
How Do Intermolecular Interactions Evolve at the Nematic to Twist–Bent Phase Transition?

 ,
,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
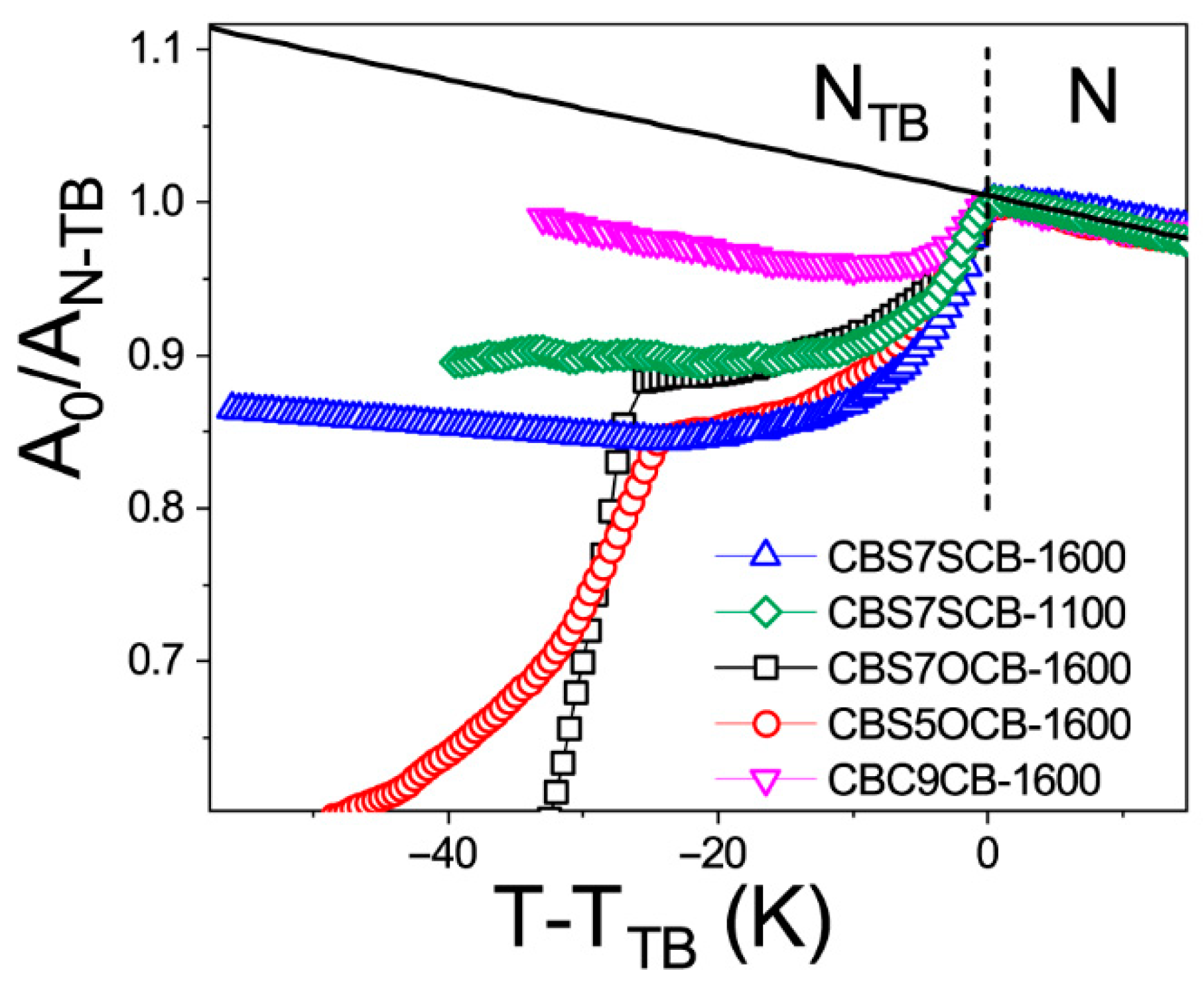
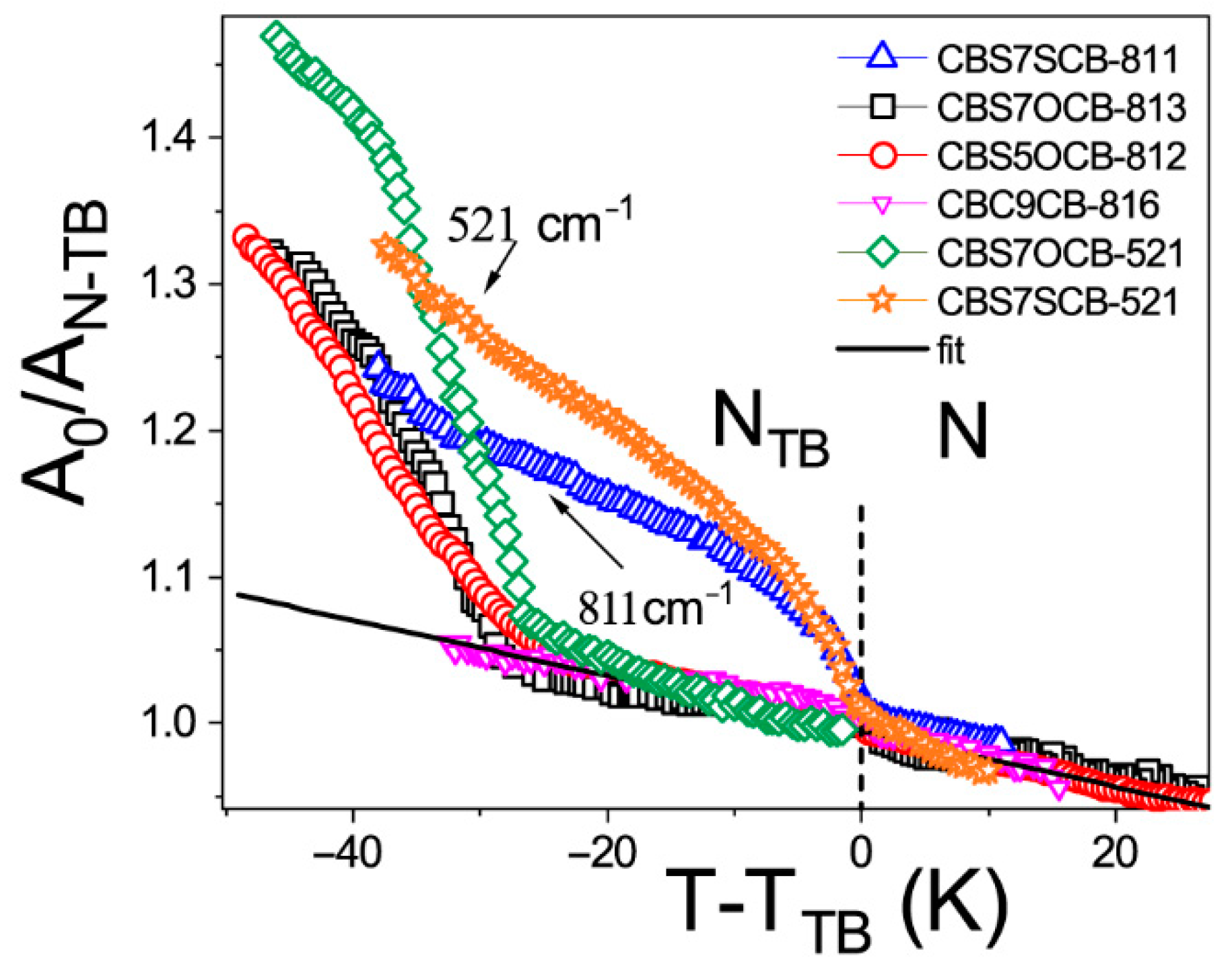
2.1. Temperature Dependencies of the Absorbance
2.2. Intermolecular Nonspecific Binding Energy in the System
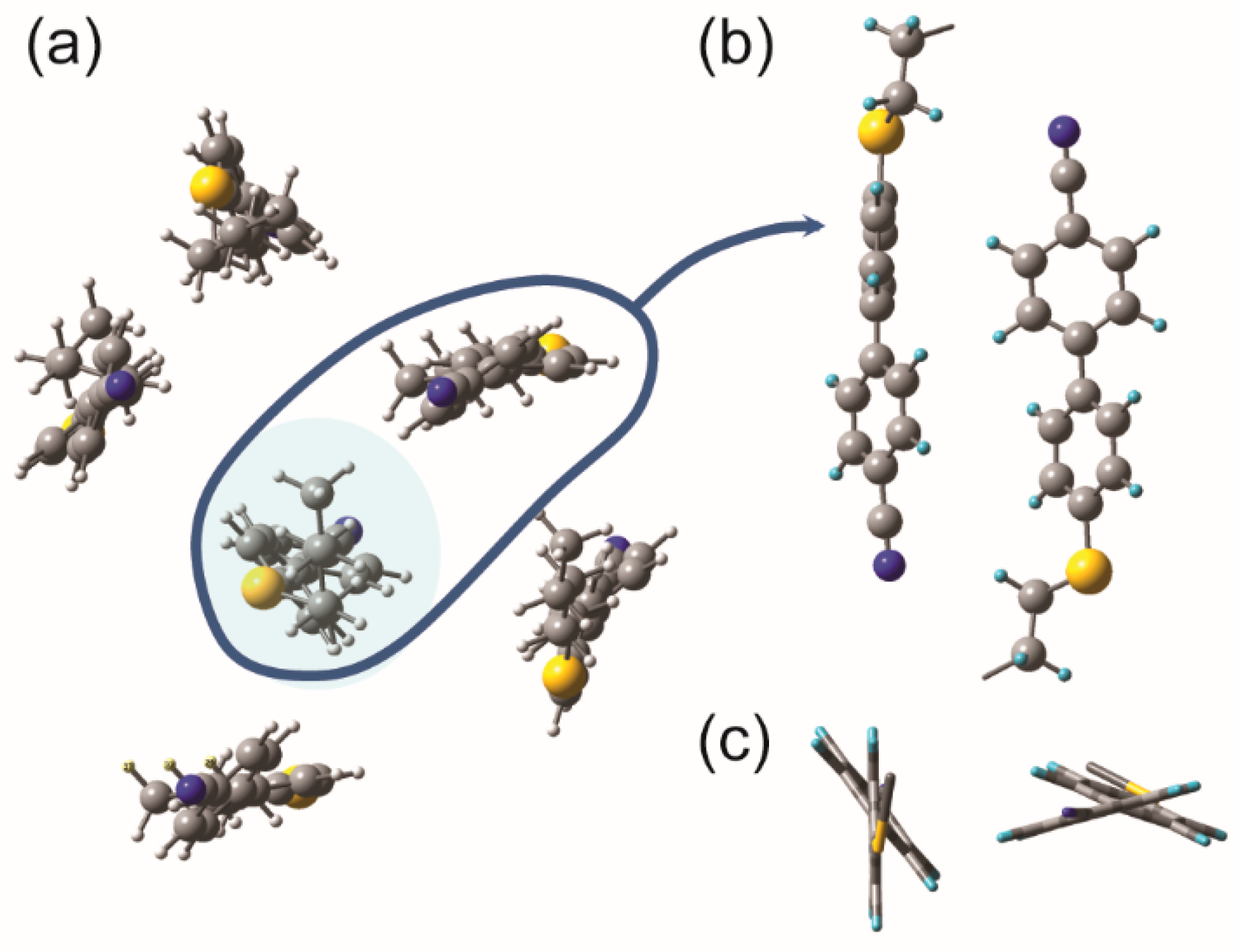
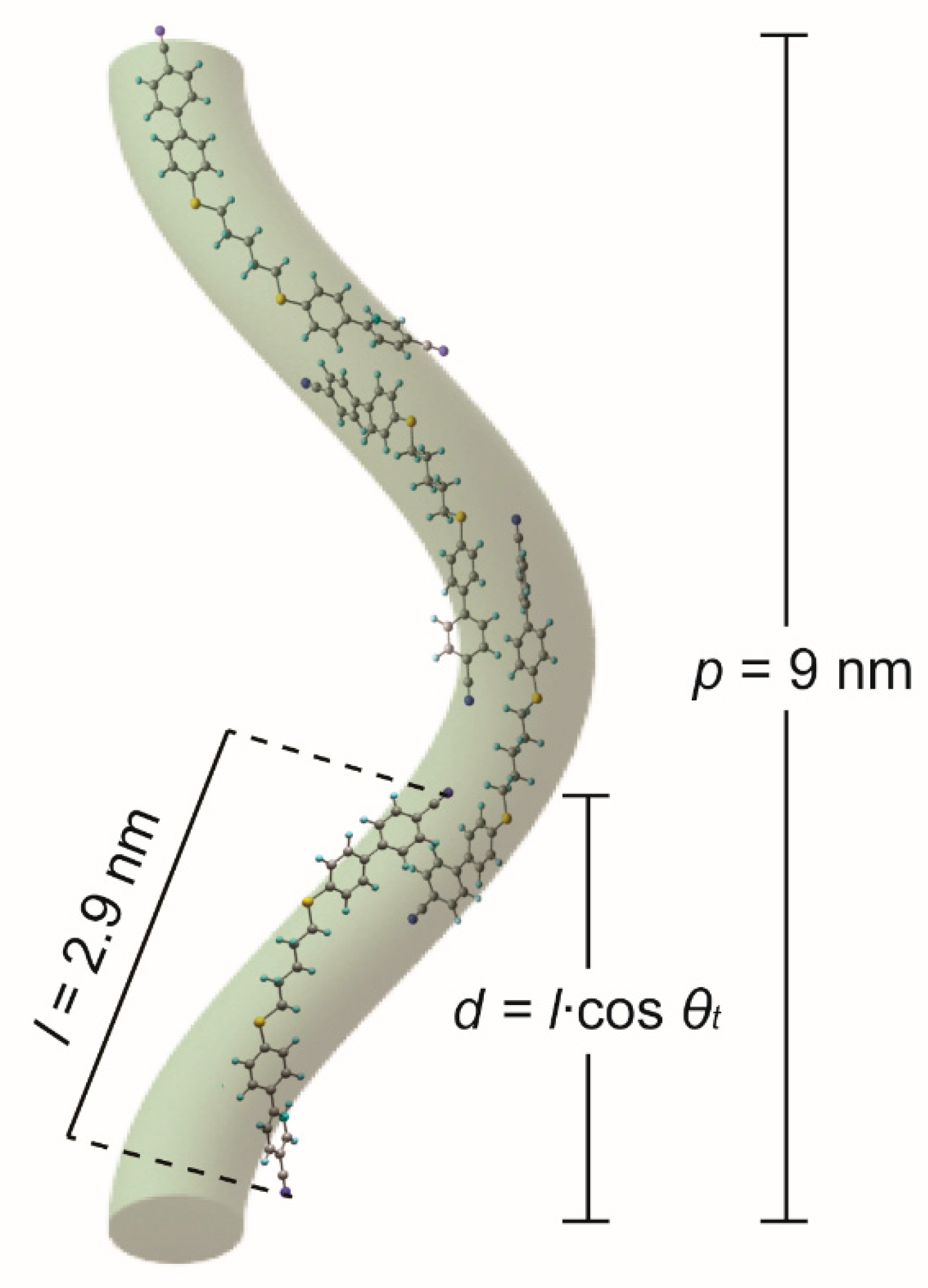
2.3. Hypothetical Arrangement of the CBSC7SCB Molecules—DFT Modeling
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Infrared Spectroscopy
4.3. Density Functional Theory Calculations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whitesides, G.M.; Grzybowski, B. Self-Assembly at All Scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef]
- Yan, F.; Hixson, C.A.; Earl, D.J. Self-Assembled Chiral Superstructures Composed of Rigid Achiral Molecules and Molecular Scale Chiral Induction by Dopants. Phys. Rev. Lett. 2008, 101, 157801–157804. [Google Scholar] [CrossRef]
- Chothia, C. Conformation of twisted β-pleated sheets in proteins. J. Mol. Biol. 1973, 75, 295–302. [Google Scholar] [CrossRef]
- Muraoka, T.; Cui, H.; Stupp, S.I. Quadruple Helix Formation of a Photoresponsive Peptide Amphiphile and Its Light-Triggered Dissociation into Single Fibers. J. Am. Chem. Soc. 2008, 130, 2946–2947. [Google Scholar] [CrossRef]
- Li, L.; Jiang, H.; Messmore, B.W.; Bull, S.R.; Stupp, S.I. A Torsional Strain Mechanism To Tune Pitch in Supramolecular Helices. Angew. Chem. Int. Ed. 2007, 119, 5977–5980. [Google Scholar] [CrossRef]
- De Gennes, P.G.; Prost, J. The Physics of Liquid Crystals, 2nd ed.; Oxford University Press: New York, NY, USA, 1993. [Google Scholar]
- Reddy, R.A.; Tschierske, C. Bent-core liquid crystals: Polar order, superstructural chirality and spontaneous desymmetrization in soft matter systems. J. Mater. Chem. 2006, 16, 907–961. [Google Scholar] [CrossRef]
- Chen, D.; Porada, J.H.; Hooper, J.B.; Klittnick, A.; Shen, Y.; Tuchband, M.R.; Korblova, E.; Bedrov, D.; Walba, D.M.; Glaser, M.A.; et al. Chiral heliconical ground state of nanoscale pitch in a nematic liquid crystal of achiral molecular dimers. Proc. Natl. Acad. Sci. USA 2013, 110, 15931–15936. [Google Scholar] [CrossRef]
- Borshch, V.; Kim, Y.K.; Xiang, J.; Gao, M.; Jákli, A.; Panov, V.P.; Vij, J.K.; Imrie, C.T.; Tamba, M.G.; Mehl, G.H.; et al. Nematic twist-bend phase with nanoscale modulation of molecular orientation. Nat. Commun. 2013, 4, 2635–2638. [Google Scholar] [CrossRef]
- Zhu, C.; Tuchband, M.R.; Young, A.; Shuai, M.; Scarbrough, A.; Walba, D.M.; Maclennan, J.E.; Wang, C.; Hexemer, A.; Clark, N.A. Resonant carbon k-edge soft x-ray scattering from lattice-free heliconical molecular ordering: Soft dilative elasticity of the twist-bend liquid crystal phase. Phys. Rev. Lett. 2016, 116, 147803–147809. [Google Scholar] [CrossRef]
