
New Organic–Inorganic Salt Based on Fluconazole Drug: TD-DFT Benchmark and Computational Insights into Halogen Substitution


Abstract
1. Introduction
2. Results and Discussion
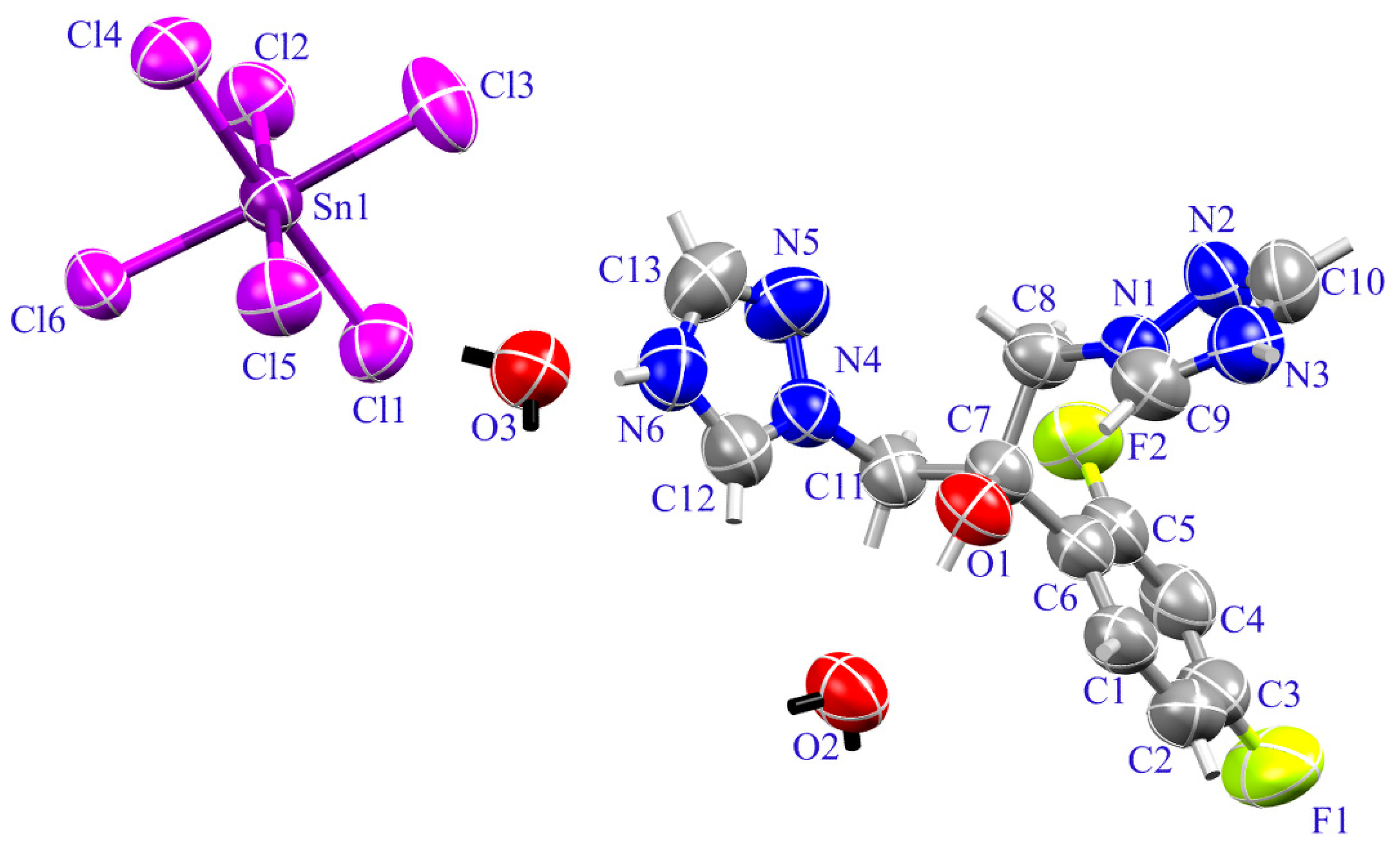
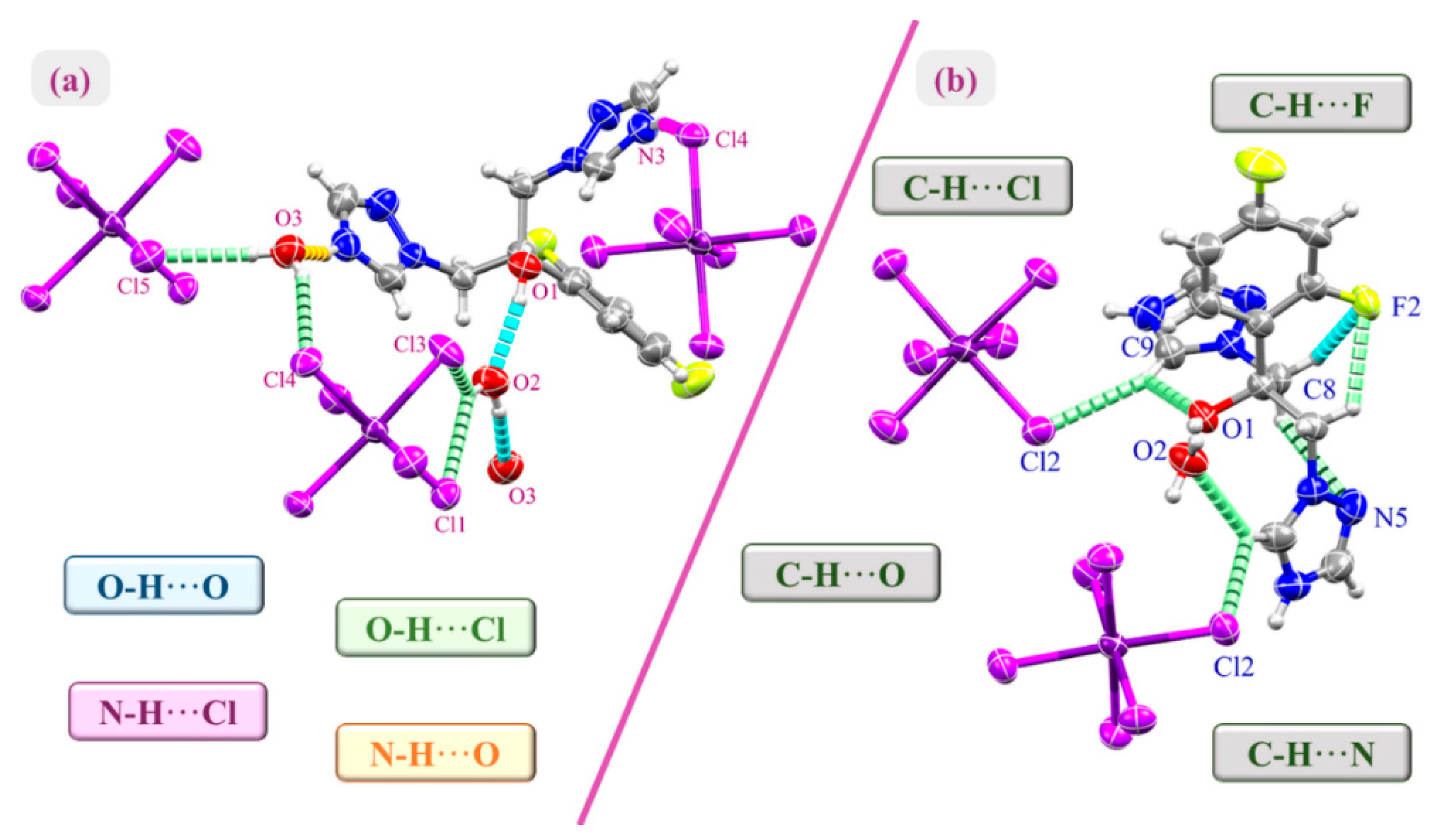
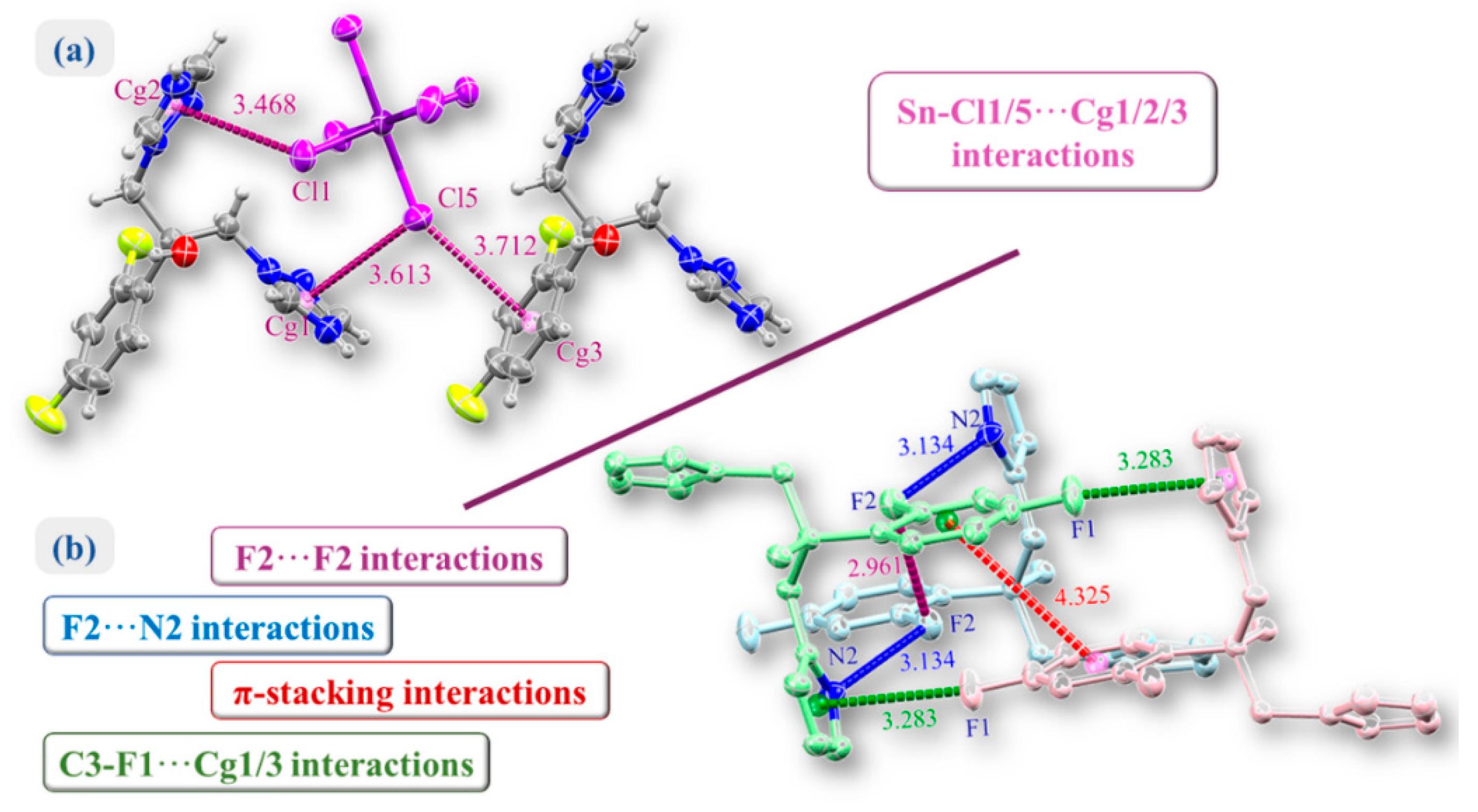
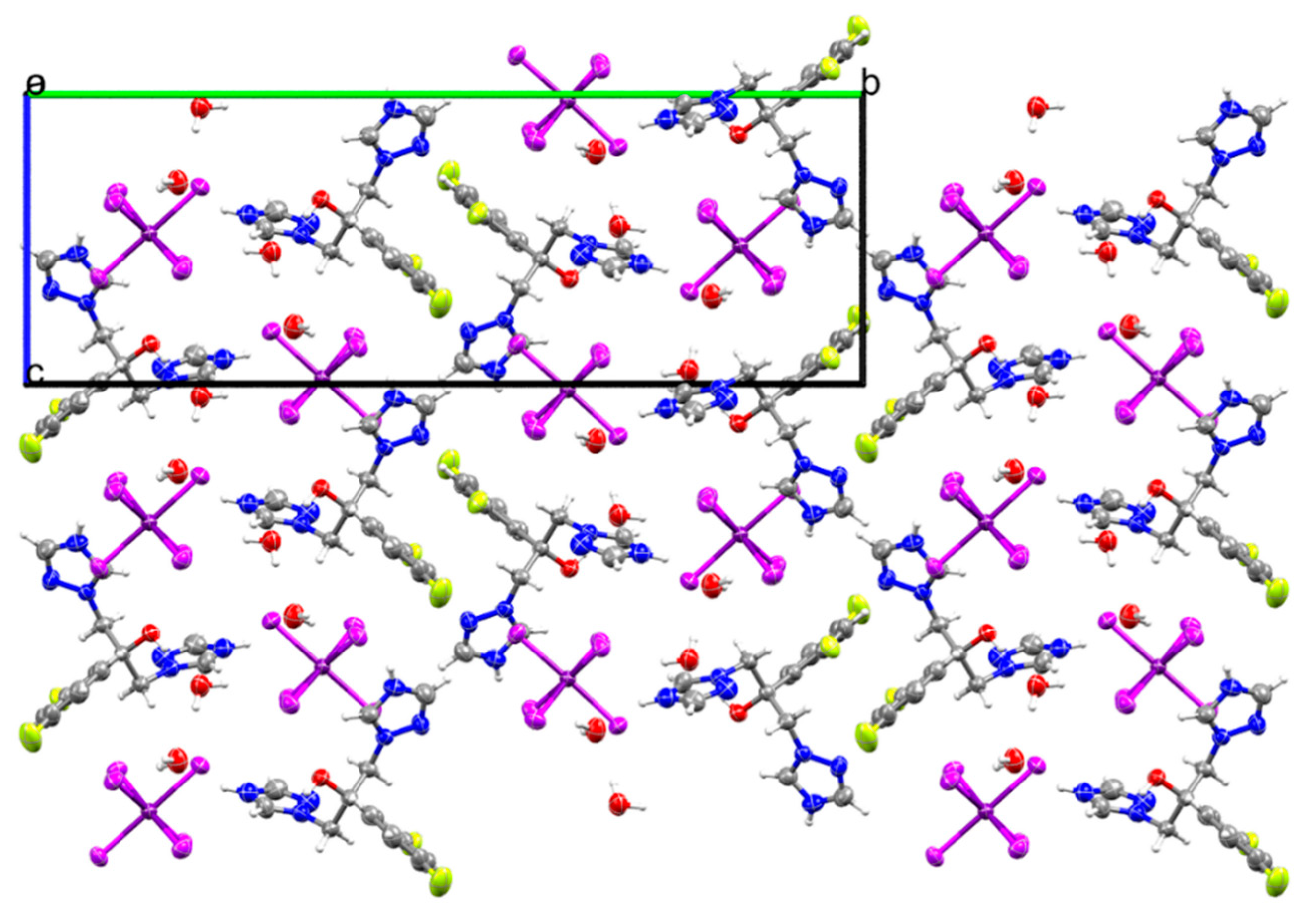
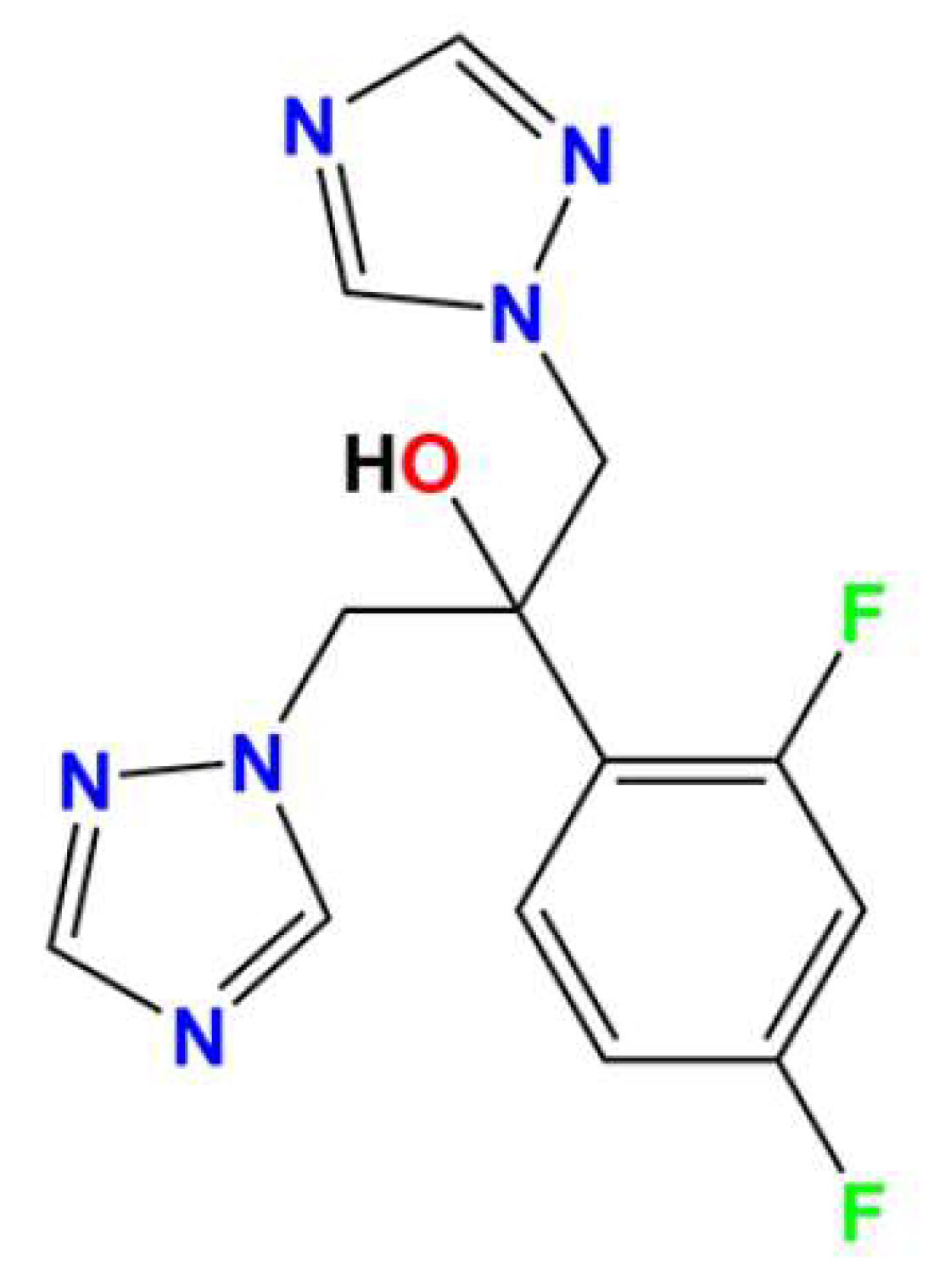
2.1. Crystal Structure of (H2Fluconazole).SnCl6.2H2O
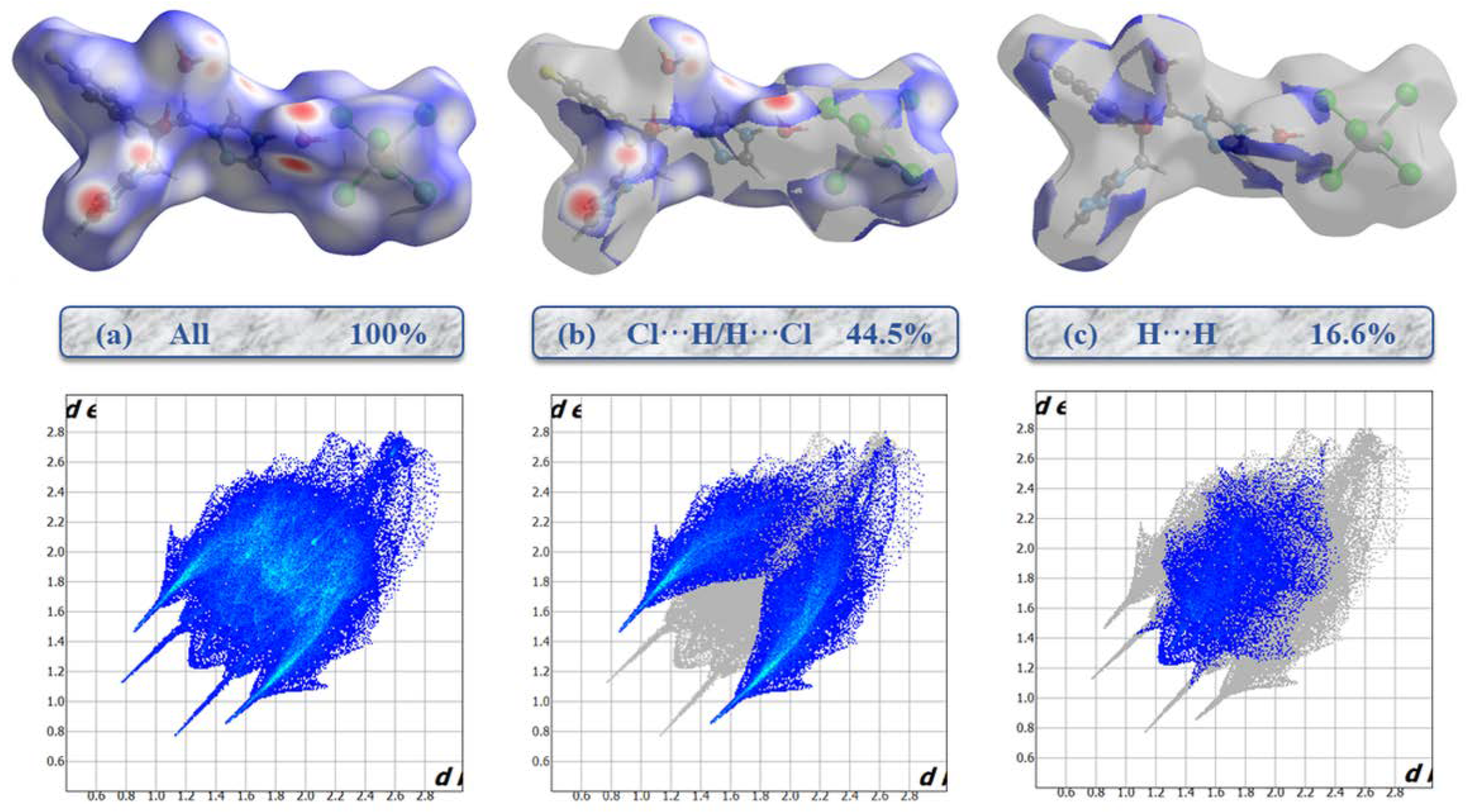
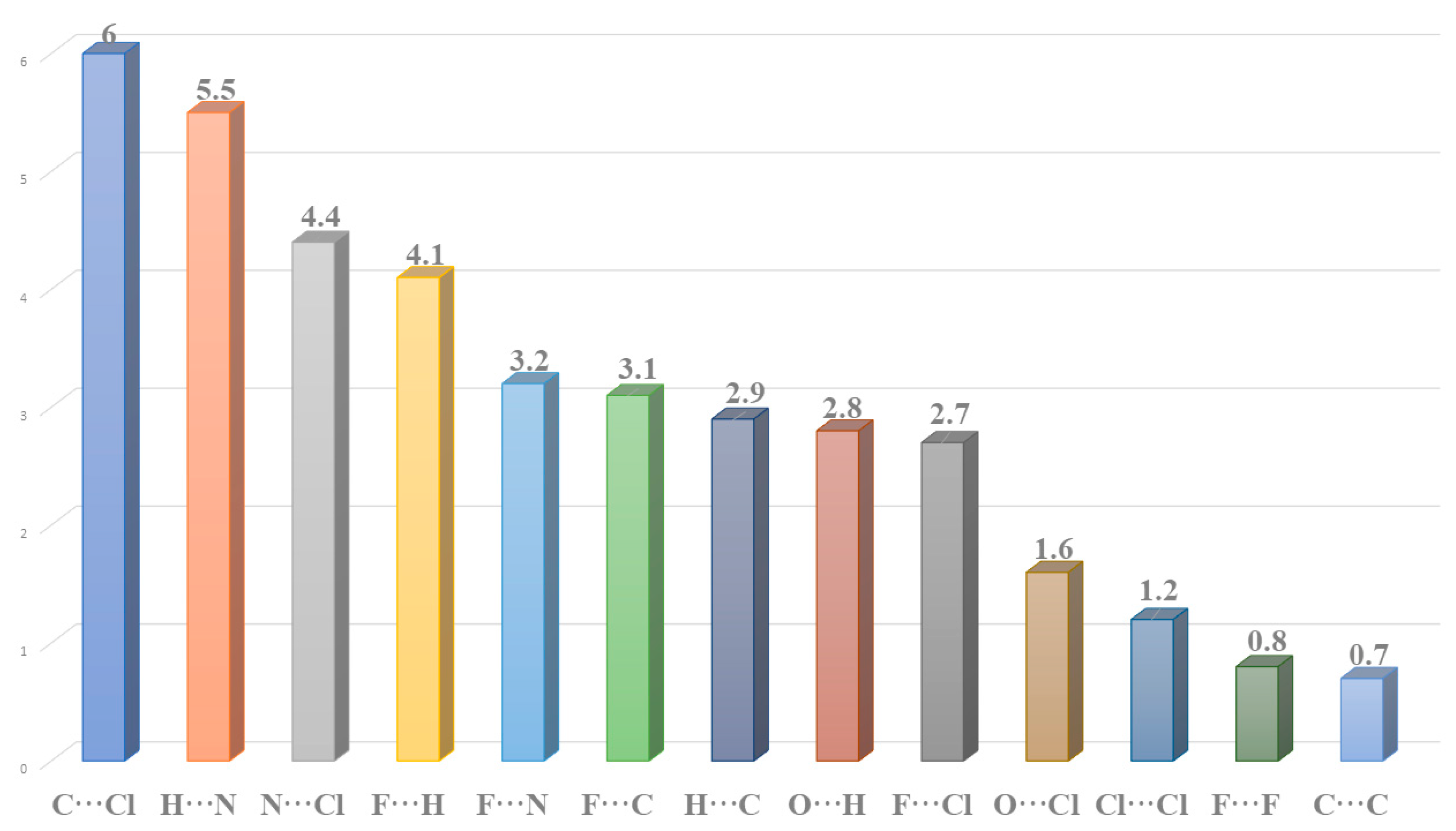
2.2. Hirshfeld Surface Analysis and Contact Enrichment Ratio
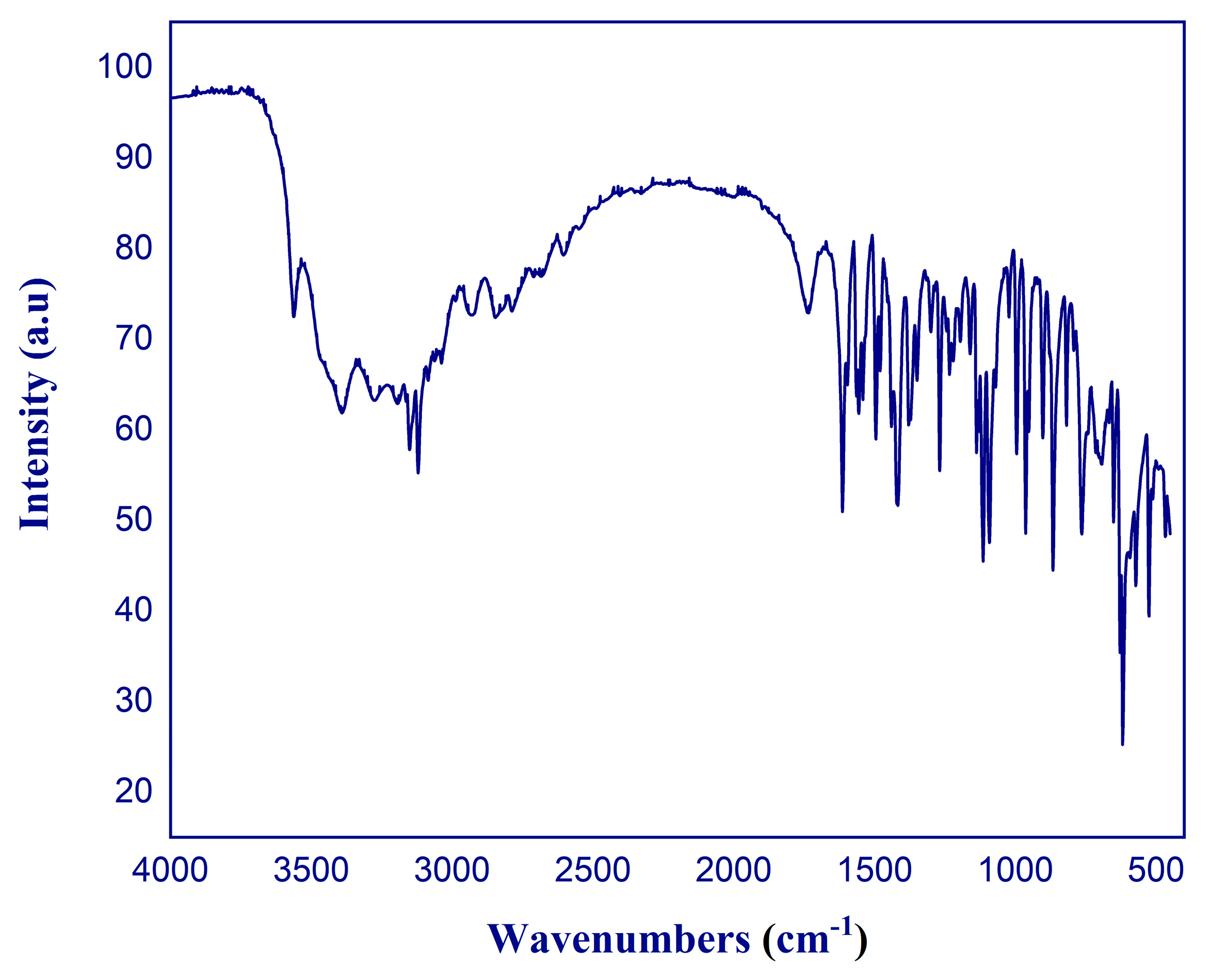
2.3. FT-IR and Raman Spectral Analysis
Internal Modes of H2Fluconazole Cation
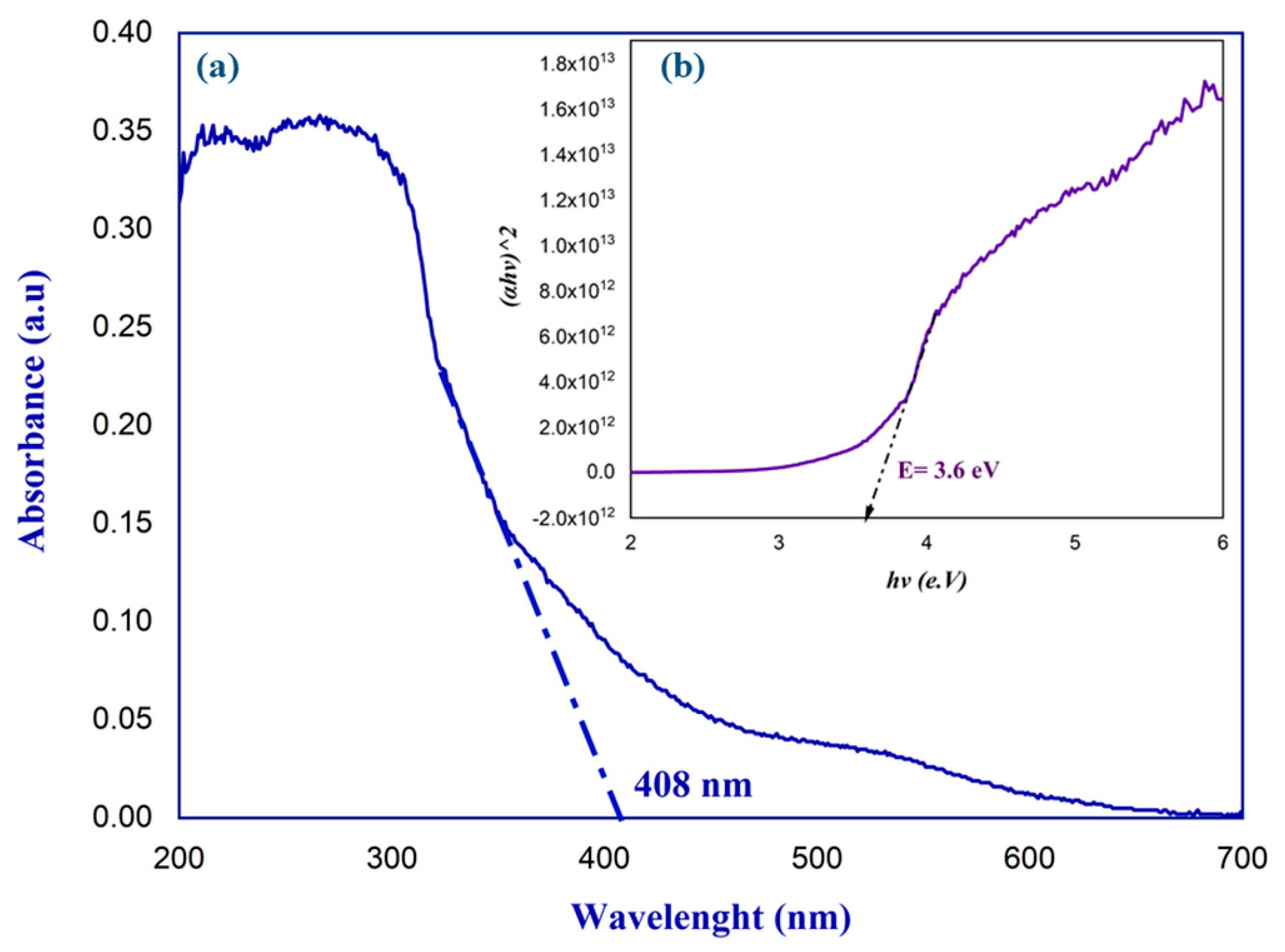
2.4. UV–Visible Spectroscopic Study
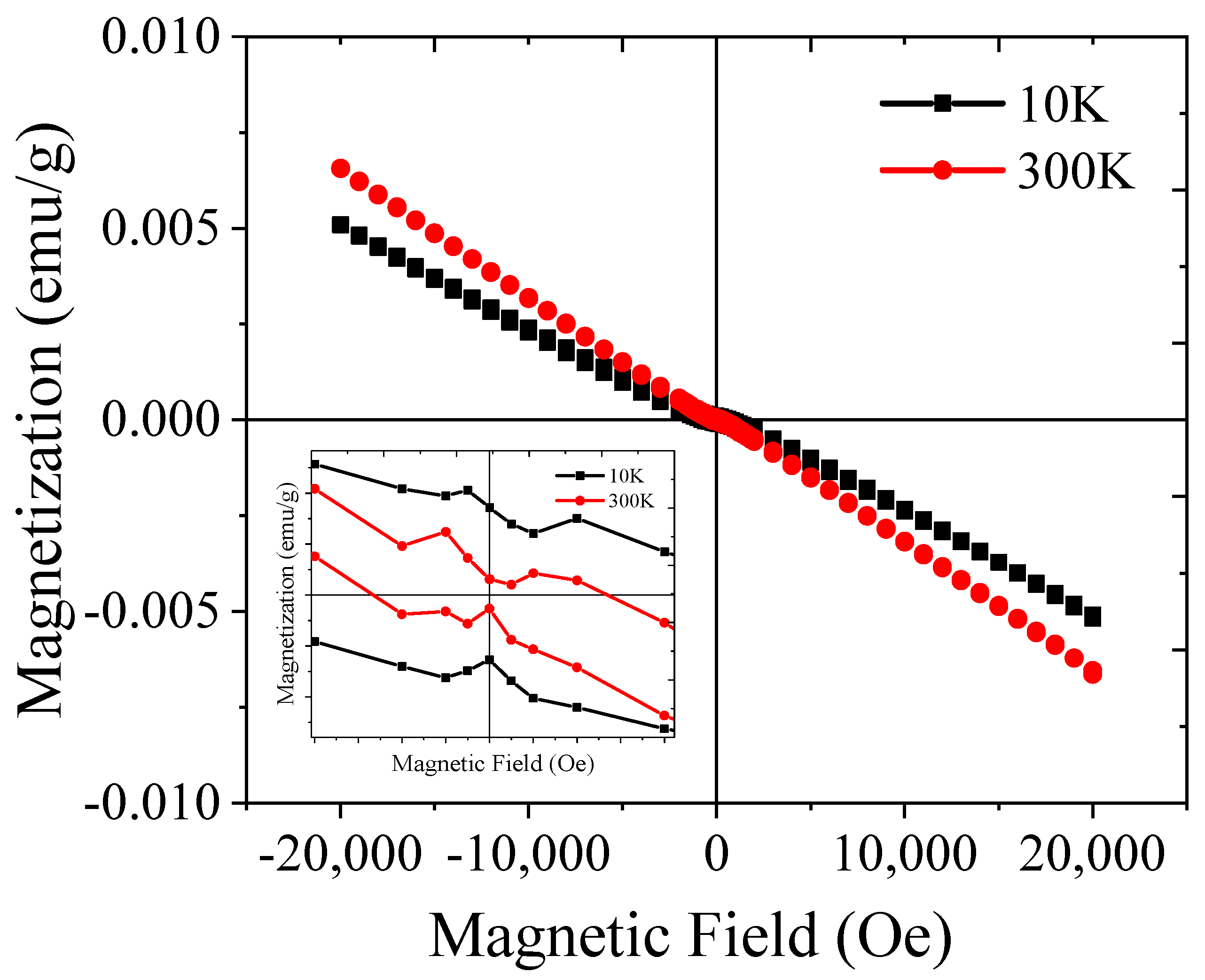
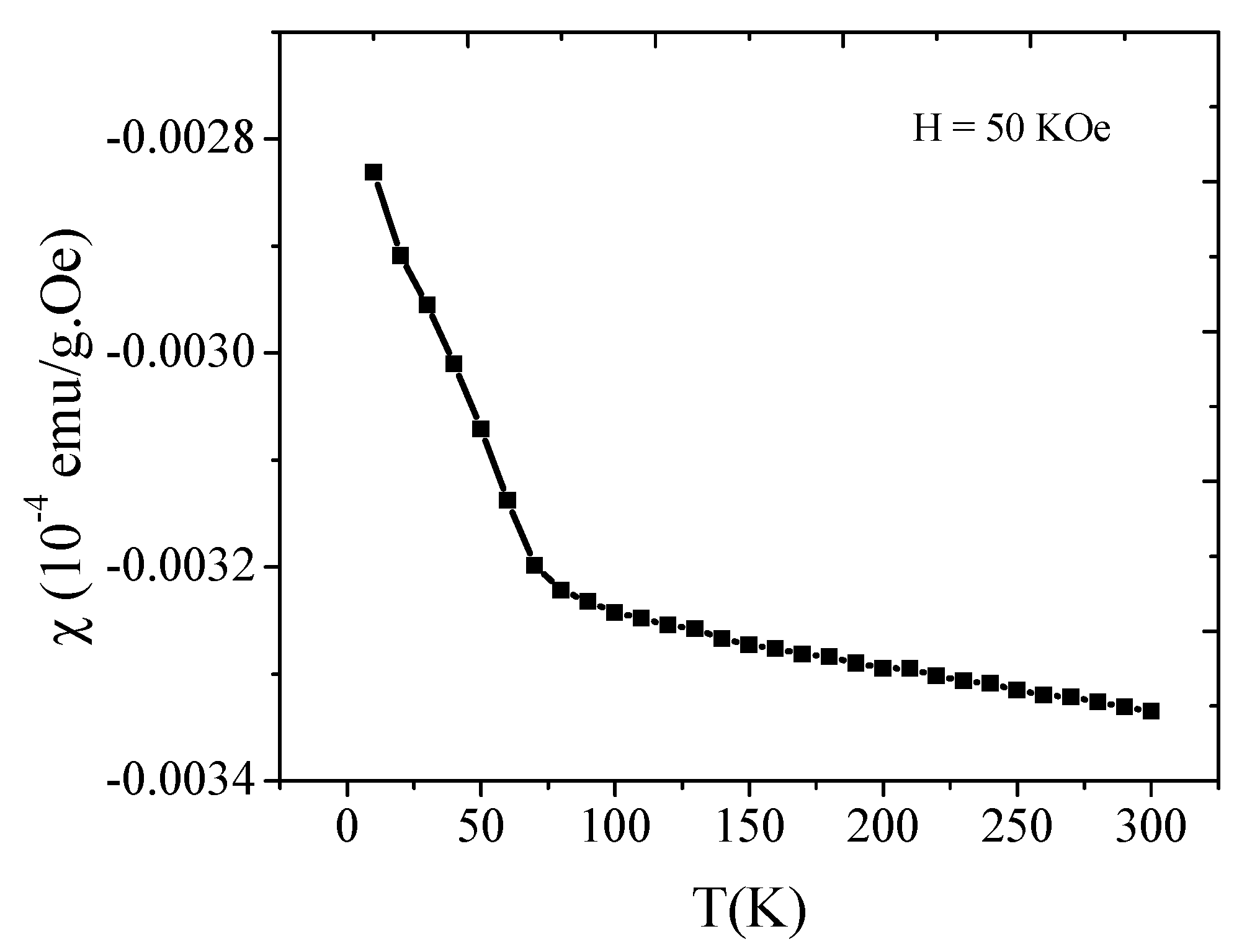
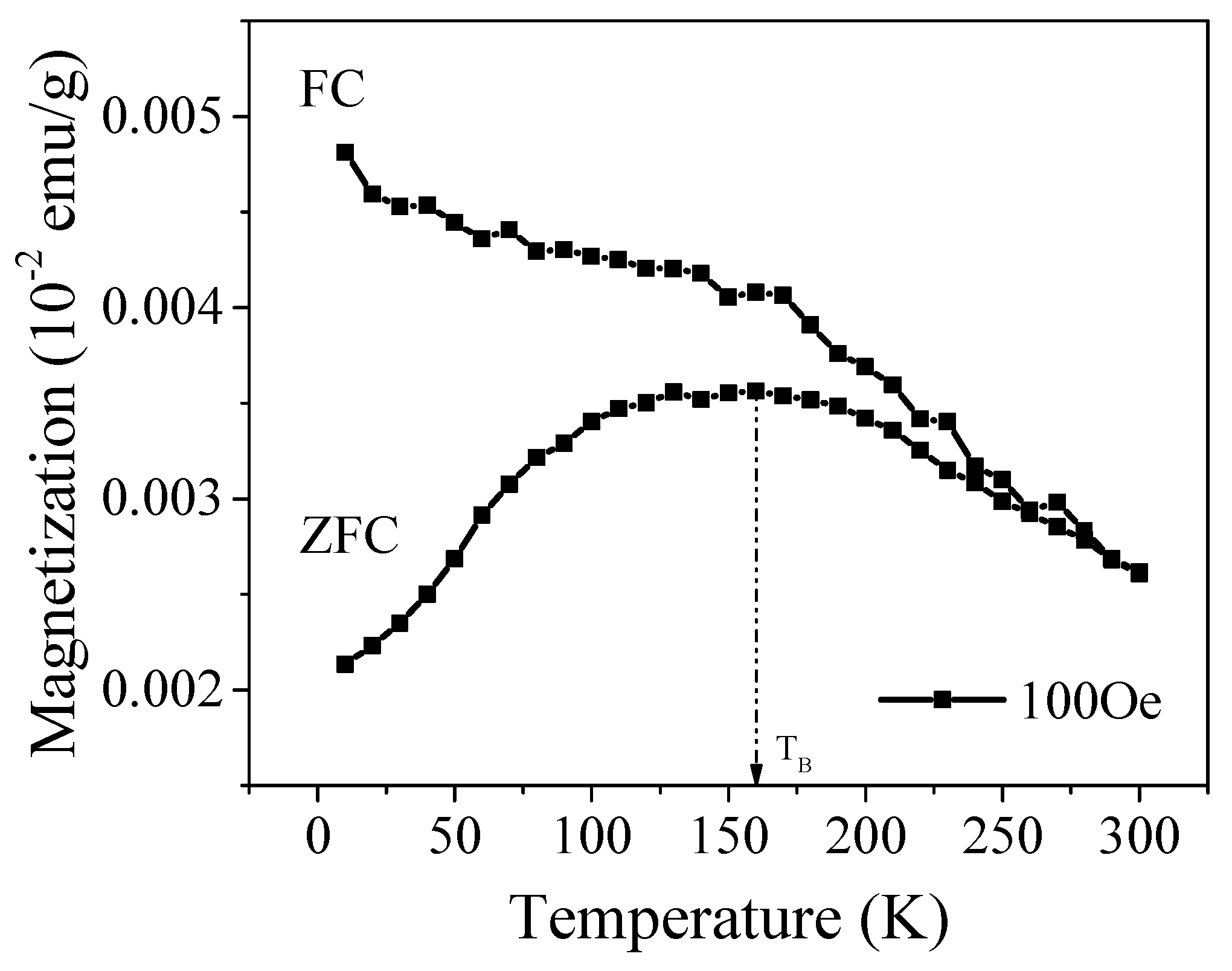
2.5. Magnetic Study
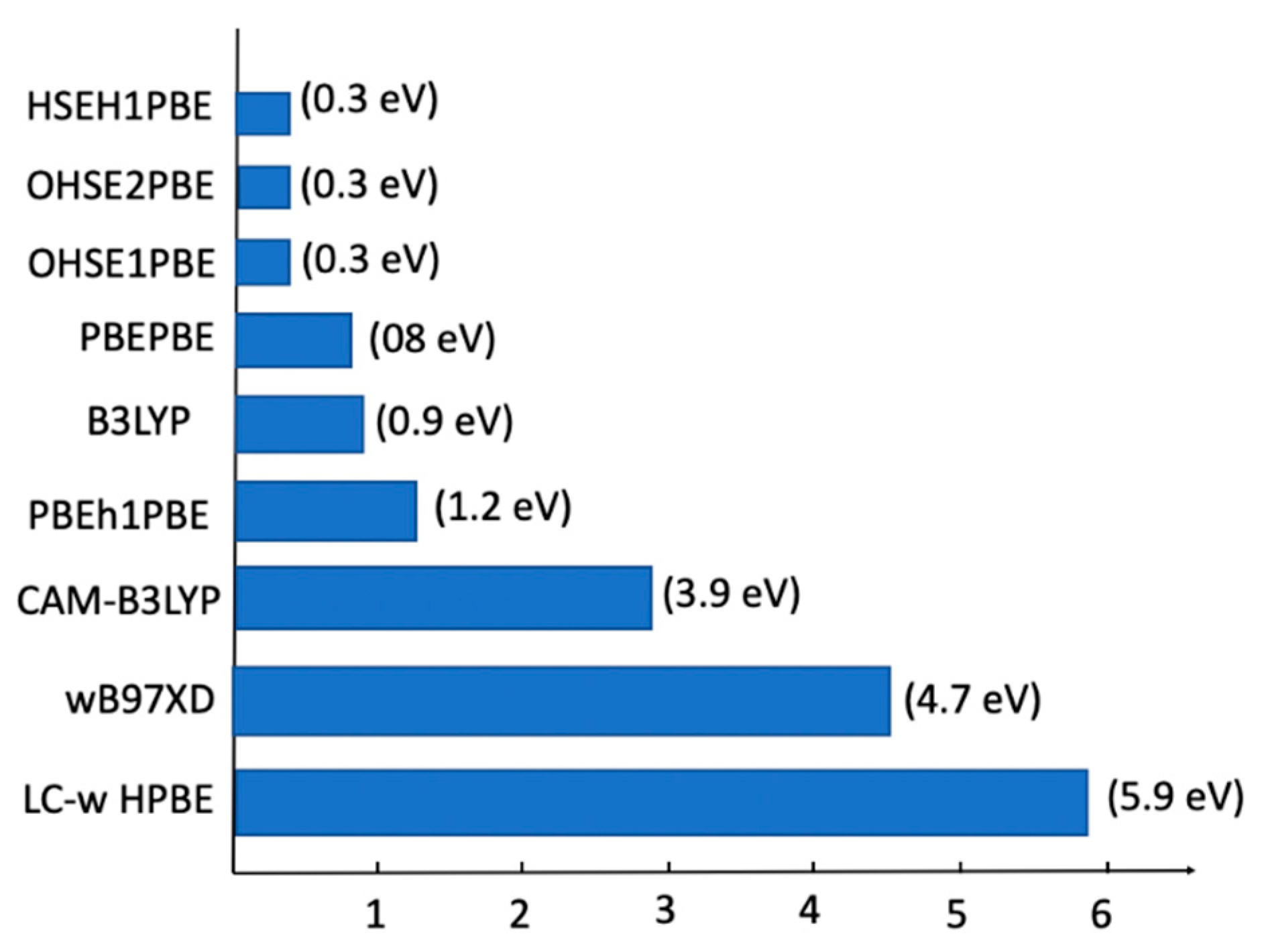
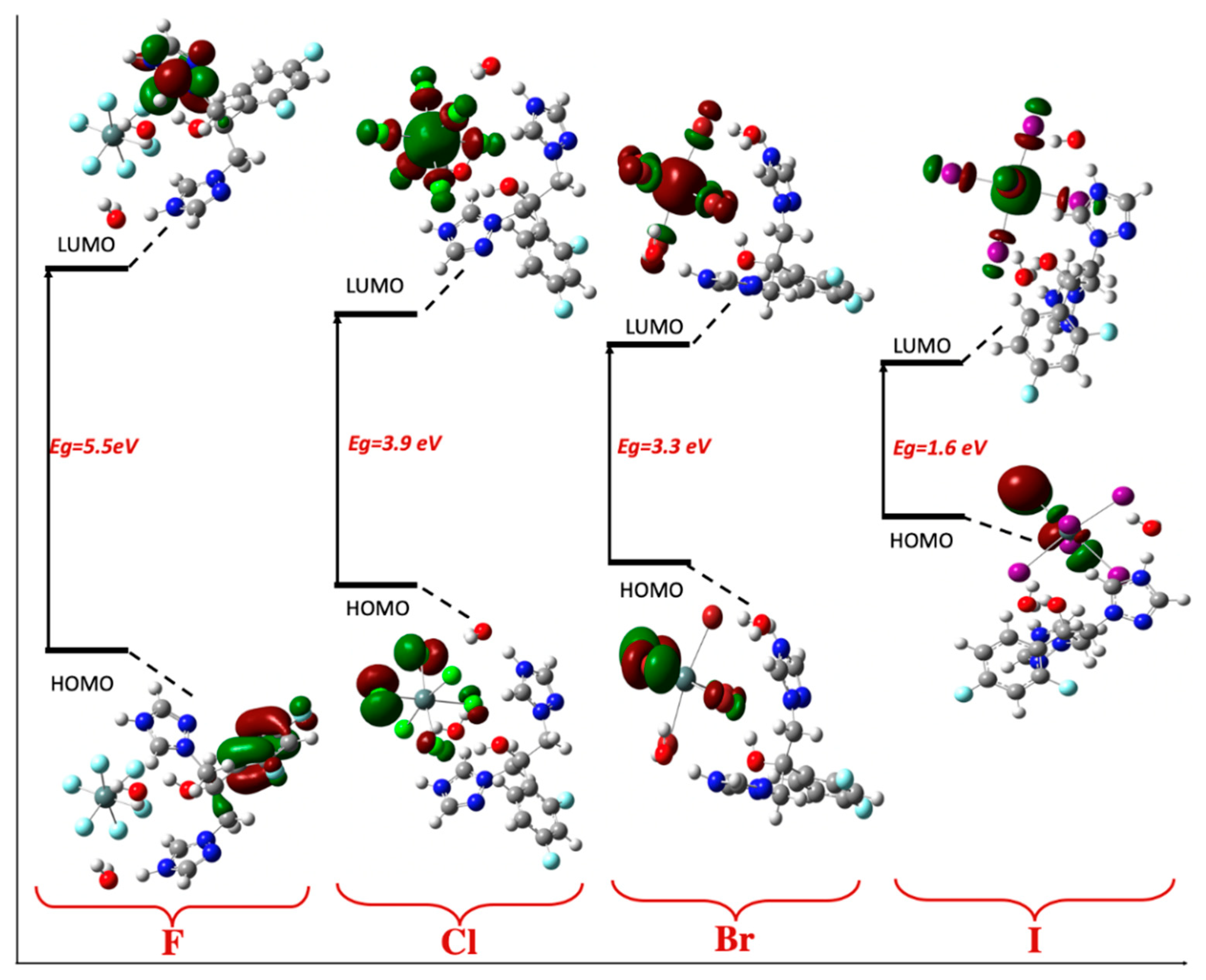
2.6. TD-DFT Study
3. Materials and Methods
3.1. Salt Preparation
3.2. X-ray Structure Determination
3.3. Physicochemical Characterization
3.4. Hirshfeld Surface Analysis (HS)
3.5. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allendorf, M.D.; Bauer, C.A.; Bhakta, R.; Houk, R. Luminescent metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1330–1352. [Google Scholar] [CrossRef]
- Saparov, B.; Mitzi, D.B. Organic–inorganic perovskites: Structural versatility for functional materials design. Chem. Rev. 2016, 116, 4558–4596. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-S.; Guo, G.-C. Inorganic-organic hybrid white light phosphors. Chem. Commun. 2016, 52, 13194–13204. [Google Scholar] [CrossRef] [PubMed]
- Shan, Q.; Song, J.; Zou, Y.; Li, J.; Xu, L.; Xue, J.; Dong, Y.; Han, B.; Chen, J.; Zeng, H. High performance metal halide perovskite light-emitting diode: From material design to device optimization. Small 2017, 13, 1701770. [Google Scholar] [CrossRef] [PubMed]
- Ferjani, H. Structural, Hirshfeld surface analysis, morphological approach, and spectroscopic study of new hybrid iodobismuthate containing tetranuclear 0D cluster Bi4I16·4(C6H9N2) 2(H2O). Crystals 2020, 10, 397. [Google Scholar] [CrossRef]
- Ferjani, H.; Boughzala, H. New hybrid material: (C3H6N3)4Bi2Cl10. Synthesis, structural study and spectroscopic behavior. Russ. J. Inorg. Chem. 2018, 63, 349–356. [Google Scholar] [CrossRef]
