
New Insights into the Pivotal Role of the Amygdala in Inflammation-Related Depression and Anxiety Disorder
Abstract
1. Introduction
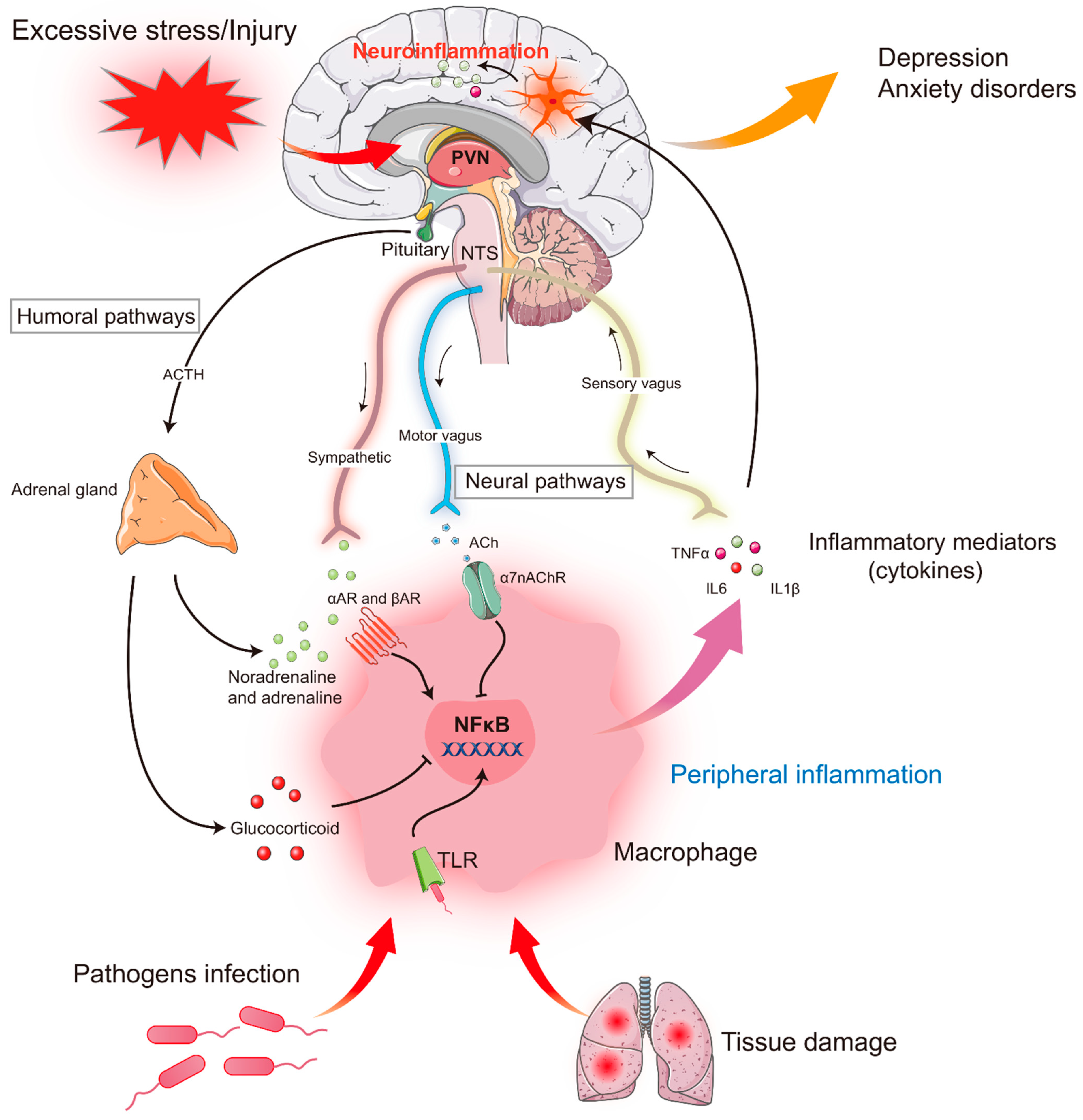
2. Stress and Inflammatory Response
2.1. Stress and Peripheral Inflammatory Response
2.2. Stress and Central Inflammatory Response
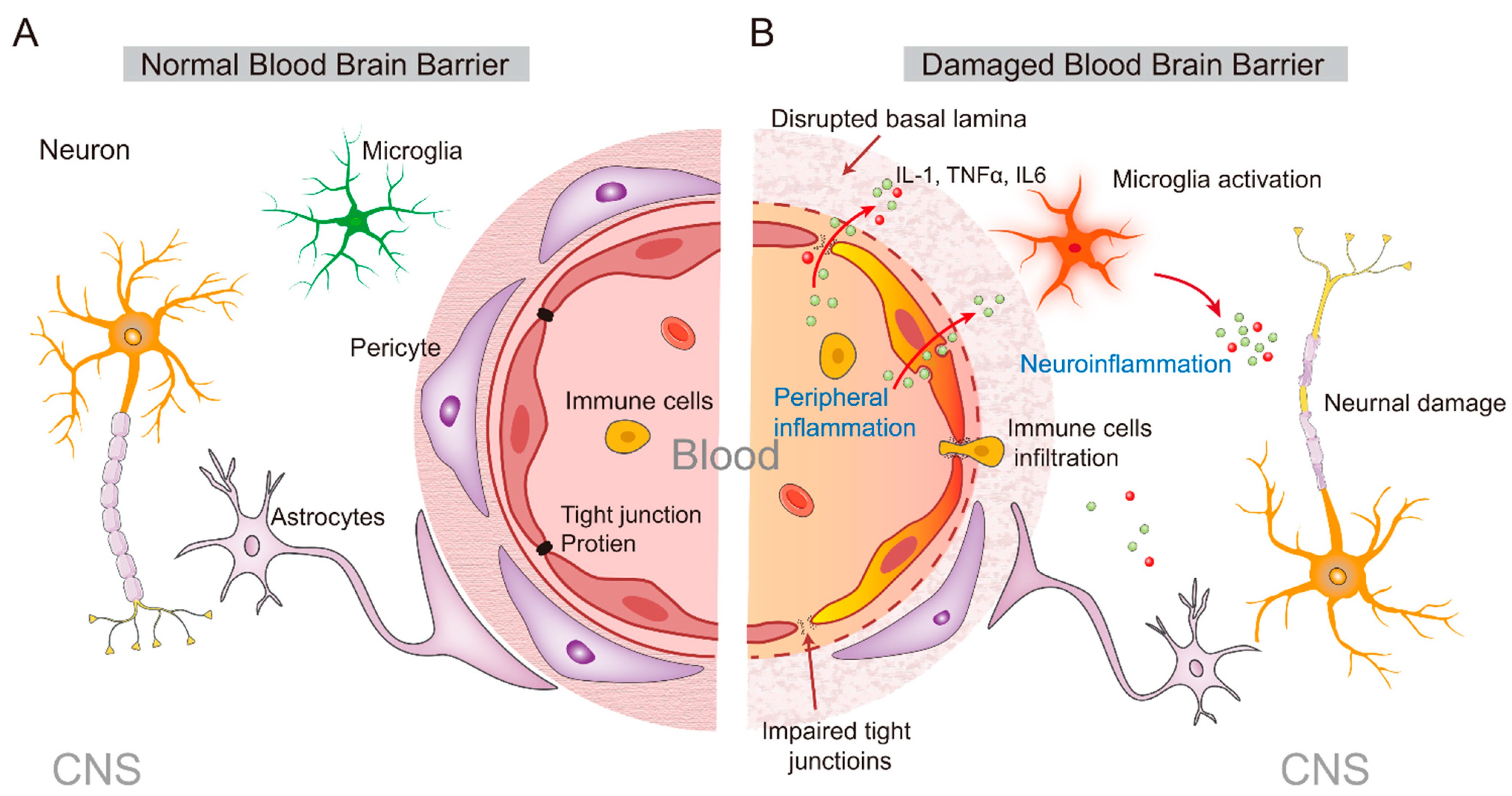
3. From the Peripheral to the Central Inflammatory Response
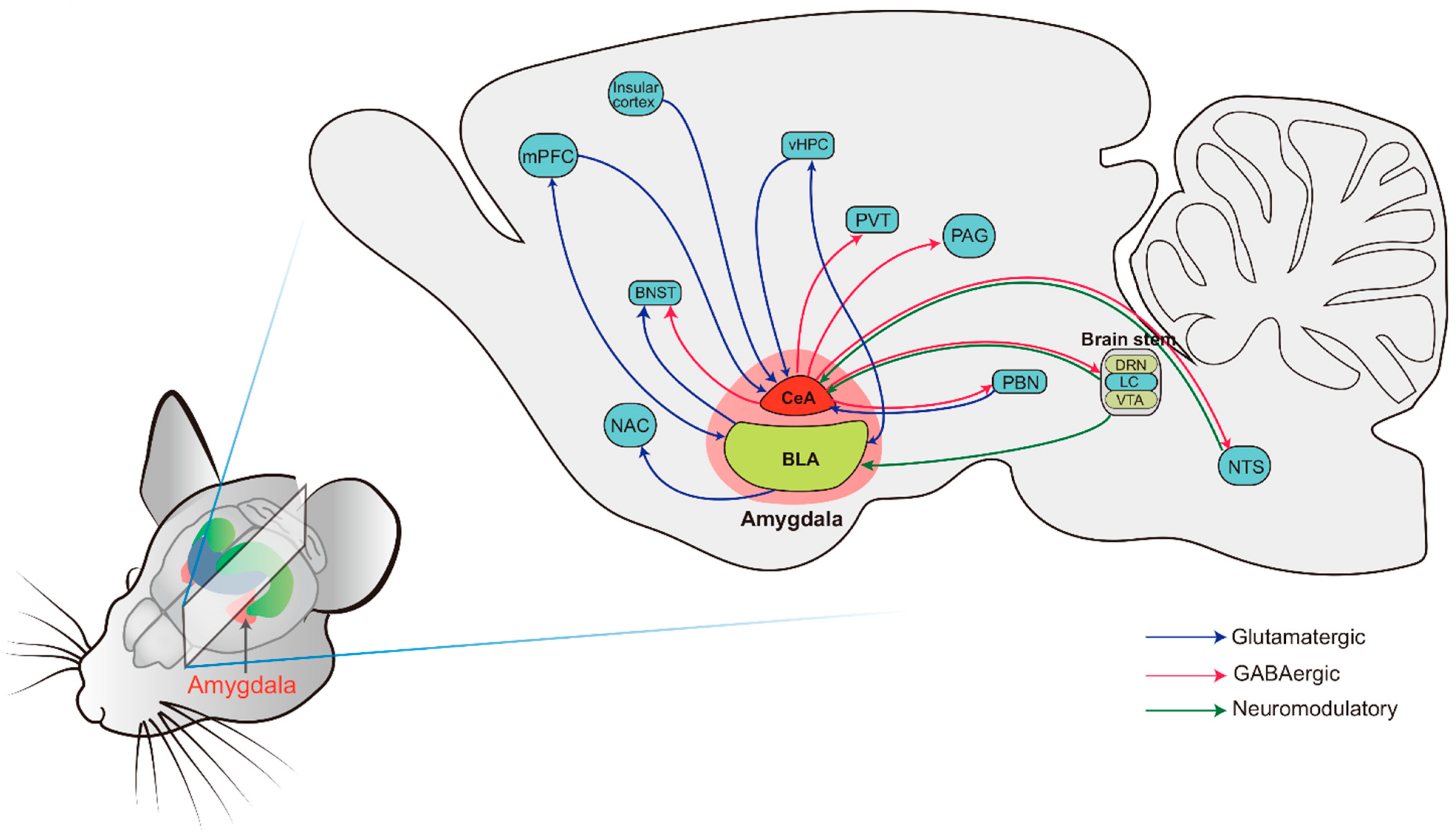
4. Stress and Amygdala
4.1. The Basic Anatomy of the Amygdala: Structure and Connectivity
4.2. Effects of Stress on Amygdala Neuronal Function
5. Amygdala Neuroinflammation and Psychiatric Disorders
5.1. Amygdala Inflammation and Anxiety Disorders
5.2. Amygdala Inflammation and Depression
5.3. Inflammation and the Amygdala Neural Circuit
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar]
- Almeida-Filho, N.; Lessa, I.; Magalhaes, L.; Arauho, M.J.; Aquino, E.; de Jesus, M.J. Co-occurrence patterns of anxiety, depression and alcohol use disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2007, 257, 423–431. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, C.-M.D. Global prevalence and burden of depressive and anxiety disorders in 204 countries and territories in 2020 due to the COVID-19 pandemic. Lancet 2021, 398, 1700–1712. [Google Scholar]
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61 (Suppl. S6), 7–11. [Google Scholar]
- Liu, B.; Liu, J.; Wang, M.; Zhang, Y.; Li, L. From serotonin to neuroplasticity: Evolvement of theories for major depressive disorder. Front. Cell. Neurosci. 2017, 11, 305. [Google Scholar] [CrossRef]
- Jakobsen, J.C.; Katakam, K.K.; Schou, A.; Hellmuth, S.G.; Stallknecht, S.E.; Leth-Moller, K.; Iversen, M.; Banke, M.B.; Petersen, I.J.; Klingenberg, S.L.; et al. Selective serotonin reuptake inhibitors versus placebo in patients with major depressive disorder. A systematic review with meta-analysis and Trial Sequential Analysis. BMC Psychiatry 2017, 17, 58. [Google Scholar]
- Westfall, S.; Caracci, F.; Estill, M.; Frolinger, T.; Shen, L.; Pasinetti, G.M. Chronic stress-induced depression and anxiety priming modulated by gut-brain-axis immunity. Front. Immunol. 2021, 12, 670500. [Google Scholar] [CrossRef]
- Risbrough, V.B.; Vaughn, M.N.; Friend, S.F. Role of inflammation in traumatic brain injury-associated risk for neuropsychiatric disorders: State of the evidence and where do we go from here. Biol. Psychiatry 2022, 91, 438–448. [Google Scholar] [CrossRef]
- Milaneschi, Y.; Kappelmann, N.; Ye, Z.; Lamers, F.; Moser, S.; Jones, P.B.; Burgess, S.; Penninx, B.; Khandaker, G.M. Association of inflammation with depression and anxiety: Evidence for symptom-specificity and potential causality from UK Biobank and NESDA cohorts. Mol. Psychiatry 2021, 26, 7393–7402. [Google Scholar] [CrossRef]
- Slavich, G.M.; Irwin, M.R. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull. 2014, 140, 774–815. [Google Scholar] [CrossRef]
- Zheng, Z.H.; Tu, J.L.; Li, X.H.; Hua, Q.; Liu, W.Z.; Liu, Y.; Pan, B.X.; Hu, P.; Zhang, W.H. Neuroinflammation induces anxiety- and depressive-like behavior by modulating neuronal plasticity in the basolateral amygdala. Brain Behav. Immun. 2021, 92, 505–518. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Derry, H.M.; Fagundes, C.P. Inflammation: Depression fans the flames and feasts on the heat. Am. J. Psychiatry 2015, 172, 1075–1091. [Google Scholar] [CrossRef]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The bidirectional relationship of depression and inflammation: Double trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef] [PubMed]
- Euesden, J.; Danese, A.; Lewis, C.M.; Maughan, B. A bidirectional relationship between depression and the autoimmune disorders—New perspectives from the National Child Development Study. PLoS ONE 2017, 12, e0173015. [Google Scholar] [CrossRef]
- Dickens, C.; McGowan, L.; Clark-Carter, D.; Creed, F. Depression in rheumatoid arthritis: A systematic review of the literature with meta-analysis. Psychosom. Med. 2002, 64, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.B.; Marrie, R.A.; Carta, M.G. Depression in multiple sclerosis. Int. Rev. Psychiatry 2017, 29, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.G.; Jorge, R.E. Post-stroke depression: A review. Am. J. Psychiatry 2016, 173, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.O.; Oriolo, G.; Bortolato, B.; Kohler, C.A.; Maes, M.; Solmi, M.; Grande, I.; Martin-Santos, R.; Vieta, E.; Carvalho, A.F. Biological mechanisms of depression following treatment with interferon for chronic hepatitis C: A critical systematic review. J. Affect. Disord. 2017, 209, 235–245. [Google Scholar] [CrossRef]
