

Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm


 ,
,  ,
,
Abstract
1. Introduction
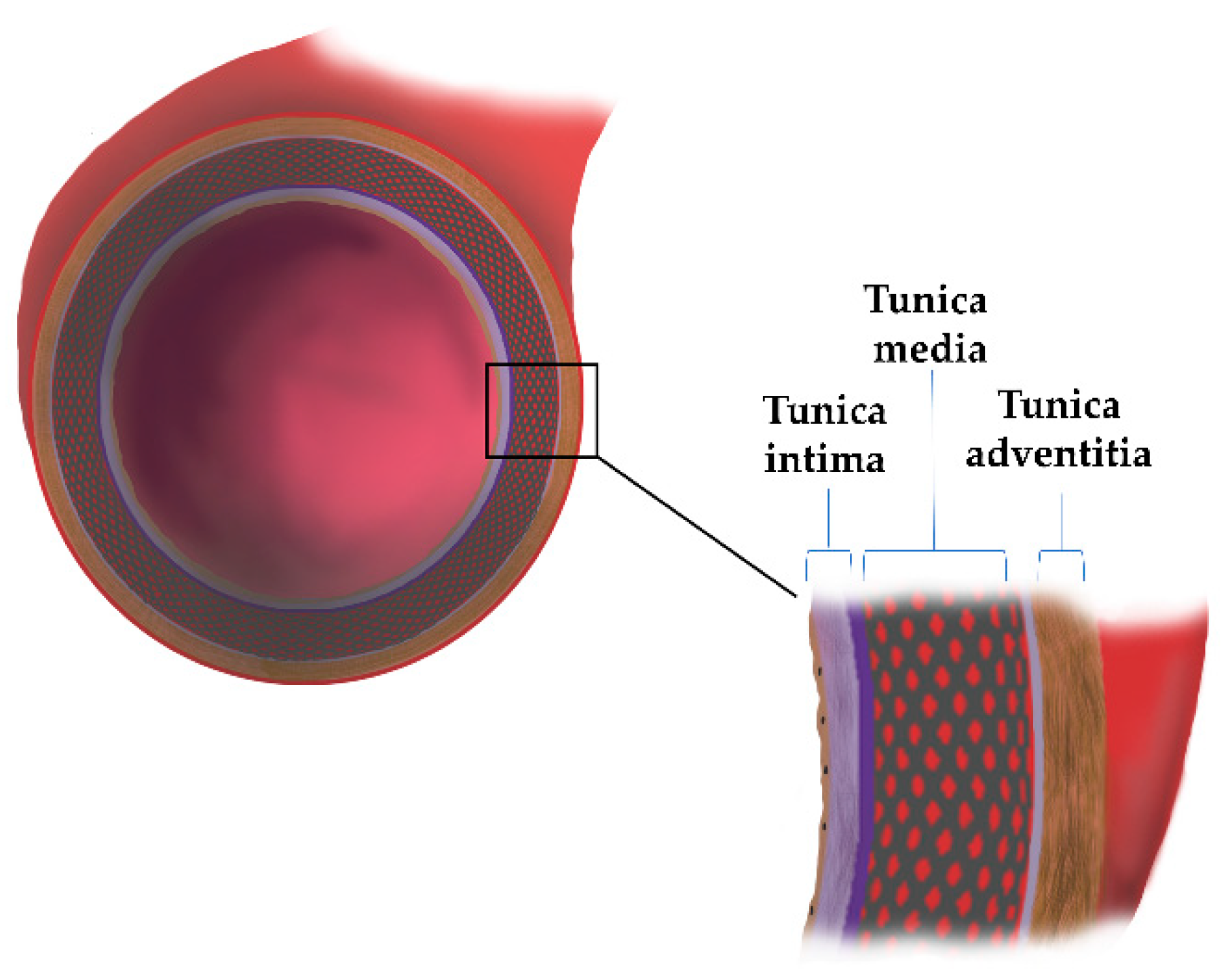
2. The Aortic Wall Structure
2.1. Fiber Type Structures Determine the Wall Properties
2.1.1. Types of Fiber Structures
2.1.2. Modifications of ECM Structural Elements
2.2. ECM Remodeling: The Major Players in This Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Metalloproteinase | Source | ECM-Substrate | Potential Role in AAA Pathogenesis | References |
|---|---|---|---|---|---|
| Tolloids | BMP-1 bone morphogenetic protein 1 (BMP-1) | osteoblasts chondroblasts | Procollagens types I and III, Laminin 5 gamma 2 chain | Converting procollagens to collagens | [39] |
| mTLD mammalian Tolloid | ECM | Procollagen types I, II, and III | Converting procollagens to collagens | [33,40] | |
| TLL-1 Tolloid-like protein 1 | Chordin, Pro-lysyl Oxidase | Crucial for activation of BMP2 and BMP4 for cell differentiation, critical for providing active Pro-lysyl Oxidase to crosslink collagen monomers and collagen fibrils | [41,42] | ||
| TLL-2 Tolloid-like protein 2 | Pro-lysyl Oxidase | Critical for providing active Pro-lysyl Oxidase to crosslink collagen monomers and collagen fibrils | [43] | ||
| ADAMTS | ADAMTS-2 A disintegrin and metalloproteinase with thrombospondin motifs 2 | fibroblasts | Procollagen types I and III | Converting procollagens to pC collagens | [44] |
| Collagenasses | Collagenase-1: MMP-1 | ECs, SMCs, FBs, macrophages, platelets, | Collagen triple helix, Versican, aggrecan, nidogen, perlacan, proteoglycan link protein | Development of the inflammatory process in the aortic wall; modulate the process of aortic rupture and dissection | [4,34,35] |
| Collagenase-2: MMP-8 | Macrophages, neutrophils, | Collagen triple helix; elastin, fibronectin, laminin, aggrecan | Significant expression in expanded and rupture AAA | [34,35] | |
| Collagenase-3: MMP-13 | VSMCs, macrophages, | Collagen triple helix; gelatin, fibronectin, laminin, tenascic | Significant expression in AAA: symptomatic and ruptured AAA | [4,34,35] | |
| Gelatinases | Gelatinase-A: MMP-2 | ECs, vascular smooth muscle (VSM), SMCs, FBs, macrophages, platelets, leukocytes, adventitia | Collagen, triple helix; gelatin, elastin, aggrecan, fibronectin, versican, proteoglycan link protein | Elevated levels in developing aneurysms | [34,35] |
| Gelatinase-B: MMP-9 | ECs, VSM, platelets, macrophages, adventitia | Collagen: IV, V, VII, X, XIV, gelatin, elastin, aggrecan, fibronectin, laminin, versican, proteoglycan link protein | High levels in developing aneurysms; stimulates the inflammatory response in AAA | [10,34] | |
| Stromelysins | Stromelysin-1: MMP-3 | ECs, FBs VSM, intima, epithelium | Collagen: II, III, IV, IX, X, XI, gelatin, aggrecan, decorin, elastin, fibronectin, versican, laminin, proteoglycan link protein | Promotes AAA | [4,34,35] |
| Stromelysin-2: MMP-10 | ECs, FBs | Elastin, fibronectin, gelatin I, link protein, casein, fibronectin | High level in atherosclerosis | [34] | |
| Matrilysins | Matrilysins- 1: MMP-7 | ECs, VSM, intima | Collagen: IV, X, gelatin, aggrecan, elastin, fibronectin, laminin, proteoglycan link protein, N-cadherin | Increase expression in AAA | [34] |
| Metalloelastase | MMP-12 | Macrophages, SMCs, FBs | Collagen IV, gelatin, elastin, fibronectin, laminin | Enhance AAA formation | [34,35] |