- Cruickshank, E.; Salamończyk, M.; Pociecha, D.; Strachan, G.J.; Storey, J.M.D.; Wang, C.; Feng, J.; Zhu, C.; Gorecka, E.; Imrie, C.T. Sulfur-linked cyanobiphenyl-based liquid crystal dimers and the twist-bend nematic phase. Liq. Cryst. 2019, 46, 1595–1609. [Google Scholar] [CrossRef]
- Cao, Y.; Feng, J.; Nallapaneni, A.; Arakawa, Y.; Zhao, K.; Zhang, H.; Mehl, G.H.; Zhu, C.; Liu, F. Deciphering helix assembly in the heliconical nematic phase via tender resonant X-ray scattering. J. Mater. Chem. C 2021, 9, 10020–10028. [Google Scholar] [CrossRef]
- Cestari, M.; Diez-Berart, S.; Dunmur, D.A.; Ferrarini, A.; de la Fuente, M.R.; Jackson, D.J.B.; Lopez, D.O.; Luckhurst, G.R.; Perez-Jubindo, M.A.; Richardson, R.M.; et al. Phase behavior and properties of the liquid-crystal dimer 1″,7″-bis(4-cyanobiphenyl-4′-yl) heptane: A twist-bend nematic liquid crystal. Phys. Rev. E 2011, 84, 031704-20. [Google Scholar] [CrossRef]
- Adlem, K.; Čopič, M.; Luckhurst, G.R.; Mertelj, A.; Parri, O.; Richardson, R.M.; Snow, B.D.; Timimi, B.A.; Tuffin, R.P.; Wilkes, D. Chemically induced twist-bend nematic liquid crystals, liquid crystal dimers, and negative elastic constants. Phys. Rev. E 2013, 88, 022503-8. [Google Scholar] [CrossRef]
- Stevenson, W.D.; Zou, H.-X.; Zeng, X.-B.; Welch, C.; Ungar, G.; Mehl, G.H. Dynamic calorimetry and XRD studies of the nematic and twist-bend nematic phase transitions in a series of dimers with increasing spacer length. Phys. Chem. Chem. Phys. 2018, 20, 25268–25274. [Google Scholar] [CrossRef]
- Salamończyk, M.; Vaupotič, N.; Pociecha, D.; Wang, C.; Zhu, C.; Gorecka, E. Structure of nanoscale-pitch helical phases: Blue phase and twist-bend nematic phase resolved by resonant soft X-ray scattering. Soft Matter 2017, 13, 6694–6699. [Google Scholar] [CrossRef]
- Stevenson, W.; Ahmed, Z.; Zeng, X.; Welch, C.; Ungar, G.; Mehl, G. Molecular organization in the twist–bend nematic phase by resonant X-ray scattering at the Se K-edge and by SAXS, WAXS and GIXRD. Phys. Chem. Chem. Phys. 2017, 19, 13449–13454. [Google Scholar] [CrossRef]
- Tuchband, M.R.; Paterson, D.A.; Salamończyk, M.; Norman, V.A.; Scarbrough, A.N.; Forsyth, E.; Garcia, E.; Wang, C.; Storey, J.M.; Walba, D.M.; et al. Distinct differences in the nanoscale behaviors of the twist–bend liquid crystal phase of a flexible linear trimer and homologous dimer. Proc. Natl Acad. Sci. USA 2019, 116, 10698–10704. [Google Scholar] [CrossRef]
- Saha, R.; Feng, C.; Welch, C.; Mehl, G.H.; Feng, J.; Zhu, C.; Gleeson, J.; Sprunt, S.; Jákli, A. The interplay between spatial and heliconical orientational order in twist-bend nematic materials. Phys. Chem. Chem. Phys. 2021, 23, 4055–4063. [Google Scholar] [CrossRef]
