxicol. Environ. Health A 2012, 75, 374–390. [Google Scholar] [CrossRef]
- Ferraro, P.M.; Costanzi, S.; Naticchia, A.; Sturniolo, A.; Gambaro, G. Low level exposure to cadmium increases the risk of chronic kidney disease: Analysis of the NHANES 1999-2006. BMC Public Health 2010, 10, 304. [Google Scholar] [CrossRef]
- Edwards, J.R.; Prozialeck, W.C. Cadmium, diabetes and chronic kidney disease. Toxicol. Appl. Pharm. 2009, 238, 289–293. [Google Scholar] [CrossRef]
- Nguyen, J.; Patel, A.; Gensburg, A.; Bokhari, R.; Lamar, P.; Edwards, J. Diabetogenic and Obesogenic Effects of Cadmium in Db/Db Mice and Rats at a Clinically Relevant Level of Exposure. Toxics 2022, 10, 107. [Google Scholar] [CrossRef]
- Li, M.; Liu, X.; Zhang, Z. Hyperglycemia exacerbates cadmium-induced glomerular nephrosis. Toxicol. Ind. Health 2021, 37, 555–563. [Google Scholar] [CrossRef]
- Aramjoo, H.; Arab-Zozani, M.; Feyzi, A.; Naghizadeh, A.; Aschner, M.; Naimabadi, A.; Farkhondeh, T.; Samarghandian, S. The association between environmental cadmium exposure, blood pressure, and hypertension: A systematic review and meta-analysis. Environ. Sci. Pollut. Res. Int. 2022, 29, 35682–35706. [Google Scholar] [CrossRef]
- Giridhar, J.; Isom, G.E. Alteration of atrial natriuretic peptide levels by short term cadmium treatment. Toxicology 1991, 70, 185–194. [Google Scholar] [CrossRef]
- Giridhar, J.; Rathinavelu, A.; Isom, G.E. Interaction of cadmium with atrial natriuretic peptide receptors: Implications for toxicity. Toxicology 1992, 75, 133–143. [Google Scholar] [CrossRef]
- Nishida, K.; Watanabe, H.; Miyahisa, M.; Hiramoto, Y.; Nosaki, H.; Fujimura, R.; Maeda, H.; Otagiri, M.; Maruyama, T. Systemic and sustained thioredoxin analogue prevents acute kidney injury and its-associated distant organ damage in renal ischemia reperfusion injury mice. Sci. Rep. 2020, 10, 20635. [Google Scholar] [CrossRef]
- Oner, G.; Senturk, U.K.; Izgut-Uysal, V.N. Role of cadmium-induced lipid peroxidation in the kidney response to atrial natriuretic hormone. Nephron 1996, 72, 257–262. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, R.; Wang, X.; Shen, X.; Wang, P.; Sun, N.; Li, X.; Li, X.; Hai, C. Effects of sub-chronic, low-dose cadmium exposure on kidney damage and potential mechanisms. Ann. Transl. Med. 2019, 7, 177. [Google Scholar] [CrossRef]
- Ikediobi, C.O.; Badisa, V.L.; Ayuk-Takem, L.T.; Latinwo, L.M.; West, J. Response of antioxidant enzymes and redox metabolites to cadmium-induced oxidative stress in CRL-1439 normal rat liver cells. Int. J. Mol. Med. 2004, 14, 87–92. [Google Scholar] [CrossRef]
- Li, J.R.; Ou, Y.C.; Wu, C.C.; Wang, J.D.; Lin, S.Y.; Wang, Y.Y.; Chen, W.Y.; Liao, S.L.; Chen, C.J. Endoplasmic reticulum stress and autophagy contributed to cadmium nephrotoxicity in HK-2 cells and Sprague-Dawley rats. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2020, 146, 111828. [Google Scholar] [CrossRef]
- Wang, Q.W.; Wang, Y.; Wang, T.; Zhang, K.B.; Yuan, Y.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; Zhu, J.Q.; Liu, Z.P. Cadmium-induced autophagy is mediated by oxidative signaling in PC-12 cells and is associated with cytoprotection. Mol. Med. Rep. 2015, 12, 4448–4454. [Google Scholar] [CrossRef][Green Version]