- Ferjani, H.; Boughzala, H.; Driss, A. Poly[bis(1-carbamoylguanidinium) [tri-μ-chlorido-dichloridobismuthate (III)]]. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, m615. [Google Scholar] [CrossRef]
- Ferjani, H.; Boughzala, H.; Driss, A. Synthesis, Crystal Structure, and Characterization of a New Organic-Inorganic Hybrid Material. J. Crystallogr. 2013, 2013, 658939. [Google Scholar] [CrossRef]
- Fu, P.; Huang, M.; Shang, Y.; Yu, N.; Zhou, H.-L.; Zhang, Y.-B.; Chen, S.; Gong, J.; Ning, Z. Organic-Inorganic layered and hollow tin bromide perovskite with tunable broadband emission. ACS Appl. Mater. Interfaces 2018, 10, 34363–34369. [Google Scholar] [CrossRef]
- Lassoued, M.S.; Osman, H.H.; Abdelbaky, M.S.M.; Lassoued, A.; Ammar, S.; Ben Salah, A.; Gadri, A.; García-Granda, S. Synthesis, crystal structure, DFT (B3LYP/LanL2DZ) and photoluminescence study of new stanate (IV) based inorgan-ic-organic hybrid. J. Phys. Chem. Solids 2018, 121, 177–185. [Google Scholar] [CrossRef]
- Spanopoulos, I.; Hadar, I.; Ke, W.; Guo, P.; Sidhik, S.; Kepenekian, M.; Even, J.; Mohite, A.D.; Schaller, R.D.; Kanatzidis, M.G. Water-Stable 1D Hybrid Tin(II) Iodide Emits Broad Light with 36% Photoluminescence Quantum Efficiency. J. Am. Chem. Soc. 2020, 142, 9028–9038. [Google Scholar] [CrossRef] [PubMed]
- Mathlouthi, M.; Valkonen, A.; Rzaigui, M.; Smirani, W. Structural characterization, spectroscopic, thermal, AC conductivity and dielectric properties and antimicrobial studies of (C8H12N)2[SnCl6]. Phase Transit. 2017, 90, 399–414. [Google Scholar] [CrossRef]
- Matela, G.; Aman, R.; Sharma, C.; Chaudhary, S. Reactions of tin- and triorganotin(IV) isopropoxides with thymol derivative: Synthesis, characterization and in vitro antimicrobial screening. J. Serb. Chem. Soc. 2013, 78, 1323–1333. [Google Scholar] [CrossRef]
- Amir, M.K.; Khan, S.; Zia-ur-Rehman; Shah, A.; Butler, I.S. Anticancer activity of organotin(IV) carboxylates. Inorg. Chim. Acta 2014, 423, 14–25. [Google Scholar] [CrossRef]
- Kaiba, A.; Geesi, M.H.; Riadi, Y.; Ibnouf, E.O.; Aljohani, T.A.; Guionneau, P. A new Organic—Inorganic hybrid compound (NH3(CH2)2C6H5)2[SnCl6]: Crystal structure, characterization, Hirshfeld surface analysis, DFT calculation, vibrational properties and biological evaluation. J. Solid State Chem. 2021, 304, 122587. [Google Scholar] [CrossRef]