- Chen, D.; Nakata, M.; Shao, R.; Tuchband, M.R.; Shuai, M.; Baumeister, U.; Weissflog, W.; Walba, D.M.; Glaser, M.A.; Maclennan, J.E.; et al. Twist-bend heliconical chiral nematic liquid crystal phase of an achiral rigid bent-core mesogen. Phys. Rev. E. 2014, 89, 022506. [Google Scholar] [CrossRef]
- Gorecka, E.; Salamonczyk, M.; Zep, A.; Pociecha, D.; Welch, C.; Ahmed, Z.; Mehl, G.H. Do the short helices exist in the nematic TB phase? Liq. Cryst. 2015, 42, 1–7. [Google Scholar] [CrossRef]
- Knežević, A.; Dokli, I.; Sapunar, M.; Sěgota, S.; Baumeister, U.; Lesac, A. Induced smectic phase in binary mixtures of twist-bend nematogens. Beilstein J. Nanotechnol. 2018, 9, 1297–1307. [Google Scholar] [CrossRef]
- Zhang, Z.; Panov, V.P.; Nagaraj, M.; Mandle, R.J.; Goodby, J.W.; Luckhurst, G.R.; Jones, J.C.; Gleeson, H.F. Raman scattering studies of order parameters in liquid crystalline dimers exhibiting the nematic and twist-bend nematic phases. J. Mater. Chem. C 2015, 3, 10007–10016. [Google Scholar] [CrossRef]
- Merkel, K.; Loska, B.; Welch, C.; Mehl, G.H.; Kocot, A. Molecular biaxiality determines the helical structure–infrared measurements of the molecular order in the nematic twist-bend phase of difluoro terphenyl dimer. Phys. Chem. Chem. Phys. 2021, 23, 4151–4160. [Google Scholar] [CrossRef]
- Merkel, K.; Loska, B.; Welch, C.; Mehl, G.H.; Kocot, A. The role of intermolecular interactions in stabilizing the structure of the nematic twist-bend phase. RSC Adv. 2021, 11, 2917–2925. [Google Scholar] [CrossRef]
- Kocot, A.; Loska, B.; Arakawa, Y.; Merkel, K. Structure of the twist-bend nematic phase with respect to the orientational molecular order of the thioether-linked dimers. Phys. Rev. E 2022, 105, 044701-10. [Google Scholar] [CrossRef]
- Beguin, L.; Emsley, J.W.; Lelli, M.; Lesage, A.; Luckhurst, G.R.; Timimi, B.A.; Zimmermann, H. The chirality of a twist–bend nematic phase identified by NMR spectroscopy. J. Phys. Chem. B 2012, 116, 7940–7951. [Google Scholar] [CrossRef]
- Emsley, J.W.; Lesot, P.; Luckhurst, G.R.; Meddour, A.; Merlet, D. Chiral solutes can seed the formation of enantiomorphic domains in a twist-bend nematic liquid crystal. Phys. Rev. E 2013, 87, 040501. [Google Scholar] [CrossRef]
- Emsley, J.W.; Lelli, M.; Lesage, A.; Luckhurst, G.R. A comparison of the conformational distributions of the achiral symmetric liquid crystal dimer cb7cb in the achiral nematic and chiral twist-bend nematic phases. J. Phys. Chem. B 2013, 117, 6547–6557. [Google Scholar] [CrossRef]
- Jokisaari, J.P.; Luckhurst, G.R.; Timimi, B.A.; Zhu, J.F.; Zimmermann, H. Twist-bend nematic phase of the liquid crystal dimer cb7cb: Orientational order and conical angle determined by xe-129 and h-2 nmr spectroscopy. Liq. Cryst. 2015, 42, 708–721. [Google Scholar]