- So, K.Y.; Park, B.H.; Oh, S.H. Cytoplasmic sirtuin 6 translocation mediated by p62 polyubiquitination plays a critical role in cadmium-induced kidney toxicity. Cell Biol. Toxicol. 2021, 37, 193–207. [Google Scholar] [CrossRef]
- Dong, W.; Liu, G.; Zhang, K.; Tan, Y.; Zou, H.; Yuan, Y.; Gu, J.; Song, R.; Zhu, J.; Liu, Z. Cadmium exposure induces rat proximal tubular cells injury via p62-dependent Nrf2 nucleus translocation mediated activation of AMPK/AKT/mTOR pathway. Ecotoxicol. Environ. Saf. 2021, 214, 112058. [Google Scholar] [CrossRef]
- Fan, R.F.; Tang, K.K.; Wang, Z.Y.; Wang, L. Persistent activation of Nrf2 promotes a vicious cycle of oxidative stress and autophagy inhibition in cadmium-induced kidney injury. Toxicology 2021, 464, 152999. [Google Scholar] [CrossRef]
- Lee, H.Y.; Oh, S.H. Autophagy-mediated cytoplasmic accumulation of p53 leads to apoptosis through DRAM-BAX in cadmium-exposed human proximal tubular cells. Biochem. Biophys. Res. Commun. 2021, 534, 128–133. [Google Scholar] [CrossRef]
- Go, Y.-M.; Orr, M.; Jones, D.P. Increased Nuclear Thioredoxin-1 Potentiates Cadmium-Induced Cytotoxicity. Toxicol. Sci. 2012, 131, 84–94. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, S.; Liu, H.; Xu, S. MAPK/iNOS pathway is involved in swine kidney necrosis caused by cadmium exposure. Environ. Pollut. 2021, 274, 116497. [Google Scholar] [CrossRef] [PubMed]
- Prozialeck, W.C.; Edwards, J.R. Mechanisms of cadmium-induced proximal tubule injury: New insights with implications for biomonitoring and therapeutic interventions. J. Pharmacol. Exp. Ther. 2012, 343, 2–12. [Google Scholar] [CrossRef]
- Kothinti, R.K.; Blodgett, A.B.; Petering, D.H.; Tabatabai, N.M. Cadmium down-regulation of kidney Sp1 binding to mouse SGLT1 and SGLT2 gene promoters: Possible reaction of cadmium with the zinc finger domain of Sp1. Toxicol. Appl. Pharmacol. 2010, 244, 254–262. [Google Scholar] [CrossRef]
- Gage, J.C. Distribution and Excretion of Methyl and Phenyl Mercury Salts. Br. J. Ind. Med. 1964, 21, 197–202. [Google Scholar] [CrossRef]
- Norseth, T.; Clarkson, T.W. Studies on the biotransformation of 203Hg-labeled methyl mercury chloride in rats. Arch. Environ. Health 1970, 21, 717–727. [Google Scholar] [CrossRef]
- Norseth, T.; Clarkson, T.W. Biotransformation of methylmercury salts in the rat studied by specific determination of inorganic mercury. Biochem. Pharmacol. 1970, 19, 2775–2783. [Google Scholar] [CrossRef]
- Omata, S.; Sato, M.; Sakimura, K.; Sugano, H. Time-dependent accumulation of inorganic mercury in subcellular fractions of kidney, liver, and brain of rats exposed to methylmercury. Arch. Toxicol. 1980, 44, 231–241. [Google Scholar] [CrossRef]
- Houser, M.T.; Berndt, W.O. The effect of unilateral nephrectomy on the nephrotoxicity of mercuric chloride in the rat. Toxicol. Appl. Pharmacol. 1986, 83, 506–515. [Google Scholar] [CrossRef]
- Ramos-Frendo, B.; Perez-Garcia, R.; Lopez-Novoa, J.M.; Hernando-Avendano, L. Increased severity of the acute renal failure induced by HgCl2 on rats with reduced renal mass. Biomedicine 1979, 31, 167–170. [Google Scholar] [PubMed]