| MEMBRANE TYPE MMPs | MT1-MMP: MMP-14 | FBs, VSM, SMCs platelets, macrophages | Collagen I, II, III, gelatin, aggrecan, elastin, fibronectin, laminin, proteoglycan, vitronectin | Direct degradation of ECM in the tunica media and adventitia in aortic wall—formation of AAA | [34,35] |
| MT1-MMP MMP-15 | FBs, leukocytes | Collagen type I, gelatin, aggrecan, fibronectin, laminin, nidogen, tenascin, perlacan | Reduction in cell adhesion | [34,37] | |
| MT1-MMP MMP-17 | VSMCs | Osteopontin in VSMCs, gelatin, fibrin | Restrain AAA formation | [34] |
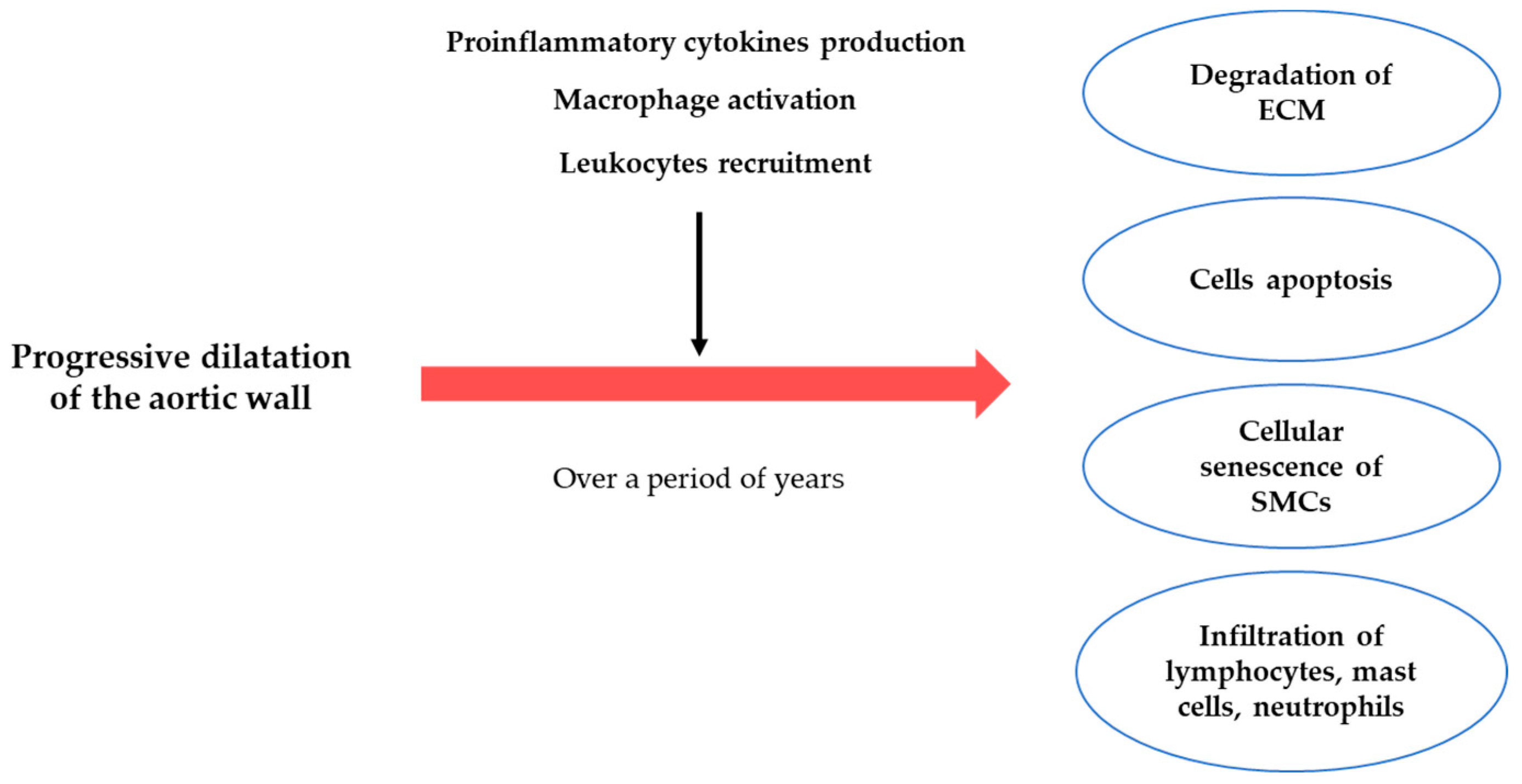
3. The Pathogenesis of the AAA
3.1. ECM Degradation
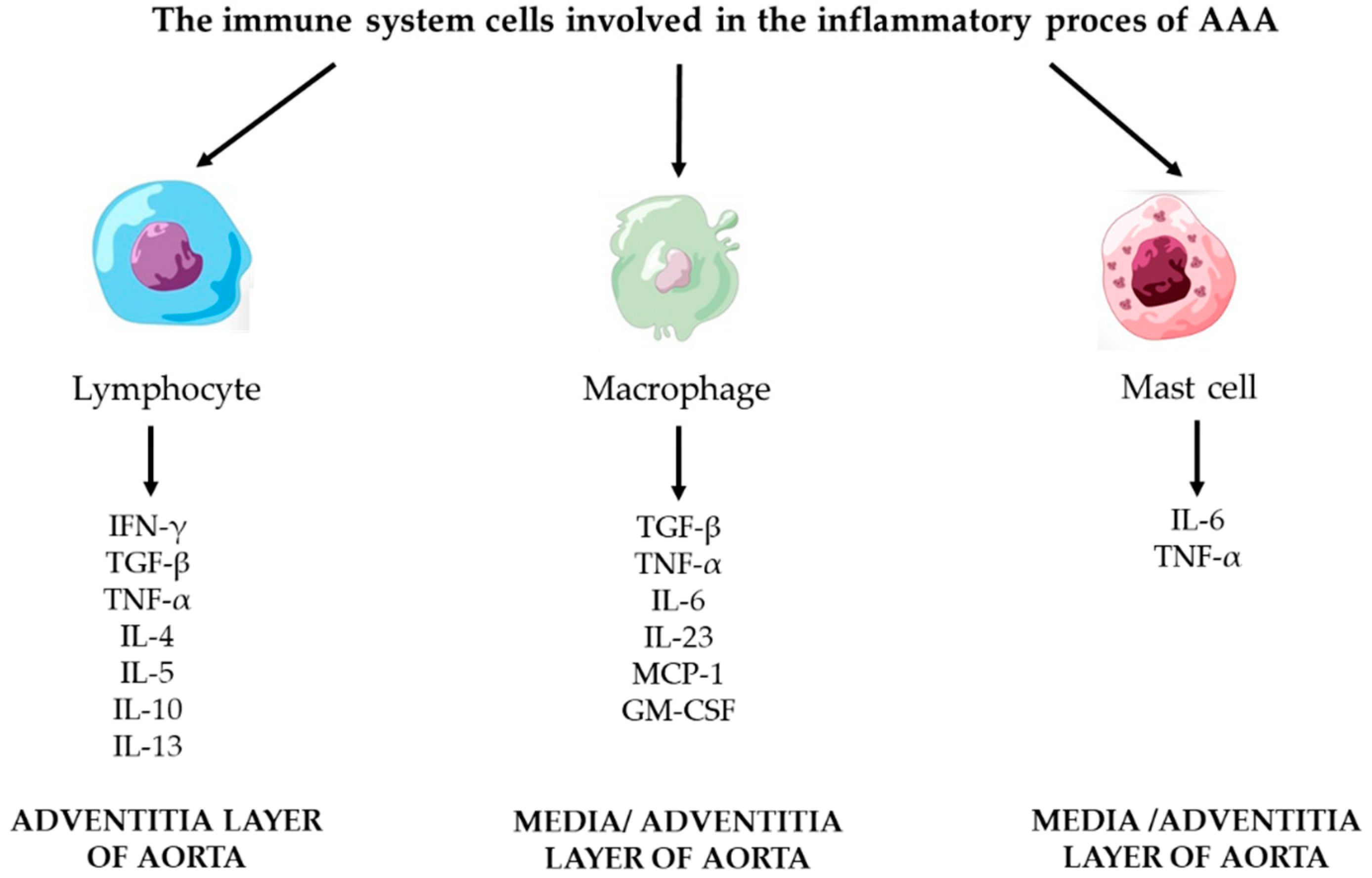
3.2. Inflammatory Process
3.2.1. Lymphocyte
3.2.2. Macrophages
3.2.3. Mast Cells
4. Potential Inflammatory Markers of AAA
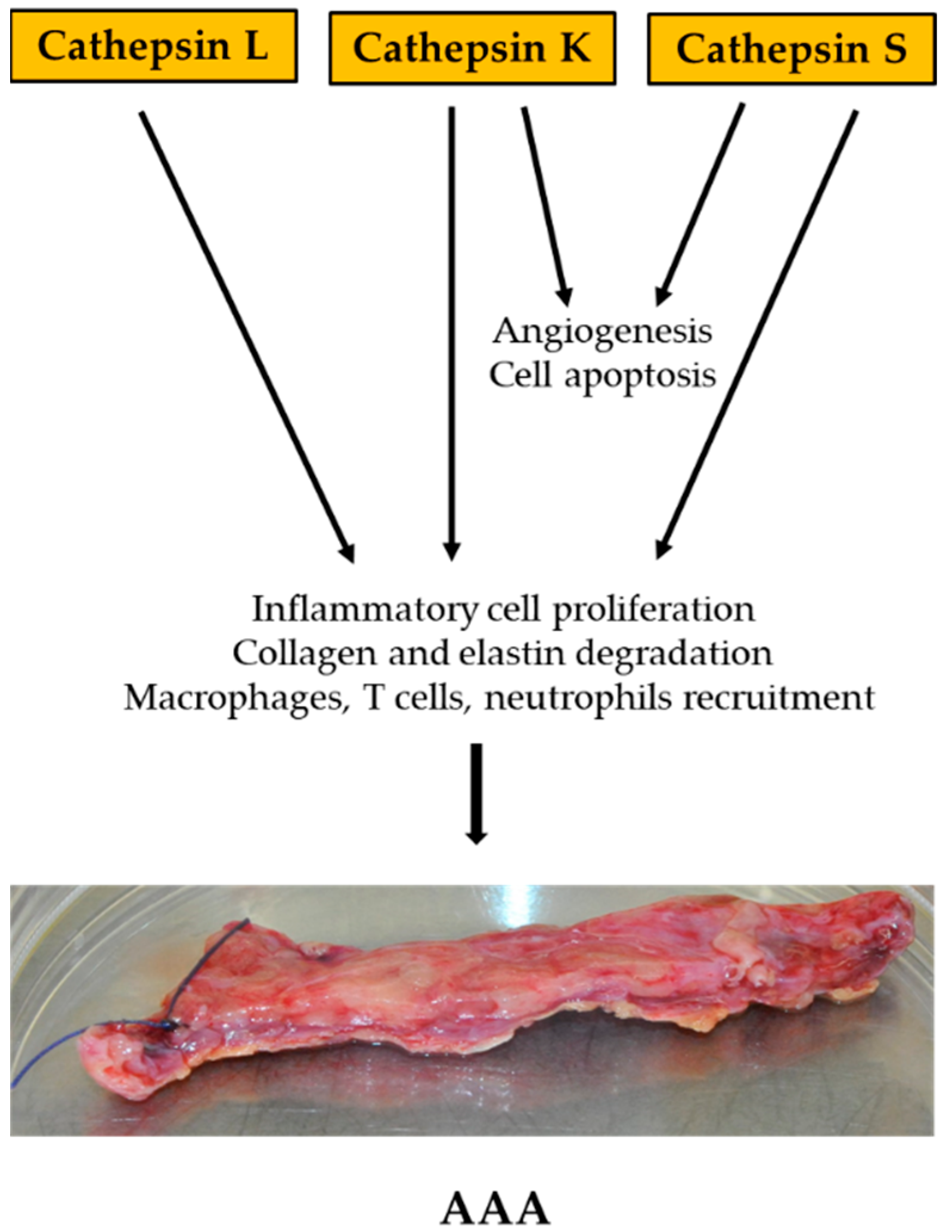
4.1. Cathepsin
4.2. Homocysteine
4.3. Osteoprotegerin
4.4. Osteopontin
5. Conclusions/Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuivaniemi, H.; Ryer, E.J.; Elmore, J.R.; Tromp, G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev. Cardiovasc. Ther. 2015, 13, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Rateri, D.L.; Daugherty, A. Abdominal aortic aneurysm: Novel mechanisms and therapies. Curr. Opin. Cardiol. 2015, 30, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Moll, F.L.; Powell, J.T.; Fraedrich, G.; Verzini, F.; Haulon, S.; Waltham, M.; van Herwaarden, J.A.; Holt, P.J.E.; van Keulen, J.W.; Rantner, B.; et al. Management of Abdominal Aortic Aneurysms Clinical Practice Guidelines of the European Society for Vascular Surgery. Eur. J. Vasc. Endovasc. Surg. 2011, 41 (Suppl. 1), S1–S58. [Google Scholar] [CrossRef] [PubMed]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef]
- Starr, J.E.; Halpern, V. Abdominal aortic aneurysms in women. J. Vasc. Surg. 2013, 57 (Suppl. S4), 3S–10S. [Google Scholar] [CrossRef] [PubMed]
- Hultgren, R.; Zommorodi, S.; Gambe, M.; Roy, J. A Majority of Admitted Patients with Ruptured Abdominal Aortic Aneurysm Undergo and Survive Corrective Treatment: A Population-Based Retrospective Cohort Study. World J. Surg. 2016, 40, 3080–3087. [Google Scholar] [CrossRef]
- Khan, S.; Verma, V.; Verma, S.; Polzer, S.; Jha, S. Assessing the potential risk of rupture of abdominal aortic aneurysms. Clin. Radiol. 2015, 70, 11–20. [Google Scholar] [CrossRef]
- Malm, I.; De Basso, R.; Blomstrand, P.; Bjarnegård, N. Increased arterial stiffness in males with abdominal aortic aneurysm. Clin. Physiol. Funct. Imaging 2021, 41, 68–75. [Google Scholar] [CrossRef]
- Jana, S.; Hu, M.; Shen, M.; Kassiri, Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef]
- Ramella, M.; Bertozzi, G.; Fusaro, L.; Talmon, M.; Manfredi, M.; Catoria, M.C.; Casella, F.; Porta, C.M.; Boldorini, R.; Fresu, L.G.; et al. Effect of Cyclic Stretch on Vascular Endothelial Cells and Abdominal Aortic Aneurysm (AAA): Role in the Inflammatory Response. Int. J. Mol. Sci. 2019, 20, 287. [Google Scholar] [CrossRef]
- Xu, J.; Shi, G. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta 2014, 1842, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. Vascular Extracellular Matrix and Arterial Mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef] [PubMed]
- Wagenseil, J.E.; Mecham, R.P. Elastin in Large Artery Stiffness and Hypertension. J. Cardiovasc. Transl. Res. 2012, 5, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Berillis, P. The Role of Collagen in the Aorta’s Structure. Open Circ. Vasc. J. 2013, 6, 1–8. [Google Scholar] [CrossRef]