- Ivšić, T.; Vinković, M.; Baumeister, U.; Mikleušević, A.; Lesac, A. Towards understanding the NTB phase: A combined experimental, computational and spectroscopic study. RSC Adv. 2016, 6, 5000–5007. [Google Scholar] [CrossRef]
- Panov, V.; Balachandran, R.; Nagaraj, M.; Vij, J.K.; Tamba, M.G.; Kohlmeier, A.; Mehl, G.H. Microsecond linear optical response in the unusual nematic phase of achiral bimesogens. Appl. Phys. Lett. 2011, 99, 261903. [Google Scholar] [CrossRef]
- Meyer, C.; Luckhurst, G.R.; Dozov, I. Flexoelectrically driven electroclinic effect in the twist-bend nematic phase of achiral molecules with bent shapes. Phys. Rev. Lett. 2013, 111, 067801. [Google Scholar] [CrossRef]
- Sebastián, N.; Tamba, M.G.; Stannarius, R.; de la Fuente, M.R.; Salamonczyk, M.; Cukrov, G.; Gleeson, J.T.; Sprunt, S.; Jakli, A.; Welch, C.; et al. Mesophase structure and behaviour in bulk and restricted geometry of a dimeric compound exhibiting a nematic–nematic transition. Phys. Chem. Chem. Phys. 2016, 18, 19299. [Google Scholar] [CrossRef]
- Merkel, K.; Kocot, A.; Vij, J.K.; Shanker, G. Distortions in structures of the twist bend nematic phase of a bent-core liquid crystal by the electric field. Phys. Rev. E 2018, 98, 022704-8. [Google Scholar] [CrossRef]
- Merkel, K.; Kocot, A.; Welch, C.; Mehl, G.H. Soft modes of the dielectric response in the twist–bend nematic phase and identification of the transition to a nematic splay bend phase in the CBC7CB dimer. Phys. Chem. Chem. Phys. 2019, 21, 22839–22848. [Google Scholar] [CrossRef]
- Merkel, K.; Welch, C.; Ahmed, Z.; Piecek, W.; Mehl, G.H. Dielectric response of electric-field distortions of the twist-bend nematic phase for LC dimers. J. Chem. Phys. 2019, 151, 114908. [Google Scholar] [CrossRef]
- Meyer, C.; Blanc, C.; Luckhurst, G.R.; Davidson, P.; Dozov, I. Biaxiality-driven twist-bend to splay-bend nematic phase transition induced by an electric field. Sci. Adv. 2020, 6, eabb8212. [Google Scholar] [CrossRef]
- Challa, P.K.; Borshch, V.; Parri, O.; Imrie, C.T.; Sprunt, S.N.; Gleeson, J.T.; Lavrentovich, O.D.; Jakli, A. Twist-bend nematic liquid crystals in high magnetic fields. Phys. Rev. E 2014, 89, 060501. [Google Scholar] [CrossRef]
- Mandle, R.J.; Goodby, J.W. Progression from nano to macro science in soft matter systems: Dimers to trimers and oligomers in twist-bend liquid crystals. RSC Adv. 2016, 6, 34885–34893. [Google Scholar] [CrossRef]
- Tuchband, M.R.; Shuai, M.; Graber, K.A.; Chen, D.; Zhu, C.; Radzihovsky, L.; Klittnick, A.; Foley, L.M.; Scarbrough, A.; Porada, J.H.; et al. Double-Helical Tiled Chain Structure of the Twist-Bend Liquid Crystal phase in CB7CB. arXiv 2017, arXiv:1703.10787v1. [Google Scholar]
- Salamończyk, M.; Vaupotič, N.; Pociecha, D.; Walker, R.; Storey, J.M.D.; Imrie, C.T.; Wang, C.; Zhu, C.; Gorecka, E. Multi-level chirality in liquid crystals formed by achiral molecules. Nat. Commun. 2019, 10, 1922. [Google Scholar] [CrossRef]