- Chiu, W.C.; Su, Y.P.; Su, K.P.; Chen, P.C. Recurrence of depressive disorders after interferon-induced depression. Transl. Psychiatry 2017, 7, e1026. [Google Scholar] [CrossRef]
- Griffiths, C.E.M.; Fava, M.; Miller, A.H.; Russell, J.; Ball, S.G.; Xu, W.; Acharya, N.; Rapaport, M.H. Impact of ixekizumab treatment on depressive symptoms and systemic inflammation in patients with moderate-to-severe psoriasis: An integrated analysis of three phase 3 clinical studies. Psychother. Psychosom. 2017, 86, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C. Imaging the role of inflammation in mood and anxiety-related disorders. Curr. Neuropharmacol. 2018, 16, 533–558. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.E. Inflammation and depression: A causal or coincidental link to the pathophysiology? Acta Neuropsychiatr. 2018, 30, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Russo, S.J.; Nestler, E.J. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013, 14, 609–625. [Google Scholar] [CrossRef]
- Price, J.L.; Drevets, W.C. Neurocircuitry of mood disorders. Neuropsychopharmacology 2010, 35, 192–216. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.K.; Gore, F.; Salzman, C.D. Basolateral amygdala circuitry in positive and negative valence. Curr. Opin. Neurobiol. 2018, 49, 175–183. [Google Scholar] [CrossRef]
- Sah, P. Fear, anxiety, and the amygdala. Neuron 2017, 96, 1–2. [Google Scholar] [CrossRef]
- Rosenkranz, J.A.; Venheim, E.R.; Padival, M. Chronic stress causes amygdala hyperexcitability in rodents. Biol. Psychiatry 2010, 67, 1128–1136. [Google Scholar] [CrossRef]
- Zhang, W.; Rosenkranz, J.A. Repeated restraint stress increases basolateral amygdala neuronal activity in an age-dependent manner. Neuroscience 2012, 226, 459–474. [Google Scholar] [CrossRef]
- Birbaumer, N.; Grodd, W.; Diedrich, O.; Klose, U.; Erb, M.; Lotze, M.; Schneider, F.; Weiss, U.; Flor, H. fMRI reveals amygdala activation to human faces in social phobics. Neuroreport 1998, 9, 1223–1226. [Google Scholar] [CrossRef]
- Phan, K.L.; Fitzgerald, D.A.; Nathan, P.J.; Tancer, M.E. Association between amygdala hyperactivity to harsh faces and severity of social anxiety in generalized social phobia. Biol. Psychiatry 2006, 59, 424–429. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.B.; Thayer, J.F.; Vedhara, K. Stress and health: A review of psychobiological processes. Annu. Rev. Psychol. 2021, 72, 663–688. [Google Scholar] [CrossRef] [PubMed]
- Ulrich-Lai, Y.M.; Herman, J.P. Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef]
- McEwen, B.S.; Gianaros, P.J. Central role of the brain in stress and adaptation: Links to socioeconomic status, health, and disease. Ann. N. Y. Acad. Sci. 2010, 1186, 190–222. [Google Scholar] [CrossRef]
- McEwen, B.S. Protective and damaging effects of stress mediators. N. Engl. J. Med. 1998, 338, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573–2589.e9. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.E.; Cohen, S.; Ritchey, A.K. Chronic psychological stress and the regulation of pro-inflammatory cytokines: A glucocorticoid-resistance model. Health Psychol. 2002, 21, 531–541. [Google Scholar] [CrossRef]
- Cohen, S.; Janicki-Deverts, D.; Miller, G.E. Psychological stress and disease. JAMA 2007, 298, 1685–1687. [Google Scholar] [CrossRef]
- Gordan, R.; Gwathmey, J.K.; Xie, L.H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204–214. [Google Scholar] [CrossRef]
- Hasko, G.; Szabo, C. Regulation of cytokine and chemokine production by transmitters and co-transmitters of the autonomic nervous system. Biochem. Pharmacol. 1998, 56, 1079–1087. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex—Linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The cholinergic anti-inflammatory pathway: A missing link in neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Nardi, D.; Zhang, M.; Yang, H.; Ombrellino, M.; Tracey, K.J. Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton Neurosci. 2000, 85, 141–147. [Google Scholar] [CrossRef]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the developing nervous system. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The complement system: An unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Baratta, M.V.; Sprunger, D.B.; Watkins, L.R.; Maier, S.F. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav. Immun. 2007, 21, 47–59. [Google Scholar] [CrossRef]