- Shen, Y.; Russo, V.; Zeglinski, M.; Sellers, S.L.; Wu, Z.; Oram, C.; Santacruz, S.; Merkulova, Y.; Turner, C.; Tauh, K.; et al. Recombinant Decorin Fusion Protein Attenuates Murine Abdominal Aortic Aneurysm Formation and Rupture. Sci. Rep. 2017, 7, 15857. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, N.; Lunde, I.G.; Andenæs, K.; Strand, M.E.; Aronsen, J.M.; Skrbic, B.; Marstein, H.S.; Bandlien, C.; Nygård, S.; Gorham, J.; et al. The extracellular matrix proteoglycan lumican improves survival and counteracts cardiac dilatation and failure in mice subjected to pressure overload. Sci. Rep. 2019, 9, 9206. [Google Scholar] [CrossRef] [PubMed]
- Nikitovic, D.; Papoutsidakis, A.; Karamanos, N.K.; Tzanakakis, G.N. Lumican affects tumor cell functions, tumor–ECM interactions, angiogenesis and inflammatory response. Matrix Biol. 2014, 35, 206–214. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.K.; Shin, J.-M.; Jeoun, U.-W.; Jang, Y.J.; Park, H.S.; Kim, J.-H.; Gong, G.-Y.; Lee, T.J.; Hong, J.P.; et al. Enhanced biglycan gene expression in the adipose tissues of obese women and its association with obesity-related genes and metabolic parameters. Sci. Rep. 2016, 6, 30609. [Google Scholar] [CrossRef]
- Wight, T.N.; Kang, I.; Evanko, S.P.; Harten, I.A.; Chang, M.Y.; Pearce, O.M.T.; Allen, C.E.; Frevert, C.W. Versican—A Critical Extracellular Matrix Regulator of Immunity and Inflammation. Front. Immunol. 2020, 11, 512. [Google Scholar] [CrossRef]
- Hatano, S.; Nagai, N.; Sugiura, N.; Tsuchimoto, J.; Isogai, Z.; Kimata, K.; Ota, A.; Karnan, S.; Hosokawa, Y.; Watanabe, H. Versican A-subdomain is required for its adequate function in dermal development. Connect Tissue Res. 2018, 59, 178–190. [Google Scholar] [CrossRef]
- Geiger, B.; Bershadsky, A.; Pankov, R.; Yamada, K.M. Transmembrane Crosstalk between the Extracellular Matrix and the Cytoskeleton. Nat. Rev. Mol. Cell Biol. 2001, 2, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of Fibronectin Extracellular Matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Labosky, P.; Furuta, Y.; Hargett, L.; Dunn, R.; Fogo, A.; Takahara, K.; Peters, D.; Greenspan, D.; Hogan, B. Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development 1996, 122, 3587–3595. [Google Scholar] [CrossRef]
- Lesiak, M.; Auguściak-Duma, A.; Szydło, A.; Sieroń, A.L. Blocking angiogenesis with peptides that inhibit the activity of procollagen C-endopeptidase. Pharmacol. Rep. 2009, 61, 468–476. [Google Scholar] [CrossRef]
- Li, S.-W.; Arita, M.; Fertala, A.; Bao, Y.; Kopen, G.C.; Långsjö, T.K.; Hyttinen, M.M.; Helminen, H.J.; Prockop, D.J. Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem. J. 2001, 355 Pt 2, 271–278. [Google Scholar] [CrossRef]
- Van Dijk, F.; Sillence, D. Osteogenesis imperfecta: Clinical diagnosis, nomenclature and severity assessment. Am. J. Med Genet. A 2014, 164, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Valadares, E.R.; Carneiro, T.B.; Santos, P.M.; Oliveira, A.C.; Zabel, B. What is new in genetics and osteogenesis imperfecta classification? J. Pediatr. 2014, 90, 536–541. [Google Scholar] [CrossRef]
- Van Damme, T.; Colige, A.; Syx, D.; Giunta, C.; Lindert, U.; Rohrbach, M.; Aryani, O.; Alanay, Y.; Simsek-Kiper, P.; Kroes, H.Y.; et al. Expanding the clinical and mutational spectrum of the Ehlers–Danlos syndrome, dermatosparaxis type. Genet. Med. 2016, 18, 882–891. [Google Scholar] [CrossRef]