- Zalups, R.K.; Diamond, G.L. Mercuric chloride-induced nephrotoxicity in the rat following unilateral nephrectomy and compensatory renal growth. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1987, 53, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K. Reductions in renal mass and the nephropathy induced by mercury. Toxicol. Appl. Pharmacol. 1997, 143, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Zalups, R.K.; Bridges, C.C. Seventy-five percent nephrectomy and the disposition of inorganic mercury in 2,3-dimercaptopropanesulfonic acid-treated rats lacking functional multidrug-resistance protein 2. J. Pharmacol. Exp. Ther. 2010, 332, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Pallan, S.; Gangji, A.S.; Lukic, D.; Clase, C.M. Mercury-associated nephrotic syndrome: A case report and systematic review of the literature. Am. J. Kidney Dis. 2013, 62, 135–138. [Google Scholar] [CrossRef]
- Gao, Z.; Wu, N.; Du, X.; Li, H.; Mei, X.; Song, Y. Toxic Nephropathy Secondary to Chronic Mercury Poisoning: Clinical Characteristics and Outcomes. Kidney Int. Rep. 2022, 7, 1189–1197. [Google Scholar] [CrossRef]
- Alok, A.; Yadav, A. Membranous Nephropathy; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Qin, A.B.; Lin, Z.S.; Wang, S.X.; Wang, H.; Cui, Z.; Zhou, F.D.; Zhao, M.H. Comparison of Ultrastructural Features between Patients with Mercury-associated Membranous Nephropathy and Idiopathic Membranous Nephropathy. Am. J. Med. Sci. 2021, 361, 327–335. [Google Scholar] [CrossRef]
- Molina, A.; Sanchez-Madrid, F.; Bricio, T.; Martin, A.; Barat, A.; Alvarez, V.; Mampaso, F. Prevention of mercuric chloride-induced nephritis in the brown Norway rat by treatment with antibodies against the alpha 4 integrin. J. Immunol. 1994, 153, 2313–2320. [Google Scholar]
- Bowman, C.; Peters, D.K.; Lockwood, C.M. Anti-glomerular basement membrane autoantibodies in the Brown Norway rat: Detection by a solid-phase radioimmunoassay. J. Immunol. Methods 1983, 61, 325–333. [Google Scholar] [CrossRef]
- Molina, A.; Sanchez-Madrid, F.; Bricio, T.; Martin, A.; Escudero, E.; Alvarez, V.; Mampaso, F. Abrogation of mercuric chloride-induced nephritis in the Brown Norway rat by treatment with antibodies against TNFalpha. Mediat. Inflamm. 1995, 4, 444–451. [Google Scholar] [CrossRef]
- Guery, J.C.; Druet, E.; Glotz, D.; Hirsch, F.; Mandet, C.; De Heer, E.; Druet, P. Specificity and cross-reactive idiotypes of anti-glomerular basement membrane autoantibodies in HgCl2-induced autoimmune glomerulonephritis. Eur. J. Immunol. 1990, 20, 93–100. [Google Scholar] [CrossRef]
- Mathieson, P.W.; Thiru, S.; Oliveira, D.B. Mercuric chloride-treated brown Norway rats develop widespread tissue injury including necrotizing vasculitis. Lab. Investig. 1992, 67, 121–129. [Google Scholar] [PubMed]
- Pelletier, L.; Rossert, J.; Pasquier, R.; Vial, M.C.; Druet, P. Role of CD8+ T cells in mercury-induced autoimmunity or immunosuppression in the rat. Scand. J. Immunol. 1990, 31, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Kosuda, L.L.; Wayne, A.; Nahounou, M.; Greiner, D.L.; Bigazzi, P.E. Reduction of the RT6.2+ subset of T lymphocytes in brown Norway rats with mercury-induced renal autoimmunity. Cell. Immunol. 1991, 135, 154–167. [Google Scholar] [CrossRef]