- Hinwood, M.; Morandini, J.; Day, T.A.; Walker, F.R. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 2012, 22, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Kreisel, T.; Frank, M.G.; Licht, T.; Reshef, R.; Ben-Menachem-Zidon, O.; Baratta, M.V.; Maier, S.F.; Yirmiya, R. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 2014, 19, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; Meyer zu Schwabedissen, L.; et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflammation 2011, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Schnieder, T.P.; Trencevska, I.; Rosoklija, G.; Stankov, A.; Mann, J.J.; Smiley, J.; Dwork, A.J. Microglia of prefrontal white matter in suicide. J. Neuropathol. Exp. Neurol. 2014, 73, 880–890. [Google Scholar] [CrossRef]
- Calcia, M.A.; Bonsall, D.R.; Bloomfield, P.S.; Selvaraj, S.; Barichello, T.; Howes, O.D. Stress and neuroinflammation: A systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016, 233, 1637–1650. [Google Scholar] [CrossRef]
- Johnson, J.D.; Cortez, V.; Kennedy, S.L.; Foley, T.E.; Hanson, H., 3rd; Fleshner, M. Role of central beta-adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav. Immun. 2008, 22, 1078–1086. [Google Scholar] [CrossRef]
- Johnson, J.D.; Zimomra, Z.R.; Stewart, L.T. Beta-adrenergic receptor activation primes microglia cytokine production. J. Neuroimmunol. 2013, 254, 161–164. [Google Scholar] [CrossRef]
- McNamee, E.N.; Griffin, E.W.; Ryan, K.M.; Ryan, K.J.; Heffernan, S.; Harkin, A.; Connor, T.J. Noradrenaline acting at beta-adrenoceptors induces expression of IL-1beta and its negative regulators IL-1ra and IL-1RII, and drives an overall anti-inflammatory phenotype in rat cortex. Neuropharmacology 2010, 59, 37–48. [Google Scholar] [CrossRef]
- Hoogland, I.C.; Houbolt, C.; van Westerloo, D.J.; van Gool, W.A.; van de Beek, D. Systemic inflammation and microglial activation: Systematic review of animal experiments. J. Neuroinflammation 2015, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Konsman, J.P.; Parnet, P.; Dantzer, R. Cytokine-induced sickness behaviour: Mechanisms and implications. Trends Neurosci. 2002, 25, 154–159. [Google Scholar] [CrossRef]
- Cao, C.; Matsumura, K.; Yamagata, K.; Watanabe, Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1 beta: A possible site of prostaglandin synthesis responsible for fever. Brain Res. 1996, 733, 263–272. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. The vagus nerve in the neuro-immune axis: Implications in the pathology of the gastrointestinal tract. Front. Immunol. 2017, 8, 1452. [Google Scholar] [CrossRef] [PubMed]
- Ek, M.; Kurosawa, M.; Lundeberg, T.; Ericsson, A. Activation of vagal afferents after intravenous injection of interleukin-1beta: Role of endogenous prostaglandins. J. Neurosci. 1998, 18, 9471–9479. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Kelley, K.W. Twenty years of research on cytokine-induced sickness behavior. Brain Behav. Immun. 2007, 21, 153–160. [Google Scholar] [CrossRef]
- Wan, W.; Janz, L.; Vriend, C.Y.; Sorensen, C.M.; Greenberg, A.H.; Nance, D.M. Differential induction of c-Fos immunoreactivity in hypothalamus and brain stem nuclei following central and peripheral administration of endotoxin. Brain Res. Bull. 1993, 32, 581–587. [Google Scholar] [CrossRef]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Wang, X.; Xue, G.X.; Liu, W.C.; Shu, H.; Wang, M.; Sun, Y.; Liu, X.; Sun, Y.E.; Liu, C.F.; Liu, J.; et al. Melatonin alleviates lipopolysaccharide-compromised integrity of blood-brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell 2017, 16, 414–421. [Google Scholar] [CrossRef]
- Qin, L.H.; Huang, W.; Mo, X.A.; Chen, Y.L.; Wu, X.H. LPS induces occludin dysregulation in cerebral microvascular endothelial cells via MAPK signaling and augmenting MMP-2 levels. Oxid. Med. Cell. Longev. 2015, 2015, 120641. [Google Scholar] [CrossRef] [PubMed]
- Salkeni, M.A.; Lynch, J.L.; Otamis-Price, T.; Banks, W.A. Lipopolysaccharide impairs blood-brain barrier P-glycoprotein function in mice through prostaglandin- and nitric oxide-independent pathways. J. Neuroimmune Pharmacol. 2009, 4, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Karahashi, H.; Michelsen, K.S.; Arditi, M. Lipopolysaccharide-induced apoptosis in transformed bovine brain endothelial cells and human dermal microvessel endothelial cells: The role of JNK. J. Immunol. 2009, 182, 7280–7286. [Google Scholar] [CrossRef] [PubMed]