- Karaa, A.; Stoler, J.M. Ehlers Danlos Syndrome: An Unusual Presentation You Need to Know about. Case Rep. Pediatr. 2013, 2013, 764659. [Google Scholar] [CrossRef]
- Kessler, E.; Takahara, K.; Biniaminov, L.; Brusel, M.; Greenspan, D.S. Bone Morphogenetic Protein-1: The Type I Procollagen C-Proteinase. Science 1996, 271, 360–362. [Google Scholar] [CrossRef]
- Li, S.W.; Sieron, A.L.; Fertala, A.; Hojima, Y.; Arnold, W.V.; Prockop, D.J. The C-proteinase that processes procollagens to fibrillar collagens is identical to the protein previously identified as bone morphogenic protein-1. Proc. Natl. Acad. Sci. USA 1996, 93, 5127–5130. [Google Scholar] [CrossRef] [PubMed]
- Colige, A.; Li, S.-W.; Sieron, A.L.; Nusgens, B.V.; Prockop, D.J.; Lapière, C.M. cDNA cloning and expression of bovine procollagen I N-proteinase: A new member of the superfamily of zinc-metalloproteinases with binding sites for cells and other matrix components. Proc. Natl. Acad. Sci. USA 1997, 94, 2374–2379. [Google Scholar] [CrossRef] [PubMed]
- Sieron, A.L.; Tretiakova, A.; Jameson, B.A.; Segall, M.L.; Lund-Katz, S.; Khan, M.T.; Li, S.-W.; Stöcker, W. Structure and Function of Procollagen C-Proteinase (mTolloid) Domains Determined by Protease Digestion, Circular Dichroism, Binding to Procollagen Type I, and Computer Modeling. Biochemistry 2000, 39, 3231–3239. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv Pharmacol. 2018, 81, 241–330. [Google Scholar]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, R.; Oo, A.Y.; Xiao, Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharmaceuticals 2019, 12, 118. [Google Scholar] [CrossRef]
- Augusciak-Duma, A.; Stepien, K.L.; Lesiak, M.; Gutmajster, E.; Fus-Kujawa, A.; Botor, M.; Sieron, A.L. Expression gradient of metalloproteinases and their inhibitors from proximal to distal segments of abdominal aortic aneurysm. J. Appl. Genet. 2021, 62, 499–506. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), mainextracellular matrix (ECM) enzymes in collagendegradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Miyahara, M.; Njieha, F.K.; Prockop, D.J. Formation of collagen fibrils in vitro by cleavage of procollagen with procollagen proteinases. J. Biol. Chem. 1982, 257, 8442–8448. [Google Scholar] [CrossRef]
- Zafarullah, K.; Brown, E.M.; Kuivaniemi, H.; Tromp, G.; Sieron, A.L.; Fertala, A.; Prockop, D.J. Synthesis and conformational properties of a recombinant C-propeptide of human type III procollagen. Matrix Biol. 1997, 16, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Sieron, L.; Lesiak, M.; Schisler, I.; Drzazga, Z.; Fertala, A.; Sieron, A.L. Functional and structural studies of tolloid-like 1 mutants associated with atrial-septal defect 6. Biosci. Rep. 2019, 39, BSR20180270. [Google Scholar] [CrossRef]
- Panchenko, M.; Stetler-Stevenson, W.G.; Trubetskoy, O.V.; Gacheru, S.N.; Kagan, H.M. Metalloproteinase Activity Secreted by Fibrogenic Cells in the Processing of Prolysyl Oxidase. J. Biol. Chem. 1996, 271, 7113–7119. [Google Scholar] [CrossRef] [PubMed]
- Uzel, M.I.; Scott, I.C.; Babakhanlou-Chase, H.; Palamakumbura, A.H.; Pappano, W.N.; Hong, H.-H.; Greenspan, D.S.; Trackman, P.C. Multiple Bone Morphogenetic Protein 1-related Mammalian Metalloproteinases Process Pro-lysyl Oxidase at the Correct Physiological Site and Control Lysyl Oxidase Activation in Mouse Embryo Fibroblast Cultures. J. Biol. Chem. 2001, 276, 22537–22543. [Google Scholar] [CrossRef] [PubMed]