- Escudero, E.; Martin, A.; Nieto, M.; Nieto, E.; Navarro, E.; Luque, A.; Cabanas, C.; Sanchez-Madrid, F.; Mampaso, F. Functional relevance of activated beta1 integrins in mercury-induced nephritis. J. Am. Soc. Nephrol. 2000, 11, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhu, Q.; Xu, Y.; Zhou, Y.; Zhu, L.; Jin, L.; Wang, W.; Gao, L.; Chen, G.; Zhao, H. Total arsenic, dimethylarsinic acid, lead, cadmium, total mercury, methylmercury and hypertension among Asian populations in the United States: NHANES 2011–2018. Ecotoxicol. Environ. Saf. 2022, 241, 113776. [Google Scholar] [CrossRef]
- Simoes, R.P.; Fardin, P.B.A.; Simoes, M.R.; Vassallo, D.V.; Padilha, A.S. Long-term Mercury Exposure Accelerates the Development of Hypertension in Prehypertensive Spontaneously Hypertensive Rats Inducing Endothelial Dysfunction: The Role of Oxidative Stress and Cyclooxygenase-2. Biol. Trace Elem. Res. 2020, 196, 565–578. [Google Scholar] [CrossRef]
- Fardin, P.B.A.; Simoes, R.P.; Schereider, I.R.G.; Almenara, C.C.P.; Simoes, M.R.; Vassallo, D.V. Chronic Mercury Exposure in Prehypertensive SHRs Accelerates Hypertension Development and Activates Vasoprotective Mechanisms by Increasing NO and H2O2 Production. Cardiovasc. Toxicol. 2020, 20, 197–210. [Google Scholar] [CrossRef]
- Vassallo, D.V.; Simoes, M.R.; Giuberti, K.; Azevedo, B.F.; Ribeiro Junior, R.F.; Salaices, M.; Stefanon, I. Effects of Chronic Exposure to Mercury on Angiotensin-Converting Enzyme Activity and Oxidative Stress in Normotensive and Hypertensive Rats. Arq. Bras. Cardiol. 2019, 112, 374–380. [Google Scholar] [CrossRef]
- Habeeb, E.; Aldosari, S.; Saghir, S.A.; Cheema, M.; Momenah, T.; Husain, K.; Omidi, Y.; Rizvi, S.A.A.; Akram, M.; Ansari, R.A. Role of environmental toxicants in the development of hypertensive and cardiovascular diseases. Toxicol. Rep. 2022, 9, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Orr, S.E.; Barnes, M.C.; Joshee, L.; Uchakina, O.; McKallip, R.J.; Bridges, C.C. Potential mechanisms of cellular injury following exposure to a physiologically relevant species of inorganic mercury. Toxicol. Lett. 2019, 304, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lund, B.O.; Miller, D.M.; Woods, J.S. Studies on Hg(II)-induced H2O2 formation and oxidative stress in vivo and in vitro in rat kidney mitochondria. Biochem. Pharmacol. 1993, 45, 2017–2024. [Google Scholar] [CrossRef]
- Ma, L.; Bi, K.D.; Fan, Y.M.; Jiang, Z.Y.; Zhang, X.Y.; Zhang, J.W.; Zhao, J.; Jiang, F.L.; Dong, J.X. In vitro modulation of mercury-induced rat liver mitochondria dysfunction. Toxicol. Res. 2018, 7, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Lv, Z.; Han, X.; Li, S.; Han, B.; Yang, Q.; Wang, X.; Wu, P.; Li, J.; Deng, N.; et al. Harmful Effects of Inorganic Mercury Exposure on Kidney Cells: Mitochondrial Dynamics Disorder and Excessive Oxidative Stress. Biol. Trace Elem. Res. 2022, 200, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Singh, R.D.; Tiwari, R.; Gangopadhyay, S.; Roy, S.K.; Singh, D.; Srivastava, V. Mercury exposure induces cytoskeleton disruption and loss of renal function through epigenetic modulation of MMP9 expression. Toxicology 2017, 386, 28–39. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, B.; Hu, S.; Wang, T.; Zhang, Y.; Wang, J.; Liu, Y.; Zhang, H. The role of endoplasmic reticulum stress in renal damage caused by acute mercury chloride poisoning. J. Toxicol. Sci. 2020, 45, 589–598. [Google Scholar] [CrossRef]