- Leadbetter, A.J.; Frost, J.C.; Gaughan, J.P.; Gray, G.W.; Mosley, A. The structure of smectic a phases of compounds with cyano end groups. J. Phys. France 1979, 40, 375–380. [Google Scholar] [CrossRef]
- Goodby, J.W.; Davis, E.J.; Mandle, R.J.; Cowling, S.J. Nano-Segregation and Directed Self-Assembly in the Formation of Functional Liquid Crystals. Isr. J. Chem. 2012, 52, 863–880. [Google Scholar] [CrossRef]
- Dwivedi, K.K.; Dwivedi, M.K.; Tiwa, S.N. Study of Intermolecular Interactions in Liquid Crystals: Para-butyl-p’-cyano-biphenyl. J. Cryst. Process Tech. 2014, 4, 31–38. [Google Scholar] [CrossRef]
- Sims, M.T.; Abbott, L.C.; Goodby, J.W.; Moore, J.N. Shape segregation in molecular organization: A combined X-ray scattering and molecular dynamics study of smectic liquid crystals. Soft Matter 2019, 15, 7722–7732. [Google Scholar] [CrossRef]
- Mandle, R.J.; Goodby, J.W. Does Topology Dictate the Incidence of the Twist-Bend Phase? Insights Gained from Novel Unsymmetrical Bimesogens. Chem. Eur. J. 2016, 22, 18456–18464. [Google Scholar] [CrossRef]
- Mandle, R.J. The dependency of twist-bend nematic liquid crystals on molecular structure: A progression from dimers to trimers, oligomers and polymers. Soft Matter 2016, 12, 7883–7901. [Google Scholar] [CrossRef]
- Paterson, D.A.; Gao, M.; Kim, Y.-K.; Jamali, A.; Finley, K.L.; Robles-Hernández, B.; Diez-Berart, S.; Salud, J.; de la Fuente, M.R.; Timimi, B.A.; et al. Understanding the twist-bend nematic phase: The characterization of 1-(4-cyanobiphenyl-40-yloxy)- 6-(4-cyanobiphenyl-40-yl)hexane (CB6OCB) and comparison with CB7CB. Soft Matter 2016, 12, 6827–6840. [Google Scholar] [CrossRef]
- Greco, C.; Marini, A.; Frezza, E.; Ferrarini, A. From the Molecular Structure to Spectroscopic and Material Properties: Computational Investigation of a Bent-Core Nematic Liquid Crystal. ChemPhysChem 2014, 15, 1336–1344. [Google Scholar] [CrossRef]
- Mandle, R.J.; Archbold, C.T.; Sarju, J.P.; Andrews, J.L.; Goodby, J.W. The Dependency of Nematic and Twist-bend Mesophase Formation on Bend Angle. Sci. Rep. 2016, 6, 36682. [Google Scholar] [CrossRef]
- Tomczyk, W.; Longa, L. Role of molecular bend angle and biaxiality in the stabilization of the twist-bend nematic phase. Soft Matter 2020, 16, 4350–4357. [Google Scholar] [CrossRef]
- Lesac, A.; Baumeister, U.; Dokli, I.; Hameršak, Z.; Ivšić, T.; Kontrec, D.; Viskić, M.; Knežević, A.; Mandle, R.J. Geometric aspects influencing N-N TB transition-implication of intramolecular torsion. Liq. Cryst. 2018, 45, 1101–1110. [Google Scholar] [CrossRef]
- Abberley, J.P.; Storey, J.M.D.; Imrie, C.T. Structure-property relationships in azobenzene-based twist-bend nematogens. Liq. Cryst. 2019, 46, 2102–2114. [Google Scholar] [CrossRef]