- van den Bulk, B.G.; Meens, P.H.; van Lang, N.D.; de Voogd, E.L.; van der Wee, N.J.; Rombouts, S.A.; Crone, E.A.; Vermeiren, R.R. Amygdala activation during emotional face processing in adolescents with affective disorders: The role of underlying depression and anxiety symptoms. Front. Hum. Neurosci. 2014, 8, 393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Song, C.; Wang, M.; He, Y.; Xu, X.B.; Pan, H.Q.; Chen, W.B.; Peng, W.J.; Pan, B.X. Chronic stress impairs GABAergic control of amygdala through suppressing the tonic GABAA receptor currents. Mol. Brain 2014, 7, 32. [Google Scholar] [CrossRef]
- Zhang, W.H.; Zhang, J.Y.; Holmes, A.; Pan, B.X. Amygdala circuit substrates for stress adaptation and adversity. Biol. Psychiatry 2021, 89, 847–856. [Google Scholar] [CrossRef]
- Qin, X.; Pan, H.Q.; Huang, S.H.; Zou, J.X.; Zheng, Z.H.; Liu, X.X.; You, W.J.; Liu, Z.P.; Cao, J.L.; Zhang, W.H.; et al. GABAA(δ) receptor hypofunction in the amygdala-hippocampal circuit underlies stress-induced anxiety. Sci. Bull. 2022, 67, 97–110. [Google Scholar] [CrossRef]
- Sah, P.; Faber, E.S.; Lopez De Armentia, M.; Power, J. The amygdaloid complex: Anatomy and physiology. Physiol. Rev. 2003, 83, 803–834. [Google Scholar] [CrossRef]
- Gilpin, N.W.; Herman, M.A.; Roberto, M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol. Psychiatry 2015, 77, 859–869. [Google Scholar] [CrossRef]
- Carlsen, J.; Heimer, L. The basolateral amygdaloid complex as a cortical-like structure. Brain Res. 1988, 441, 377–380. [Google Scholar] [CrossRef]
- Keifer, O.P., Jr.; Hurt, R.C.; Ressler, K.J.; Marvar, P.J. The physiology of fear: Reconceptualizing the role of the central amygdala in fear learning. Physiology 2015, 30, 389–401. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.J. Cytoarchitecture of the central amygdaloid nucleus of the rat. J. Comp. Neurol. 1982, 208, 401–418. [Google Scholar] [CrossRef] [PubMed]
- LeDoux, J.E.; Cicchetti, P.; Xagoraris, A.; Romanski, L.M. The lateral amygdaloid nucleus: Sensory interface of the amygdala in fear conditioning. J. Neurosci. 1990, 10, 1062–1069. [Google Scholar] [CrossRef]
- Tully, K.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 14146–14150. [Google Scholar] [CrossRef]
- Thomas, K.M.; Drevets, W.C.; Dahl, R.E.; Ryan, N.D.; Birmaher, B.; Eccard, C.H.; Axelson, D.; Whalen, P.J.; Casey, B.J. Amygdala response to fearful faces in anxious and depressed children. Arch. Gen. Psychiatry 2001, 58, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Felmingham, K.; Kemp, A.; Williams, L.; Das, P.; Hughes, G.; Peduto, A.; Bryant, R. Changes in anterior cingulate and amygdala after cognitive behavior therapy of posttraumatic stress disorder. Psychol. Sci. 2007, 18, 127–129. [Google Scholar] [CrossRef]
- Shin, L.M.; Wright, C.I.; Cannistraro, P.A.; Wedig, M.M.; McMullin, K.; Martis, B.; Macklin, M.L.; Lasko, N.B.; Cavanagh, S.R.; Krangel, T.S.; et al. A functional magnetic resonance imaging study of amygdala and medial prefrontal cortex responses to overtly presented fearful faces in posttraumatic stress disorder. Arch. Gen. Psychiatry 2005, 62, 273–281. [Google Scholar] [CrossRef]
- Drevets, W.C.; Bogers, W.; Raichle, M.E. Functional anatomical correlates of antidepressant drug treatment assessed using PET measures of regional glucose metabolism. Eur. Neuropsychopharmacol. 2002, 12, 527–544. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Gotlib, I.H. Neural substrates of increased memory sensitivity for negative stimuli in major depression. Biol. Psychiatry 2008, 63, 1155–1162. [Google Scholar] [CrossRef]
- Yang, T.T.; Simmons, A.N.; Matthews, S.C.; Tapert, S.F.; Frank, G.K.; Max, J.E.; Bischoff-Grethe, A.; Lansing, A.E.; Brown, G.; Strigo, I.A.; et al. Adolescents with major depression demonstrate increased amygdala activation. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 42–51. [Google Scholar]
- Froemke, R.C. Plasticity of cortical excitatory-inhibitory balance. Annu. Rev. Neurosci. 2015, 38, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.Z.; Zhang, W.H.; Zheng, Z.H.; Zou, J.X.; Liu, X.X.; Huang, S.H.; You, W.J.; He, Y.; Zhang, J.Y.; Wang, X.D.; et al. Identification of a prefrontal cortex-to-amygdala pathway for chronic stress-induced anxiety. Nat. Commun. 2020, 11, 2221. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Liu, X.X.; Wang, Y.; Wang, D.; Song, Y.; Zou, J.X.; Pan, H.Q.; Zhai, X.Z.; Zhang, Y.M.; Zhang, Y.B.; et al. Early life stress induces anxiety-like behavior during adulthood through dysregulation of neuronal plasticity in the basolateral amygdala. Life Sci. 2021, 285, 119959. [Google Scholar] [CrossRef]