- Sedaghat, T.; Naseh, M.; Khavasi, H.R.; Motamedi, H. Synthesis, spectroscopic investigations, crystal structures and antibacterial activity of 3-(3-hydroxypyridin-2-ylamino)-1-phenylbut-2-en-1-one and its diorganotin (IV) complexes. Polyhedron 2012, 33, 435–440. [Google Scholar] [CrossRef]
- Joshi, R.; Kumari, A.; Singh, K.; Mishra, H.; Pokharia, S. New diorganotin(IV) complexes of Schiff base derived from 4-amino-3-hydrazino-5-mercapto-4H1,2,4-triazole: Synthesis, structural characterization, density functional theory studies, atoms-in-molecules analysis and antifungal activity. Appl. Organomet. Chem. 2019, 33, 4894. [Google Scholar] [CrossRef]
- Shah, F.A.; Sabir, S.; Fatima, K.; Ali, S.; Qadri, I.; Rizzoli, C. Organotin(iv) based anti-HCV drugs: Synthesis, characterization and biochemical activity. Dalton Trans. 2015, 44, 10467–10478. [Google Scholar] [CrossRef]
- Nath, M.; Vats, M.; Roy, P. Design, spectral characterization, anti-tumor and anti-inflammatory activity of trorganotin (IV) hydroxycarboxulates, apoptosis inducers: In vitro assessment of inductiom of apoptosis by enzyme, DNA fragmentation, acridin orange and comet assays. Inorg. Chim. Acta. 2014, 423, 70–82. [Google Scholar] [CrossRef]
- Mjos, K.D.; Orvig, C. Metallodrugs in medicinal inorganic chemistry. Chem. Rev. 2014, 114, 4540–4563. [Google Scholar] [CrossRef]
- Gasser, G.; Otto, I.; Metzler-Nolte, N.J. Organometallic Anticancer Compounds. Med. Chem. 2011, 54, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Lippard, S.J. New metal complexes as potential therapeutics. Curr. Opin. Chem. Biol. 2003, 7, 481–489. [Google Scholar] [CrossRef]
- Dayo Owoyemi, B.C.; da Silva, C.C.; Souza, M.S.; Diniz, L.F.; Ellena, J.; Carneiro, R.L. Fluconazole: Synthesis and structural characterization of four new pharmaceutical cocrystal forms. Cryst. Growth Des. 2019, 19, 648–657. [Google Scholar] [CrossRef]
- Gonnade, R.G.; Sangtani, E. Polymorphs and cocrystals: A comparative analysis. J. Indian Inst. Sci. 2017, 97, 193–226. [Google Scholar] [CrossRef]
- Ferjani, H.; Bechaieb, R.; El-Fattah, W.A.; Fettouhi, M. Broad-band luminescence involving fluconazole antifungal drug in a lead-free bismuth iodide perovskite: Combined experimental and computational insights. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 237, 118354. [Google Scholar] [CrossRef]