- Rojas-Franco, P.; Franco-Colin, M.; Torres-Manzo, A.P.; Blas-Valdivia, V.; Thompson-Bonilla, M.D.R.; Kandir, S.; Cano-Europa, E. Endoplasmic reticulum stress participates in the pathophysiology of mercury-caused acute kidney injury. Ren. Fail. 2019, 41, 1001–1010. [Google Scholar] [CrossRef]
- Vieira, J.; Marques, V.B.; Vieira, L.V.; Crajoinas, R.O.; Shimizu, M.H.M.; Seguro, A.C.; Carneiro, M.; Girardi, A.C.C.; Vassallo, D.V.; Dos Santos, L. Changes in the renal function after acute mercuric chloride exposure in the rat are associated with renal vascular endothelial dysfunction and proximal tubule NHE3 inhibition. Toxicol. Lett. 2021, 341, 23–32. [Google Scholar] [CrossRef]
- Kramer, H.J.; Gonick, H.C.; Lu, E. In vitro inhibition of Na-K-ATPase by trace metals: Relation to renal and cardiovascular damage. Nephron 1986, 44, 329–336. [Google Scholar] [CrossRef]
- Savage, D.F.; Stroud, R.M. Structural basis of aquaporin inhibition by mercury. J. Mol. Biol. 2007, 368, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Burrow, C.R.; Smith, B.L.; Agre, P.; Knepper, M.A.; Wilson, P.D. Expression of aquaporins-1 and -2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am. J. Physiol. 1996, 271 Pt 2, F169–F183. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Cao, R.; Zhang, X.Y.; Guan, Y. Aquaporins in the kidney: Physiology and pathophysiology. Am. J. Physiol. Ren. Physiol. 2020, 318, F193–F203. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, M.; Nichols, L.; Dave, A.A.; Pittman, E.H.; Cheek, J.P.; Caroland, A.J.V.; Lotwala, P.; Drummond, J.; Bridges, C.C. Molecular Mechanisms of Cellular Injury and Role of Toxic Heavy Metals in Chronic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 11105. https://doi.org/10.3390/ijms231911105
Mishra M, Nichols L, Dave AA, Pittman EH, Cheek JP, Caroland AJV, Lotwala P, Drummond J, Bridges CC. Molecular Mechanisms of Cellular Injury and Role of Toxic Heavy Metals in Chronic Kidney Disease. International Journal of Molecular Sciences. 2022; 23(19):11105. https://doi.org/10.3390/ijms231911105
Chicago/Turabian StyleMishra, Manish, Larry Nichols, Aditi A. Dave, Elizabeth H Pittman, John P. Cheek, Anasalea J. V. Caroland, Purva Lotwala, James Drummond, and Christy C. Bridges. 2022. "Molecular Mechanisms of Cellular Injury and Role of Toxic Heavy Metals in Chronic Kidney Disease" International Journal of Molecular Sciences 23, no. 19: 11105. https://doi.org/10.3390/ijms231911105
APA StyleMishra, M., Nichols, L., Dave, A. A., Pittman, E. H., Cheek, J. P., Caroland, A. J. V., Lotwala, P., Drummond, J., & Bridges, C. C. (2022). Molecular Mechanisms of Cellular Injury and Role of Toxic Heavy Metals in Chronic Kidney Disease. International Journal of Molecular Sciences, 23(19), 11105. https://doi.org/10.3390/ijms231911105