- Qin, X.; He, Y.; Wang, N.; Zou, J.X.; Zhang, Y.M.; Cao, J.L.; Pan, B.X.; Zhang, W.H. Moderate maternal separation mitigates the altered synaptic transmission and neuronal activation in amygdala by chronic stress in adult mice. Mol. Brain 2019, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.Y.; Huang, L.; Lin, S.; Yin, Y.N.; Jie, W.; Hu, N.Y.; Hu, Y.Y.; Guan, Y.F.; Liu, J.H.; You, Q.L.; et al. Erbin in amygdala parvalbumin-positive neurons modulates anxiety-like behaviors. Biol. Psychiatry 2020, 87, 926–936. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Liu, T.H.; He, Y.; Pan, H.Q.; Zhang, W.H.; Yin, X.P.; Tian, X.L.; Li, B.M.; Wang, X.D.; Holmes, A.; et al. Chronic stress remodels synapses in an amygdala circuit-specific manner. Biol. Psychiatry 2019, 85, 189–201. [Google Scholar] [CrossRef]
- Miller, A.H.; Haroon, E.; Raison, C.L.; Felger, J.C. Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depress. Anxiety 2013, 30, 297–306. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Nazir, S.; Farooq, R.K.; Nasir, S.; Hanif, R.; Javed, A. Therapeutic effect of Thymoquinone on behavioural response to UCMS and neuroinflammation in hippocampus and amygdala in BALB/c mice model. Psychopharmacology 2022, 239, 47–58. [Google Scholar] [CrossRef]
- Meng, Y.; Zhuang, L.; Xue, Q.; Zhang, J.; Yu, B. NLRP3-mediated Neuroinflammation Exacerbates Incisional Hyperalgesia and Prolongs Recovery After Surgery in Chronic Stressed Rats. Pain Physician 2021, 24, E1099–E1108. [Google Scholar]
- Inagaki, T.K.; Muscatell, K.A.; Irwin, M.R.; Cole, S.W.; Eisenberger, N.I. Inflammation selectively enhances amygdala activity to socially threatening images. Neuroimage 2012, 59, 3222–3226. [Google Scholar] [CrossRef] [PubMed]
- Harrison, N.A.; Brydon, L.; Walker, C.; Gray, M.A.; Steptoe, A.; Critchley, H.D. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol. Psychiatry 2009, 66, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mohammed, Z.; Singh, I. Bruton’s tyrosine kinase drives neuroinflammation and anxiogenic behavior in mouse models of stress. J. Neuroinflammation 2021, 18, 289. [Google Scholar] [CrossRef] [PubMed]
- Avolio, E.; Fazzari, G.; Mele, M.; Alo, R.; Zizza, M.; Jiao, W.; Di Vito, A.; Barni, T.; Mandala, M.; Canonaco, M. Unpredictable chronic mild stress paradigm established effects of pro- and anti-inflammatory cytokine on neurodegeneration-linked depressive states in hamsters with brain endothelial damages. Mol. Neurobiol. 2017, 54, 6446–6458. [Google Scholar] [CrossRef]
- Kim, J.; Suh, Y.H.; Chang, K.A. Interleukin-17 induced by cumulative mild stress promoted depression-like behaviors in young adult mice. Mol. Brain 2021, 14, 11. [Google Scholar] [CrossRef]
- Nozaki, K.; Ito, H.; Ohgidani, M.; Yamawaki, Y.; Sahin, E.H.; Kitajima, T.; Katsumata, S.; Yamawaki, S.; Kato, T.A.; Aizawa, H. Antidepressant effect of the translocator protein antagonist ONO-2952 on mouse behaviors under chronic social defeat stress. Neuropharmacology 2020, 162, 107835. [Google Scholar] [CrossRef]
- Patki, G.; Solanki, N.; Atrooz, F.; Allam, F.; Salim, S. Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res. 2013, 1539, 73–86. [Google Scholar] [CrossRef]
- Li, S.; Liao, Y.; Dong, Y.; Li, X.; Li, J.; Cheng, Y.; Cheng, J.; Yuan, Z. Microglial deletion and inhibition alleviate behavior of post-traumatic stress disorder in mice. J. Neuroinflammation 2021, 18, 7. [Google Scholar] [CrossRef]
- Guan, X.T.; Lin, W.J.; Tang, M.M. Comparison of stress-induced and LPS-induced depressive-like behaviors and the alterations of central proinflammatory cytokines mRNA in rats. Psych. J. 2015, 4, 113–122. [Google Scholar] [CrossRef]
- Hu, C.; Yang, H.; Zhao, Y.; Chen, X.; Dong, Y.; Li, L.; Dong, Y.; Cui, J.; Zhu, T.; Zheng, P.; et al. The role of inflammatory cytokines and ERK1/2 signaling in chronic prostatitis/chronic pelvic pain syndrome with related mental health disorders. Sci. Rep. 2016, 6, 28608. [Google Scholar] [CrossRef]
- Zhong, H.; Rong, J.; Yang, Y.; Liang, M.; Li, Y.; Zhou, R. Neonatal inflammation via persistent TGF-beta1 downregulation decreases GABAAR expression in basolateral amygdala leading to the imbalance of the local excitation-inhibition circuits and anxiety-like phenotype in adult mice. Neurobiol. Dis. 2022, 169, 105745. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhan, B.; Jia, Y.; Li, C.; Wu, N.; Zhao, M.; Chen, N.; Guo, Y.; Du, Y.; Zhang, Y.; et al. IL-33 in the basolateral amygdala integrates neuroinflammation into anxiogenic circuits via modulating BDNF expression. Brain Behav. Immun. 2022, 102, 98–109. [Google Scholar] [CrossRef] [PubMed]