- Ali, M.M.; Ali, M.; Gaballa, A.S.; El-Korashy, S.A.; Teleb, S.M. Preparation, spectroscopic, characterizations and biological studies of new charge transfer complexes formed between fluconazole drug with various acceptors. Bioorganic Chem. 2021, 115, 105190. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, H. Tetrakis (4-chloroanilinium) hexachloridostannate (IV) dichloride. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, m782. [Google Scholar] [CrossRef]
- Surov, A.O.; Voronin, A.P.; Vasilev, N.A.; Churakov, A.V.; Perlovich, G.L. Cocrystals of fluconazole with aromatic carboxylic acids: Competition between anhydrous and hydrated solid forms. Cryst. Growth Des. 2019, 20, 1218–1228. [Google Scholar] [CrossRef]
- Ferjani, H. Crystal structure, optical property, and Hirshfeld surface analysis of bis [1-(prop-2-en-1-yl)-1H-imidazol-3-ium] hexachloridostannate (IV). Acta Crystallogr. Sect. E Crystallogr. Commun. 2020, 76, 1624–1628. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Scholfield, M.R.; Vander Zanden, C.M.; Carter, M.; Ho, P.S. Halogen bonding (X-bonding): A biological perspective. Protein Sci. 2013, 22, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Chopra, D.; Row, T.N.G. Role of organic fluorine in crystal engineering. Cryst. Eng. Comm. 2011, 13, 2175. [Google Scholar] [CrossRef]
- Scheiner, S. The F-Halogen Bond: Conditions for its Existence. J. Phys. Chem. A 2020, 124, 7290–7299. [Google Scholar] [CrossRef]
- Eskandari, K.; Lesani, M. Does Fluorine Participate in Halogen Bonding? Chem. A Eur. J. 2015, 21, 4739–4746. [Google Scholar] [CrossRef]
- Pavan, M.S.; Prasad, K.D.; Row, T.N.G. Halogen bonding in fluorine: Experimental charge density study on intermolecular F⋯F and F⋯S donor–acceptor contacts. Chem. Commun. 2013, 49, 7558. [Google Scholar] [CrossRef]
- Gupta, A.K.; Foley, K.A.; Versteeg, S.G. New antifungal agents and new formulations against dermatophytes. Mycopathologia 2017, 182, 127–141. [Google Scholar] [CrossRef]
- Naveed, S. UV spectrophotometric assay method for the determination of fluconazole capsules. Open Access Libr. J. 2015, 2, 68301. [Google Scholar] [CrossRef]