- Horlein, D.; Fietzek, P.P.; Kohn, K. Pro-Gln: The procollagen peptidase cleavage site in the α1 (I) chain of dermatosparactic calf skin procollagen. FEBS Lett. 1978, 89, 279–282. [Google Scholar] [CrossRef]
- Grodin, J.L.; Powell-Wiley, T.M.; Ayers, C.R.; Kumar, D.S.; Rohatgi, A.; Khera, A.; McGuire, D.K.; de Lemos, J.A.; Das, S.R. Circulating levels of matrix metalloproteinase-9 and abdominal aortic pathology: From the Dallas Heart Study. Vasc. Med. 2011, 16, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Sagan, A.; Mikolajczyk, T.P.; Mrowiecki, W.; MacRitchie, N.; Daly, K.; Meldrum, A.; Migliarino, S.; Delles, C.; Urbanski, K.; Filip, G.; et al. T Cells Are Dominant Population in Human Abdominal Aortic Aneurysms and Their Infiltration in the Perivascular Tissue Correlates With Disease Severity. Front. Immunol. 2019, 10, 1979. [Google Scholar] [CrossRef]
- Dale, M.A.; Ruhlman, M.K.; Baxter, B.T. Inflammatory Cell Phenotypes in AAAs: Their role and potential as targets for therapy. Arter. Thromb. Vasc. Biol. 2015, 35, 1746–1755. [Google Scholar] [CrossRef]
- Schönbeck, U.; Sukhova, G.K.; Gerdes, N.; Libby, P. TH2 Predominant Immune Responses Prevail in Human Abdominal Aortic Aneurysm. Am. J. Pathol. 2002, 161, 499–506. [Google Scholar] [CrossRef]
- Shimizu, K.; Shichiri, M.; Libby, P.; Lee, R.T.; Mitchell, R.N. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J. Clin. Investig. 2004, 114, 300–308, Erratum in 2004, 114, 739. [Google Scholar] [CrossRef]
- Wei, Z.; Wang, Y.; Zhang, K.; Liao, Y.; Ye, P.; Wu, J.; Wang, Y.; Li, F.; Yao, Y.; Zhou, Y.; et al. Inhibiting the Th17/IL-17A–Related Inflammatory Responses with Digoxin Confers Protection against Experimental Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2014, 34, 2429–2438. [Google Scholar] [CrossRef]
- Madhur, M.S.; Funt, S.A.; Li, L.; Vinh, A.; Chen, W.; Lob, H.E.; Iwakura, Y.; Blinder, Y.; Rahman, A.; Quyyumi, A.A.; et al. Role of Interleukin 17 in Inflammation, Atherosclerosis, and Vascular Function in Apolipoprotein E–Deficient Mice. Arter. Thromb. Vasc. Biol. 2011, 31, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Rateri, D.L.; Daugherty, A. Mechanisms of aortic aneurysm formation: Translating preclinical studies into clinical therapies. Heart 2014, 100, 1498–1505. [Google Scholar] [CrossRef]
- Li, H.; Bai, S.; Ao, Q.; Wang, X.; Tian, X.; Li, X.; Tong, H.; Hou, W.; Fan, J. Modulation of Immune-Inflammatory Responses in Abdominal Aortic Aneurysm: Emerging Molecular Targets. J. Immunol. Res. 2018, 2018, 7213760. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Hu, Y.; Akk, A.; Ye, K.; Bacon, J.; Pham, C.T.N. Interleukin-12 and -23 blockade mitigates elastase-induced abdominal aortic aneurysm. Sci. Rep. 2019, 9, 10447. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C.; Smith, A.J.; Jones, G.T.; Swerdlow, D.I.; Rampuri, R.; Bown, M.J.; Folkersen, L.; Baas, A.F.; de Borst, G.J.; Blankensteijn, J.D.; et al. Interleukin-6 receptor pathways in abdominal aortic aneurysm. Eur. Heart J. 2013, 34, 3707–3716. [Google Scholar] [CrossRef] [PubMed]
- Son, B.-K.; Sawaki, D.; Tomida, S.; Fujita, D.; Aizawa, K.; Aoki, H.; Akishita, M.; Manabe, I.; Komuro, I.; Friedman, S.L.; et al. Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat. Commun. 2015, 6, 6994. [Google Scholar] [CrossRef]