- O’Loughlin, E.; Pakan, J.M.P.; Yilmazer-Hanke, D.; McDermott, K.W. Acute in utero exposure to lipopolysaccharide induces inflammation in the pre- and postnatal brain and alters the glial cytoarchitecture in the developing amygdala. J. Neuroinflammation 2017, 14, 212. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, J.; Lin, Q.; Mao, K.; Tian, F.; Jing, C.; Wang, C.; Ding, L.; Pang, C. Proanthocyanidin prevents lipopolysaccharide-induced depressive-like behavior in mice via neuroinflammatory pathway. Brain Res. Bull. 2017, 135, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, X.; Xu, Y.; Hao, Y.; Meng, X. Effects of metformin on lipopolysaccharide-induced depressive-like behavior in mice and its mechanisms. Neuroreport 2020, 31, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Araki, R.; Hiraki, Y.; Nishida, S.; Inatomi, Y.; Yabe, T. Gomisin N ameliorates lipopolysaccharide-induced depressive-like behaviors by attenuating inflammation in the hypothalamic paraventricular nucleus and central nucleus of the amygdala in mice. J. Pharmacol. Sci. 2016, 132, 138–144. [Google Scholar] [CrossRef]
- Araki, R.; Hiraki, Y.; Yabe, T. Genipin attenuates lipopolysaccharide-induced persistent changes of emotional behaviors and neural activation in the hypothalamic paraventricular nucleus and the central amygdala nucleus. Eur. J. Pharmacol. 2014, 741, 1–7. [Google Scholar] [CrossRef]
- Hashimoto, O.; Kuniishi, H.; Nakatake, Y.; Yamada, M.; Wada, K.; Sekiguchi, M. Early life stress from allergic dermatitis causes depressive-like behaviors in adolescent male mice through neuroinflammatory priming. Brain Behav. Immun. 2020, 90, 319–331. [Google Scholar] [CrossRef]
- Zhang, J.; Lin, W.; Tang, M.; Zhao, Y.; Zhang, K.; Wang, X.; Li, Y. Inhibition of JNK ameliorates depressive-like behaviors and reduces the activation of pro-inflammatory cytokines and the phosphorylation of glucocorticoid receptors at serine 246 induced by neuroinflammation. Psychoneuroendocrinology 2020, 113, 104580. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Wang, H.Y.; Zhang, C.; Liu, B.P.; Peng, Z.L.; Li, Y.Y.; Liu, F.M.; Song, C. Mifepristone attenuates depression-like changes induced by chronic central administration of interleukin-1beta in rats. Behav. Brain Res. 2018, 347, 436–445. [Google Scholar] [CrossRef]
- Chen, J.; Song, Y.; Yang, J.; Zhang, Y.; Zhao, P.; Zhu, X.J.; Su, H.C. The contribution of TNF-alpha in the amygdala to anxiety in mice with persistent inflammatory pain. Neurosci. Lett. 2013, 541, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Sun, T.; Yang, L.; Liu, A.; Liu, Q.Q.; Tian, Q.Q.; Wang, Y.; Zhao, M.G.; Yang, Q. Scopoletin ameliorates anxiety-like behaviors in complete Freund’s adjuvant-induced mouse model. Mol. Brain 2020, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Suhett, C.P.; Graham, A.; Chen, Y.; Deuster, P. Behavioral changes in male mice fed a high-fat diet are associated with IL-1beta expression in specific brain regions. Physiol. Behav. 2017, 169, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Akter, S.; Uddin, K.R.; Sasaki, H.; Shibata, S. Gamma oryzanol alleviates high-fat diet-induced anxiety-like behaviors through downregulation of dopamine and inflammation in the amygdala of mice. Front. Pharmacol. 2020, 11, 330. [Google Scholar] [CrossRef]
- Sasaki, A.; de Vega, W.C.; St-Cyr, S.; Pan, P.; McGowan, P.O. Perinatal high fat diet alters glucocorticoid signaling and anxiety behavior in adulthood. Neuroscience 2013, 240, 1–12. [Google Scholar] [CrossRef]
- Sasaki, A.; de Vega, W.; Sivanathan, S.; St-Cyr, S.; McGowan, P.O. Maternal high-fat diet alters anxiety behavior and glucocorticoid signaling in adolescent offspring. Neuroscience 2014, 272, 92–101. [Google Scholar] [CrossRef]
- Yue, J.; Wang, X.S.; Guo, Y.Y.; Zheng, K.Y.; Liu, H.Y.; Hu, L.N.; Zhao, M.G.; Liu, S.B. Anxiolytic effect of CPEB1 knockdown on the amygdala of a mouse model of inflammatory pain. Brain Res. Bull. 2018, 137, 156–165. [Google Scholar] [CrossRef]
- Wang, X.S.; Guan, S.Y.; Liu, A.; Yue, J.; Hu, L.N.; Zhang, K.; Yang, L.K.; Lu, L.; Tian, Z.; Zhao, M.G.; et al. Anxiolytic effects of Formononetin in an inflammatory pain mouse model. Mol. Brain 2019, 12, 36. [Google Scholar] [CrossRef]