- Nalintya, E.; Kiggundu, R.; Meya, D. Evolution of cryptococcal antigen testing: What is new? Curr. Fungal Infect. Rep. 2016, 10, 62–67. [Google Scholar] [CrossRef]
- Zayed, M.; El-Shall, M.; Azime, M.A. Spectrophotometric Determination of Fluconazole, Voriconazole and Butoconazole nitrate by Ion-Pair Formation with Rose Bengal Reagent. Egypt. J. Chem. 2017, 60, 1177–1188. [Google Scholar] [CrossRef]
- Derbel, M.A.; Turnbull, M.M.; Naïli, H.; Rekik, W. A new mixed halide 2D hybrid perovskite: Structural, thermal, optic and magnetic properties. Polyhedron 2020, 175, 114220. [Google Scholar] [CrossRef]
- Bochalya, M.; Kumar, S. Magnetocaloric effect in 2D-alkylammonium copper halides layered inorganic-organic systems. J. Appl. Phys. 2020, 127, 055501. [Google Scholar] [CrossRef]
- Zárate, X.; Schott, E.; Carey, D.M.-L.; Bustos, C.; Arratia-Pérez, R. DFT study on the electronic structure, energetics and spectral properties of several bis (organohydrazido(2-)) molybdenum complexes containing substituted phosphines and chloro atoms as ancillary ligands. J. Mol. Struct. Theochem 2010, 957, 126–132. [Google Scholar] [CrossRef]
- Xu, Z.; Li, Y.; Zhang, W.; Yuan, S.; Hao, L.; Xu, T.; Lu, X. DFT/TD-DFT study of novel T shaped phenothiazine-based organic dyes for dye-sensitized solar cells applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 212, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Song, P.; Ma, F.; Saputra, R.M.; Li, Y. Tunable linear and nonlinear optical properties of chromophores containing 3, 7-(di) vinylquinoxalinone core by modification of receptors moieties. Opt. Mater. 2020, 99, 109580. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, J.; Duan, Y.-C.; Pan, Q.-Q.; Wu, Y.; Geng, Y.; Su, Z.-M. Theoretical insights on the rigidified dithiophene effects on the performance of near-infrared cis-squaraine-based dye-sensitized solar cells with panchromatic absorption. J. Photochem. Photobiol. A Chem. 2019, 369, 150–158. [Google Scholar] [CrossRef]
- Ferjani, H.; Bechaieb, R.; Dege, N.; El-Fattah, W.A.; Elamin, N.Y.; Frigui, W. Stabilization of supramolecular network of fluconazole drug polyiodide: Synthesis, computational and spectroscopic studies. J. Mol. Struct. 2022, 1263, 133192. [Google Scholar] [CrossRef]