- Paige, E.; Clément, M.; Lareyre, F.; Sweeting, M.; Raffort, J.; Grenier, C.; Finigan, A.; Harrison, J.; Peters, J.E.; Sun, B.B.; et al. Interleukin-6 Receptor Signaling and Abdominal Aortic Aneurysm Growth Rates. Circ. Genom. Precis. Med. 2019, 12, e002413. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krishna, S.; Walker, P.J.; Norman, P.; Golledge, J. Transforming growth factor-β and abdominal aortic aneurysms. Cardiovasc. Pathol. 2013, 22, 126–132. [Google Scholar] [CrossRef]
- Tsuruda, T.; Kato, J.; Hatakeyama, K.; Kojima, K.; Yano, M.; Yano, Y.; Nakamura, K.; Nakamura-Uchiyama, F.; Matsushima, Y.; Imamura, T.; et al. Adventitial Mast Cells Contribute to Pathogenesis in the Progression of Abdominal Aortic Aneurysm. Circ. Res. 2008, 102, 1368–1377. [Google Scholar] [CrossRef]
- Sun, J.; Sukhova, G.K.; Yang, M.; Wolters, P.J.; MacFarlane, L.A.; Libby, P.; Sun, C.; Zhang, Y.; Liu, J.; Ennis, T.L.; et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J. Clin. Investig. 2007, 117, 3359–3368. [Google Scholar] [CrossRef]
- Qin, Y.; Cao, X.; Yang, Y.; Shi, G.-P. Cysteine protease cathepsins and matrix metalloproteinases in the development of abdominal aortic aneurysms. Future Cardiol. 2013, 9, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Conus, S.; Simon, H.-U. Cathepsins: Key modulators of cell death and inflammatory responses. Biochem. Pharmacol. 2008, 76, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.-P.; Sukhova, G.K.; Grubb, A.; Ducharme, A.; Rhode, L.H.; Lee, R.T.; Ridker, P.M.; Libby, P.; Chapman, H.A. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J. Clin. Investig. 1999, 104, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.-L.; Lindholt, J.S.; Shi, G.-P.; Zhang, J. Plasma Cystatin B Association with Abdominal Aortic Aneurysms and Need for Later Surgical Repair: A Sub-study of the VIVA Trial. Eur. J. Vasc. Endovasc. Surg. 2018, 56, 826–832. [Google Scholar] [CrossRef]
- Lok, Z.S.Y.; Lyle, A. Osteopontin in Vascular Disease. Arter. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef]
- Wolak, T. Osteopontin—A multi-modal marker and mediator in atherosclerotic vascular disease. Atherosclerosis 2014, 236, 327–337. [Google Scholar] [CrossRef]
- Migacz, M.; Janoska-Gawrońska, A.; Holecki, M.; Chudek, J. The role of osteoprotegerin in the development, progression and management of abdominal aortic aneurysms. Open Med. 2020, 15, 457–463. [Google Scholar] [CrossRef]
- Kiechl, S.; Schett, G.; Wenning, G.; Redlich, K.; Oberhollenzer, M.; Mayr, A.; Santer, P.; Smolen, J.; Poewe, W.; Willeit, J. Osteoprotegerin Is a Risk Factor for Progressive Atherosclerosis and Cardiovascular Disease. Circulation 2004, 109, 2175–2180. [Google Scholar] [CrossRef]
- Martinez-Pinna, R.; Lindholt, J.; Blanco-Colio, L.; Dejouvencel, T.; Madrigal-Matute, J.; Ramos-Mozo, P.; de Ceniga, M.V.; Michel, J.; Egido, J.; Meilhac, O.; et al. Increased levels of thioredoxin in patients with abdominal aortic aneurysms (AAAs). A potential link of oxidative stress with AAA evolution. Atherosclerosis 2010, 212, 333–338. [Google Scholar] [CrossRef]
- Licholai, S.; Szczeklik, W.; Sanak, M. miR-29c-3p is an Effective Biomarker of Abdominal Aortic Aneurysm in Patients Undergoing Elective Surgery. MicroRNA 2016, 5, 124–131. [Google Scholar] [CrossRef]