- Sebastián, N.; Robles-Hernández, B.; Diez-Berart, S.; Salud, J.; Luckhurst, G.R.; Dunmur, D.A.; López, D.O.; de la Fuente, M.R. Distinctive dielectric properties of nematic liquid crystal dimers. Liq. Cryst. 2017, 44, 177–190. [Google Scholar] [CrossRef]
- Archbold, C.T.; Mandle, R.J.; Andrews, J.L.; Cowling, S.J.; Goodby, J.W. Conformational landscapes of bimesogenic compounds and their implications for the formation of modulated nematic phases. Liq. Cryst. 2017, 44, 2079–2088. [Google Scholar] [CrossRef]
- Pocock, E.E.; Mandle, R.J.; Goodby, J.W. Molecular shape as a means to control the incidence of the nanostructured twist bend phase. Soft Matter 2018, 14, 2508–2514. [Google Scholar] [CrossRef]
- Pocock, E.E.; Mandle, R.J.; Goodby, J.W. Experimental and Computational Study of a Liquid Crystalline Dimesogen Exhibiting Nematic, Twist-Bend Nematic, Intercalated Smectic, and Soft Crystalline Mesophases. Molecules 2021, 26, 532–544. [Google Scholar] [CrossRef]
- Ferrarini, A. The twist-bend nematic phase: Molecular insights from a generalized Maier–Saupe theory. Liq. Cryst. 2017, 44, 45–57. [Google Scholar] [CrossRef]
- Mandle, R.J.; Goodby, J.W. A twist-bend nematic to an intercalated, anticlinic, biaxial phase transition in liquid crystal bimesogens. Soft Matter 2016, 12, 1436–1443. [Google Scholar] [CrossRef]
- Goodby, J.W. Free volume, molecular grains, self-organization, and anisotropic entropy: Machining materials. Liq. Cryst. 2017, 44, 1755–1763. [Google Scholar]
- Jansze, S.M.; Martínez-Felipe, A.; Storey, J.M.D.; Marcelis, A.T.M.; Imrie, C.T. A Twist-Bend Nematic Phase Driven by Hydrogen Bonding. Angew. Chem. Int. Ed. 2015, 54, 643–646. [Google Scholar] [CrossRef]
- Martinez-Felipe, A.; Cook, G.; Abberley, J.P.; Walker, R.; Storey, J.M.D.; Imrie, C.T. An FT-IR spectroscopic study of the role of hydrogen bonding in the formation of liquid crystallinity for mixtures containing bipyridines and 4-pentoxybenzoic acid. RSC Adv. 2016, 6, 108164–108179. [Google Scholar] [CrossRef]
- Walker, R.; Pociecha, D.; Abberley, J.P.; Martinez-Felipe, A.; Paterson, D.A.; Forsyth, E.; Lawrence, G.B.; Henderson, P.A.; Storey, J.M.D.; Gorecka, E.; et al. Spontaneous chirality through mixing achiral components: A twist-bend nematic phase driven by hydrogen-bonding between unlike components. Chem. Commun. 2018, 54, 3383–3386. [Google Scholar] [CrossRef]
- Knežević, A.; Sapunar, M.; Buljan, A.; Dokli, I.; Hameršak, Z.; Kontrec, D.; Lesac, A. Fine-tuning the effect of p–p interactions on the stability of the NTB phase. Soft Matter 2018, 14, 8466–8474. [Google Scholar] [CrossRef]
- Alaasar, M.; Tschierske, C. Nematic phases driven by hydrogen-bonding in liquid crystalline nonsymmetric dimers. Liq. Cryst. 2019, 49, 124–130. [Google Scholar] [CrossRef]