- Yang, L.; Wang, M.; Guo, Y.Y.; Sun, T.; Li, Y.J.; Yang, Q.; Zhang, K.; Liu, S.B.; Zhao, M.G.; Wu, Y.M. Systemic inflammation induces anxiety disorder through CXCL12/CXCR4 pathway. Brain Behav. Immun. 2016, 56, 352–362. [Google Scholar] [CrossRef]
- Doenni, V.M.; Song, C.M.; Hill, M.N.; Pittman, Q.J. Early-life inflammation with LPS delays fear extinction in adult rodents. Brain Behav. Immun. 2017, 63, 176–185. [Google Scholar] [CrossRef]
- Xu, Y.; Day, T.A.; Buller, K.M. The central amygdala modulates hypothalamic-pituitary-adrenal axis responses to systemic interleukin-1beta administration. Neuroscience 1999, 94, 175–183. [Google Scholar] [CrossRef]
- Weidenfeld, J.; Itzik, A.; Goshen, I.; Yirmiya, R.; Ben-Hur, T. Role of the central amygdala in modulating the pituitary-adrenocortical and clinical responses in experimental herpes simplex virus-1 encephalitis. Neuroendocrinology 2005, 81, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Sawchenko, P.E.; Li, H.Y.; Ericsson, A. Circuits and mechanisms governing hypothalamic responses to stress: A tale of two paradigms. Prog. Brain Res. 2000, 122, 61–78. [Google Scholar]
- Thrivikraman, K.V.; Su, Y.; Plotsky, P.M. Patterns of Fos-Immunoreactivity in the CNS Induced by Repeated Hemorrhage in Conscious Rats: Correlations with Pituitary-Adrenal Axis Activity. Stress 1997, 2, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Dayas, C.V.; Buller, K.M.; Day, T.A. Neuroendocrine responses to an emotional stressor: Evidence for involvement of the medial but not the central amygdala. Eur. J. Neurosci. 1999, 11, 2312–2322. [Google Scholar] [CrossRef] [PubMed]
- Prewitt, C.M.; Herman, J.P. Hypothalamo-Pituitary-Adrenocortical Regulation Following Lesions of the Central Nucleus of the Amygdala. Stress 1997, 1, 263–280. [Google Scholar] [CrossRef]
- Frenois, F.; Moreau, M.; O’Connor, J.; Lawson, M.; Micon, C.; Lestage, J.; Kelley, K.W.; Dantzer, R.; Castanon, N. Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology 2007, 32, 516–531. [Google Scholar] [CrossRef]
- Marvel, F.A.; Chen, C.C.; Badr, N.; Gaykema, R.P.; Goehler, L.E. Reversible inactivation of the dorsal vagal complex blocks lipopolysaccharide-induced social withdrawal and c-Fos expression in central autonomic nuclei. Brain Behav. Immun. 2004, 18, 123–134. [Google Scholar] [CrossRef]
- Miller, A.H. Norman cousins lecture. mechanisms of cytokine-induced behavioral changes: Psychoneuroimmunology at the translational interface. Brain Behav. Immun. 2009, 23, 149–158. [Google Scholar] [CrossRef]
- Modoux, M.; Rolhion, N.; Mani, S.; Sokol, H. Tryptophan metabolism as a pharmacological target. Trends Pharmacol. Sci. 2021, 42, 60–73. [Google Scholar] [CrossRef]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The role of IFN-gamma and TNF-alpha-responsive regulatory elements in the synergistic induction of indoleamine dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.C.; Andre, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef]
- O’Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Moron, J.A.; Zakharova, I.; Ferrer, J.V.; Merrill, G.A.; Hope, B.; Lafer, E.M.; Lin, Z.C.; Wang, J.B.; Javitch, J.A.; Galli, A.; et al. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transport capacity. J. Neurosci. 2003, 23, 8480–8488. [Google Scholar] [CrossRef]
- Duval, E.R.; Javanbakht, A.; Liberzon, I. Neural circuits in anxiety and stress disorders: A focused review. Ther. Clin. Risk Manag. 2015, 11, 115–126. [Google Scholar]
- Tovote, P.; Fadok, J.P.; Luthi, A. Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci. 2015, 16, 317–331. [Google Scholar] [CrossRef]
- Kim, M.J.; Loucks, R.A.; Palmer, A.L.; Brown, A.C.; Solomon, K.M.; Marchante, A.N.; Whalen, P.J. The structural and functional connectivity of the amygdala: From normal emotion to pathological anxiety. Behav. Brain Res. 2011, 223, 403–410. [Google Scholar] [CrossRef]
- Muscatell, K.A.; Dedovic, K.; Slavich, G.M.; Jarcho, M.R.; Breen, E.C.; Bower, J.E.; Irwin, M.R.; Eisenberger, N.I. Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav. Immun. 2015, 43, 46–53. [Google Scholar] [CrossRef]
- Dehdar, K.; Mahdidoust, S.; Salimi, M.; Gholami-Mahtaj, L.; Nazari, M.; Mohammadi, S.; Dehghan, S.; Jamaati, H.; Khosrowabadi, R.; Nasiraei-Moghaddam, A.; et al. Allergen-induced anxiety-like behavior is associated with disruption of medial prefrontal cortex—Amygdala circuit. Sci. Rep. 2019, 9, 19586. [Google Scholar] [CrossRef] [PubMed]