- Boon, R.A.; Seeger, T.; Heydt, S.; Fischer, A.; Hergenreider, E.; Horrevoets, A.J.; Vinciguerra, M.; Rosenthal, N.; Sciacca, S.; Pilato, M.; et al. MicroRNA-29 in Aortic Dilation: Implications for Aneurysm Formation. Circ. Res. 2011, 109, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Halazun, K.; Bofkin, K.; Asthana, S.; Evans, C.; Henderson, M.; Spark, J. Hyperhomocysteinaemia is Associated with the Rate of Abdominal Aortic Aneurysm Expansion. Eur. J. Vasc. Endovasc. Surg. 2007, 33, 391–394; discussion 395–396. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Smith, A.D.; Ueland, P.M.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and Recommendations about Total Homocysteine Determinations: An Expert Opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.T.; Cheng, S.W.K. Elevated homocysteine in human abdominal aortic aneurysmal tissues. Vasc. Med. 2017, 22, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Moroz, P.; Le, M.T.Q.; Norman, P.E. Homocysteine and abdominal aortic aneurysms. ANZ J. Surg. 2007, 77, 329–332. [Google Scholar] [CrossRef]
- Cao, H.; Hu, X.; Zhang, Q.; Li, J.; Wang, J.; Shao, Y.; Liu, B.; Xin, S. Homocysteine Level and Risk of Abdominal Aortic Aneurysm: A Meta-Analysis. PLoS ONE 2014, 9, e85831. [Google Scholar] [CrossRef]
- Qin, Y.; Shi, G.-P. Cysteinyl cathepsins and mast cell proteases in the pathogenesis and therapeutics of cardiovascular diseases. Pharmacol. Ther. 2011, 131, 338–350. [Google Scholar] [CrossRef]
- Sukhova, G.K.; Shi, G.P.; Simon, D.I.; Chapman, H.A.; Libby, P. Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells. J. Clin. Investig. 1998, 102, 576–583. [Google Scholar] [CrossRef]
- Lv, B.-J.; Lindholt, J.S.; Cheng, X.; Wang, J.; Shi, G.-P. Plasma Cathepsin S and Cystatin C Levels and Risk of Abdominal Aortic Aneurysm: A Randomized Population–Based Study. PLoS ONE 2012, 7, e41813. [Google Scholar] [CrossRef]
- Sun, J.; Sukhova, G.K.; Zhang, J.; Chen, H.; Sjöberg, S.; Libby, P.; Xia, M.; Xiong, N.; Gelb, B.D.; Shi, G.-P. Cathepsin K Deficiency Reduces Elastase Perfusion–Induced Abdominal Aortic Aneurysms in Mice. Arter. Thromb. Vasc. Biol. 2012, 32, 15–23. [Google Scholar] [CrossRef]
- Qin, Y.; Cao, X.; Guo, J.; Zhang, Y.; Pan, L.; Zhang, H.; Li, H.; Tang, C.; Du, J.; Shi, G.-P. Deficiency of cathepsin S attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Cardiovasc. Res. 2012, 96, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Lesiak, M.; Augusciak-Duma, A.; Stepien, K.L.; Fus-Kujawa, A.; Botor, M.; Sieron, A.L. Searching for new molecular markers for cells obtained from abdominal aortic aneurysm. J. Appl. Genet. 2021, 62, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lesiak, M.; Stepien, K.L.; Fus-Kujawa, A.; Augusciak-Duma, A.; Gutmajster, E.; Ziaja, D.; Sznapka, M.; Ziaja, K.; Kuczmik, W.; Sieron, A.L. Characteristic of cells isolated from human Abdominal Aortic Aneurysm samples cultured in vitro. Acta Angiol. 2021, 27, 120–129. [Google Scholar] [CrossRef]
- Augusciak-Duma, A.; Lesiak, M.; Stepien, K.L.; Gutmajster, E.; Sieron, A.L. mRNA Expression of thrombospondin 1, 2 and 3 from proximal to distal in human abdominal aortic aneurysm—Preliminary report. Acta Biochim. Pol. 2021, 68, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Miyake, T.; Kurashiki, T.; Morishita, R. Molecular Pharmacological Approaches for Treating Abdominal Aortic Aneurysm. Ann. Vasc. Dis. 2019, 12, 137–146. [Google Scholar] [CrossRef]
- Morishita, R.; Higaki, J.; Tomita, N.; Ogihara, T. Application of Transcription Factor “Decoy” Strategy as Means of Gene Therapy and Study of Gene Expression in Cardiovascular Disease. Circ. Res. 1998, 82, 1023–1028. [Google Scholar] [CrossRef]