- Gümüş, H.P.; Tamer, Ö.; Avcı, D.; Atalay, Y. Quantum chemical calculations on the geometrical, conformational, spectroscopic and nonlinear optical parameters of 5-(2-Chloroethyl)-2, 4-dichloro-6-methylpyrimidine. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 129, 219–226. [Google Scholar] [CrossRef]
- Tamer, Ö.; Bhatti, M.H.; Yunus, U.; Avcı, D.; Atalay, Y.; Nadeem, M.; Shah, S.R.; Helliwell, M. Structural, spectroscopic, nonlinear optical and electronic properties of calcium N-phthaloylglycinate: A combined experimental and theoretical study. J. Mol. Struct. 2016, 1125, 315–322. [Google Scholar] [CrossRef]
- Hajji, M.; Kouraichi, C.; Guerfel, T. Modelling, structural, thermal, optical and vibrational studies of a new organic–inorganic hybrid material (C5H16N2)Cd1.5Cl5. Bull. Mater. Sci. 2017, 40, 55–66. [Google Scholar] [CrossRef]
- Stoe, C. X-Area (Version 1.18) and X-Red32 (Version 1.04); Stoe & Cie: Darmstadt, Germany, 2002. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Comm. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.; McKinnon, J.; Wolff, S.; Grimwood, D.; Spackman, P.; Jayatilaka, D.; Spackman, M. CrystalExplorer17; The University of Western Australia: Perth, Australia, 2017. [Google Scholar]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H. Gaussian 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E.; Perdew, J.P. Tests of functionals for systems with fractional electron number. J. Chem. Phys. 2007, 126, 154109. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Ernzerhof, M.; Perdew, J.P. Generalized gradient approximation to the angle-and system-averaged exchange hole. J. Chem. Phys. 1998, 109, 3313–3320. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E. Efficient hybrid density functional calculations in solids: Assessment of the Heyd-Scuseria-Ernzerhof screened Coulomb hybrid functional. J. Chem. Phys. 2004, 121, 1187–1192. [Google Scholar] [CrossRef]
- Heyd, J.; Peralta, J.E.; Scuseria, G.E.; Martin, R.L. Energy band gaps and lattice parameters evaluated with the Heyd-Scuseria-Ernzerhof screened hybrid functional. J. Chem. Phys. 2005, 123, 174101. [Google Scholar] [CrossRef]
- Henderson, T.M.; Izmaylov, A.F.; Scalmani, G.; Scuseria, G.E. Can short-range hybrids describe long-range-dependent properties? J. Chem. Phys. 2009, 131, 044108. [Google Scholar] [CrossRef]
- Izmaylov, A.F.; Scuseria, G.E.; Frisch, M.J. Efficient evaluation of short-range Hartree-Fock exchange in large molecules and periodic systems. J. Chem. Phys. 2006, 125, 104103. [Google Scholar] [CrossRef]