- Gholami-Mahtaj, L.; Mooziri, M.; Dehdar, K.; Abdolsamadi, M.; Salimi, M.; Raoufy, M.R. ACC-BLA functional connectivity disruption in allergic inflammation is associated with anxiety. Sci. Rep. 2022, 12, 2731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.H.; Liu, W.Z.; He, Y.; You, W.J.; Zhang, J.Y.; Xu, H.; Tian, X.L.; Li, B.M.; Mei, L.; Holmes, A.; et al. Chronic stress causes projection-specific adaptation of amygdala neurons via small-conductance calcium-activated potassium channel downregulation. Biol. Psychiatry 2019, 85, 812–828. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Li, C.; Wang, J.; Zhang, X.; Li, M.; Zhang, R.; Huang, Z.; Zhang, Y. Amygdala-hippocampal innervation modulates stress-induced depressive-like behaviors through AMPA receptors. Proc. Natl. Acad. Sci. USA 2021, 118, e2019409118. [Google Scholar] [CrossRef]
- Bourhy, L.; Mazeraud, A.; Costa, L.H.A.; Levy, J.; Rei, D.; Hecquet, E.; Gabanyi, I.; Bozza, F.A.; Chretien, F.; Lledo, P.M.; et al. Silencing of amygdala circuits during sepsis prevents the development of anxiety-related behaviours. Brain 2022, 145, 1391–1409. [Google Scholar] [CrossRef]
- Zhou, W.; Jin, Y.; Meng, Q.; Zhu, X.; Bai, T.; Tian, Y.; Mao, Y.; Wang, L.; Xie, W.; Zhong, H.; et al. A neural circuit for comorbid depressive symptoms in chronic pain. Nat Neurosci 2019, 22, 1649–1658. [Google Scholar] [CrossRef]
- Yuan, T.; Orock, A.; Greenwood-Van Meerveld, B. Amygdala microglia modify neuronal plasticity via complement C1q/C3-CR3 signaling and contribute to visceral pain in a rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G1081–G1092. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, B.; Yuan, Y.; Zhang, L.; Hu, L.; Jin, S.; Kang, B.; Liao, X.; Sun, W.; Xu, F.; et al. Brain control of humoral immune responses amenable to behavioural modulation. Nature 2020, 581, 204–208. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Stress Type | Inflammatory in Amygdala | Species | Reference |
|---|---|---|---|
| Chronic unpredictable mild stress | IL-1β↑ 1, IL-6↑, TNF-α↑ | Mouse | [99] |
| Chronic unpredictable mild stress | IL-1β↑, IL-10↑ | Hamsters | [104] |
| Chronic restrain stress | BLA: NLRP3↑, IL-1β↑ | Rat | [100] |
| Cumulative mild stress | TGF-β↑, IL-1β↑, IL-17↑, IL-18↑, IL-6↑ | Mouse | [105] |
| Chronic social defeat stress | BLA: IL-1β↑, IL-6↑, TNF-α↑, IL-12↑ | Mouse | [106] |
| Modified social defeat stress | IL-6 – 2 | Rat | [107] |
| Foot-shock stress | IL-4 –, IL-10 –, IL-1β –, TNF-α –, INF-γ –, IL-6↓ 3 | Mouse | [108] |
| Forced swim stress | TNF-α↑, IL-6–, IL-1β – | Rats | [109] |
| Physical restraint stress with brief underwater submersion, and predator odor stress | IL-6↑, IL-1β↑, Caspase 1↑, NLRP3↑ | Mouse | [103] |
| Chronic prostatitis/chronic pelvic pain syndrome | BLA: IL-1R↓, IL-4R↓, IL-13R↓, TNFR↓ | Rats | [110] |
| LPS | BLA: TGF-β1↓, TNF-α↑, IL-1β↑; IL-33↑ | Mouse | [111,112] |
| LPS | IL-6↑, IL-1β↑, TNF-α↑, IL-10↑; IL-6↑, IL-1β↑, vWF↑; IL-6↑, IL-1β↑, TNF-α↑; IL-6↑, IL-1β↑ | Mouse | [113,114,115,116,117] |
| LPS + oxazolone (Ox)-induced AD model | IL-6↑ | Mouse | [118] |
| Central LPS infusion | IL-1β↑, TNF-α↑ | Rats | [119] |
| Intracerebroventricular(i.c.v.) injection of IL-1β | IL-6↑, TNF-α↑ | Mouse | [120] |
| Injection of complete Freund’s adjuvant (CFA) | BLA: TNF-α↑IL-1β↑, IL-6↑, TNF-α↑ | Mouse | [121,122] |
| High-fat diet | IL-1β↑, TNF-α↑; TNF-α↑ | Mouse | [123,124] |
| High-fat diet | IL-6↑, CD11b –; IL-1Ra↑; IL-6 –, CD11b –, IL-1Rα↓ | Rats | [125,126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, P.; Lu, Y.; Pan, B.-X.; Zhang, W.-H. New Insights into the Pivotal Role of the Amygdala in Inflammation-Related Depression and Anxiety Disorder. Int. J. Mol. Sci. 2022, 23, 11076. https://doi.org/10.3390/ijms231911076
Hu P, Lu Y, Pan B-X, Zhang W-H. New Insights into the Pivotal Role of the Amygdala in Inflammation-Related Depression and Anxiety Disorder. International Journal of Molecular Sciences. 2022; 23(19):11076. https://doi.org/10.3390/ijms231911076
Chicago/Turabian StyleHu, Ping, Ying Lu, Bing-Xing Pan, and Wen-Hua Zhang. 2022. "New Insights into the Pivotal Role of the Amygdala in Inflammation-Related Depression and Anxiety Disorder" International Journal of Molecular Sciences 23, no. 19: 11076. https://doi.org/10.3390/ijms231911076
APA StyleHu, P., Lu, Y., Pan, B.-X., & Zhang, W.-H. (2022). New Insights into the Pivotal Role of the Amygdala in Inflammation-Related Depression and Anxiety Disorder. International Journal of Molecular Sciences, 23(19), 11076. https://doi.org/10.3390/ijms231911076