- Nakashima, H.; Aoki, M.; Miyake, T.; Kawasaki, T.; Iwai, M.; Jo, N.; Oishi, M.; Kataoka, K.; Ohgi, S.; Ogihara, T.; et al. Inhibition of experimental abdominal aortic aneurysm in the rat by use of decoy oligodeoxynucleotides suppressing activity of nuclear factor κB and ets transcription factors. Circulation 2004, 109, 132–138. [Google Scholar] [CrossRef]
- Egashira, K.; Suzuki, J.I.; Ito, H.; Aoki, M.; Isobe, M.; Morishita, R. Long-term follow up of initial clinical cases with NF-κB decoy oligodeoxynucleotide transfection at the site of coronary stenting. J. Gene Med. 2008, 10, 805–809. [Google Scholar] [CrossRef]





| Aortic Wall | Components | Functions | Reference |
|---|---|---|---|
| Tunica intima | Endothelial cells (ECs) Extracellular matrix network (ECM): laminin, collagen type IV, fibronectin, perlecan, heparan sulfate, proteoglycans, nidogen | Synthesis and release of inflammatory mediators, hormones, and factors that contract and relax arteries (NO, PGI2, E-selectin, ICAM-1) | [9,10,11] |
| Tunica media | Smooth muscle cells (SMCs); ECM proteins: Proteoglycans (PGs), Glycoproteins, Glycosaminoglycans (GAGs), collagen, elastin | Compliance and recoil properties | [9] |
| Tunica adventitia | Fibroblast (FBs), collagen type I and III, elastic fibers; chondroitin sulfate, dermatan sulfate Proteoglycans | Tensile strength | [9,11] |
| Biomarker | Type of Molecule | Role in AAA | References |
|---|---|---|---|
| Cathepsin (Cat) | Protein | Vascular remodeling, cell apoptosis, cell signalling | [61,62] |
| Cystatin B, C | Protein | Aortic wall proteolysis contributing to AAA enlargement and rupture | [63,64] |
| MMP-2, -9 | Protein | Degradation of ECM | [34,35] |
| Osteopontin (OPN) | Protein | Activation of immune cells; inflammation process | [65,66] |
| Osteoprotegerin (OPG) | Protein | Activation of immune cells; inflammation process | [67,68] |
| Thierodoxin (TRX) | Protein | Increases oxidative stress in the aorta wall | [69] |
| IL-1α | Protein | Inflammation proces | [47,50,51] |
| MiRNA712/205 | Micro RNA | Enhances the secretion of MMP (MMP-3); development of inflammation; degradation of connective tissue and ECM | [52,70,71] |
| MiRNA 29C-3P | Micro RNA | Lowers the expression of genes encoding ELN, COL4A1, VEGFA | [70,71] |
| Homocysteine (Hcy) | Amino acid | Development of a blood clot in aneurysm, degradation of elastin in the inner membrane, fibrosis, and calcification processes | [72,73,74,75,76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepien, K.L.; Bajdak-Rusinek, K.; Fus-Kujawa, A.; Kuczmik, W.; Gawron, K. Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2022, 23, 11078. https://doi.org/10.3390/ijms231911078
Stepien KL, Bajdak-Rusinek K, Fus-Kujawa A, Kuczmik W, Gawron K. Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm. International Journal of Molecular Sciences. 2022; 23(19):11078. https://doi.org/10.3390/ijms231911078
Chicago/Turabian StyleStepien, Karolina L., Karolina Bajdak-Rusinek, Agnieszka Fus-Kujawa, Wacław Kuczmik, and Katarzyna Gawron. 2022. "Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm" International Journal of Molecular Sciences 23, no. 19: 11078. https://doi.org/10.3390/ijms231911078
APA StyleStepien, K. L., Bajdak-Rusinek, K., Fus-Kujawa, A., Kuczmik, W., & Gawron, K. (2022). Role of Extracellular Matrix and Inflammation in Abdominal Aortic Aneurysm. International Journal of Molecular Sciences, 23(19), 11078. https://doi.org/10.3390/ijms231911078