- Lee, Y.S.; Ermler, W.C.; Pitzer, K.S. Ab initio effective core potentials including relativistic effects. I. Formalism and applications to the Xe and Au atoms. In Molecular Structure and Statistical Thermodynamics: Selected Papers of Kenneth S Pitzer; World Scientific: Singapore, 1993; pp. 112–127. [Google Scholar]
- Chiodo, S.; Russo, N.; Sicilia, E. LANL2DZ basis sets recontracted in the framework of density functional theory. J. Chem. Phys. 2006, 125, 104107. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | (C13H14F2N6O).SnCl6.2H2O |
|---|---|
| Formula weight (g/mol) | 675.72 |
| Crystal system, space group | monoclinic, P21/n |
| a (Å) | 8.0328 (5) |
| b (Å) | 29.4011 (13) |
| c (Å) | 10.3323 (7) |
| β (°) | 100.901 (5) |
| V (Å3) | 2396.2 (2) |
| Z | 4 |
| µ (mm−1) | 1.78 |
| Dx (Mg m−3) | 1.873 |
| F(000) | 1328 |
| Crystal size (mm) | 0.72 × 0.64 × 0.58 |
| Crystal habit | Colorless, Prism |
| θmin/θmax (deg) | 1.4/26.8 |
| Measured reflections | 13,858 |
| Independent reflections | 5043 |
| Observed refl. with I > 2σ(I) | 4495 |
| Rint | 0.046 |
| Data/restraints/parameters | 5043/0/289 |
| R[F2 > 2σ(F2)] | 0.037 |
| wR (F2) | 0.088 |
| GooF = S | 1.12 |
| Δρmax/Δρmin (e.Å−3) | 0.53/−0.95 |
| D-H···A | D-H (Å) | H···A (Å) | D···A (Å) | D-H···A(°) |
|---|---|---|---|---|
| O1-H1···O2 | 0.82 | 1.94 | 2.750 (3) | 169 |
| O2-H2A···O3 ii | 0.85 | 2.01 | 2.820(4) | 160 |
| O2-H2A···Cl1 ii | 0.85 | 2.81 | 3.522(3) | 142 |
| O2-H2B···Cl3 i | 0.85 | 2.77 | 3.395(3) | 132 |
| O3-H3A···Cl5 | 0.85 | 2.37 | 3.214(3) | 177 |
| C12-H12···Cl2 i | 0.93 | 2.72 | 3.359 (4) | 127 |
| C12-H12···O2 | 0.93 | 2.52 | 3.284 (4) | 140 |
| C11-H11A···F2 | 0.97 | 2.29 | 2.921 (4) | 122 |
| C11-H11B···Cl3 ii | 0.97 | 2.93 | 3.546 (3) | 122 |
| C8-H8A···F2 | 0.97 | 2.40 | 3.038 (4) | 123 |
| C8-H8B···Cl6 iii | 0.97 | 2.86 | 3.471 (3) | 122 |
| C8-H8B···Cl1 iii | 0.97 | 2.90 | 3.704 (3) | 141 |
| C8-H8B···N5 | 0.97 | 2.57 | 3.221 (5) | 124 |
| C9-H9···Cl2 iv | 0.93 | 2.70 | 3.566 (3) | 155 |
| C9-H9···O1 | 0.93 | 2.34 | 2.813 (4) | 111 |
| C10-H10···Cl6 v | 0.93 | 2.93 | 3.855 (4) | 175 |
| N6-H6···O3 | 0.85 (5) | 1.91 (5) | 2.736 (4) | 162 (6) |
| N3-H3···Cl4 iv | 0.85 (5) | 2.44 (5) | 3.240 (3) | 157 (4) |