- Cragg, P.J. Supramolecular Chemistry: From Biological Inspiration to Biomedical Applications, 1st ed.; Springer: Dordrecht, The Netherlands, 2010. [Google Scholar]
- Asensio, J.L.; Arda, A.; Canada, F.J.; Jimenez-Barbero, J. Carbohydrate-aromatic interactions. Acc. Chem. Res. 2013, 46, 946–954. [Google Scholar] [CrossRef]
- Mingos, D.M.P. Structure and Bonding. In Liquid Crystalline Functional Assemblies and Their Supramolecular Structures; Kato, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 128. [Google Scholar] [CrossRef]
- Kato, T.; Gupta, M.; Yamaguchi, D.; Kian, P.G.; Nakayama, M. Supramolecular Association and Nanostructure Formation of Liquid Crystals and Polymers for New Functional Materials. Bull. Chem. Soc. Jpn. 2021, 94, 357–376. [Google Scholar] [CrossRef]
- Walker, R.; Pociecha, D.; Salamończyk, M.; Storey, J.M.D.; Górecka, E.; Imrie, C.T. Supramolecular liquid crystals exhibiting a chiral twist-bend nematic phase. Mater. Adv. 2020, 1, 1622–1630. [Google Scholar] [CrossRef]
- Vanakaras, A.G.; Photinos, D.J. A molecular theory of nematic–nematic phase transitions in mesogenic dimers. Soft Matter 2016, 12, 2208–2220. [Google Scholar] [CrossRef]
- Lavrentovich, O.D. Ferroelectric nematic liquid crystal, a century in waiting. Proc. Natl. Acad. Sci. USA 2020, 117, 14629–14631. [Google Scholar] [CrossRef]
- Chen, X.; Korblova, E.; Dong, D.; Wei, X.; Shao, R.; Radzihovsky, L.; Glaser, M.A.; Maclennan, J.E.; Bedrov, D.; Walba, D.M.; et al. First-principles experimental demonstration of ferroelectricity in a thermotropic nematic liquid crystal: Polar domains and striking electro-optics. Proc. Natl. Acad. Sci. USA 2020, 117, 14021–14031. [Google Scholar] [CrossRef]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations; McGraw-Hill: New York, NY, USA, 1955. [Google Scholar]
- Kocot, A.; Loska, B.; Arakawa, Y.; Mehl, G.H.; Merkel, K. Study of the experimental and simulated vibrational spectra together with conformational analysis for thioether cyanobiphenyl-based liquid crystal dimers. Int. J. Mol. Sci. 2022, 23, 8005–8016. [Google Scholar] [CrossRef]
- Barnes, P.J.; Douglass, A.G.; Heeks, S.K.; Luckhurst, G.R. An enhanced odd-even effect of liquid crystal dimers Orientational order in the α,ω-bis(4′-cyanobiphenyl-4-yl)alkanes. Liq. Cryst. 1993, 13, 603–613. [Google Scholar] [CrossRef]
- Carvalho, J.; Cruz, C.; Figueirinhas, J.L.; Tamba, M.G.; Kohlmeier, A.; Mehl, G.H. Proton and deuterium NMR study of the CBC9CB dimer system. J. Phys. Chem. B 2019, 123, 1442–1451. [Google Scholar] [CrossRef]
- Aluculesei, A.; Cachitas, H.; Carvalho, J.; Chavez, F.V.; Figueirinhas, J.L.; Sebastião, P.J.; Cruz, C.; Tamba, M.G.; Kohlemeir, A.; Mehl, G.H. 1H NMR study of molecular order and dynamics in the liquid crystal CB-C9-CB. Phys. Chem. Chem. Phys. 2019, 21, 4523–4537. [Google Scholar] [CrossRef]
- Krzyżewska, K.; Jaroch, T.; Maranda-Niedbała, A.; Pociecha, D.; Górecka, E.; Ahmed, Z.; Welch, C.; Mehl, G.H.; Proń, A.; Nowakowski, R. Supramolecular organization of liquid-crystal dimers-bis-cyanobiphenyl alkanes on HOPG by scanning tunneling microscopy. Nanoscale 2018, 10, 16201–16210. [Google Scholar] [CrossRef]
- Arakawa, Y.; Komatsu, K.; Tsuji, H. Twist-bend nematic liquid crystals based on thioether linkage. New J. Chem. 2019, 43, 6786–6793. [Google Scholar] [CrossRef]
- Arakawa, Y.; Ishida, Y.; Tsuji, H. Ether- and Thioether-Linked Naphthalene-Based Liquid-Crystal Dimers: Influence of Chalcogen Linkage and Mesogenic-Arm Symmetry on the Incidence and Stability of the Twist–Bend Nematic Phase. Chem. Eur. J. 2020, 26, 3767–3775. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Hertwig, R.H.; Koch, W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vibration Frequency (cm−1) | A0/AN-TB | * | A0/AN-TB | A0/AN-TB | * | A0/AN-TB |
|---|---|---|---|---|---|---|
| CBSC7SCB | CBSC7SCB | CBC5OCB | CBC7OCB | CBC7OCB | CBC9CB | |
| 520 | 1.25 | 1.60 | 1.42 | 1.48 | 1.54 | -- |
| 810 | 1.22 | 1.64 | 1.32 | 1.33 | 1.41 | 1.05 |
| 1100 | 0.94 | 0.65 | 0.94 | 0.84 | 0.84 | -- |
| 1250 | -- | -- | 0.89 | 0.71 | 0.80 | -- |
| 1485 | 0.89 | 0.73 | 0.90 | 0.66 | 0.70 | 0.94 |
| 1600 | 0.86 | 0.63 | 0.78 | 0.60 | 0.61 | 0.93 |
| 2220 | 0.80 | 0.93 | 0.85 | 0.68 | 0.95 | 0.93 |
| CBACnBCB | θt (°) | p (nm) | l (nm) * | d = lcos θt | p/d |
|---|---|---|---|---|---|
| CBC9CB | 25.6 | 8.15 [10] | 2.88 | 2.6 | 3.1 |
| CBSC7SCB | 33 | 9.1 [11,12] | 2.90 | 2.43 | 3.7 |
| CBSC7OCB | 15.6 | 11.5 [12] | 2.90 | 2.79 | 4.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merkel, K.; Loska, B.; Arakawa, Y.; Mehl, G.H.; Karcz, J.; Kocot, A. How Do Intermolecular Interactions Evolve at the Nematic to Twist–Bent Phase Transition? Int. J. Mol. Sci. 2022, 23, 11018. https://doi.org/10.3390/ijms231911018
Merkel K, Loska B, Arakawa Y, Mehl GH, Karcz J, Kocot A. How Do Intermolecular Interactions Evolve at the Nematic to Twist–Bent Phase Transition? International Journal of Molecular Sciences. 2022; 23(19):11018. https://doi.org/10.3390/ijms231911018
Chicago/Turabian StyleMerkel, Katarzyna, Barbara Loska, Yuki Arakawa, Georg H. Mehl, Jakub Karcz, and Antoni Kocot. 2022. "How Do Intermolecular Interactions Evolve at the Nematic to Twist–Bent Phase Transition?" International Journal of Molecular Sciences 23, no. 19: 11018. https://doi.org/10.3390/ijms231911018
APA StyleMerkel, K., Loska, B., Arakawa, Y., Mehl, G. H., Karcz, J., & Kocot, A. (2022). How Do Intermolecular Interactions Evolve at the Nematic to Twist–Bent Phase Transition? International Journal of Molecular Sciences, 23(19), 11018. https://doi.org/10.3390/ijms231911018