| Atoms | Cl | H | C | N | O | F |
|---|---|---|---|---|---|---|
| (%) Contacts from fingerprint plots | ||||||
| Cl | 1.2 | - | - | - | - | - |
| H | 44.5 | 16.6 | - | - | - | - |
| C | 6 | 2.9 | 0.7 | - | - | - |
| N | 4.4 | 5.5 | 0 | 0 | - | - |
| O | 1.6 | 2.8 | 0 | 0 | 0 | - |
| F | 2.7 | 4.1 | 3.1 | 3.2 | 0 | 0.8 |
| Surface (%) | 30.8 | 46.5 | 6.7 | 6.54 | 2.2 | 7.35 |
| Random contacts % (RXX and RXY) | ||||||
| Cl | 9.84 | - | - | - | - | - |
| H | 28.64 | 21.6 | - | - | - | -- |
| C | 4.13 | 6.32 | 0.45 | - | - | - |
| N | 4.03 | 6.09 | 0.88 | 0.043 | - | - |
| O | 1.35 | 2.05 | 0.29 | 0.28 | 0.048 | - |
| F | 4.52 | 6.84 | 0.98 | 0.96 | 0.32 | 0.54 |
| Enrichment ratio E (EXX and EXY) | ||||||
| Cl | 0.13 | - | - | - | - | - |
| H | 1.55 | 0.77 | - | - | - | - |
| C | 1.45 | 0.46 | 1.55 | - | - | - |
| N | 1.09 | 0.9 | 0 | 0 | - | - |
| O | 1.18 | 1.36 | 0 | 0 | 0 | - |
| F | 0.59 | 0.6 | 3.16 | 3.33 | 0 | |
| Experimental | HSEH1PBE | Attribution |
|---|---|---|
| 495-547 | 450-572 | skeletal deformation |
| 613 | 611 | C-H out of plane |
| 677 | 695 | O-H out of plane |
| 749 | 755 | N-H out of plane of triazole group |
| 837 | 818 | C-H bending of triazole |
| 904 | 897 | C-H bending of benzene |
| 948 | 965 | C-C bending |
| 1004 | 1001 | C-N bending |
| 1071 | 1085 | C-O stretch mode |
| 1103 | 1115 | C-H and N-H bending of triazole |
| 1257 | 1278 | C-F bending mode |
| 1408 | 1415 | C-N stretch of triazole ring |
| 1479 | 1490 | C = C stretch 2,4-difluorobenzyl group |
| 1530 | 1554 | C = N stretch of triazole ring |
| 1620 | 1607 | C = N stretch of triazole ring |
| 1631 | 1740 | Benzene ring deformation |
| 2411 | 2850 | N-H stretch of triazole ring |
| 2989 | 2928 | C-H stretch of propane backbone |
| 3156 | 3120 | C-H stretch of propane backbone |
| 3339 | 3150 | C-H stretch (antisymmetric) of propane backbone |
| 3412 | 3190 | N-H stretch of triazole |
| 3497 | 3274 | O-H stretch mode |
| 3603 | 3390 | νs(OH) of crystallization water |
| 3607 | 3560 | νas(OH) of crystallization water |
| F (Z = 9) | Cl (Z = 17) | Br (Z = 35) | I (Z = 53) | |
|---|---|---|---|---|
| ELUMO | −1.9 | −3.2 | −3.4 | −4.3 |
| EHOMO | −7.4 | −7.2 | −6.5 | −6.0 |
| Eg | 5.5 | 3.9 | 3.1 | 1.6 |
| Ionis | 7.4 | 7.2 | 6.5 | 6.0 |
| Elec. Aff | 1.9 | 3.2 | 3.4 | 4.3 |
| Hardness | 2.7 | 2.0 | 1.6 | 0.8 |
| Softness | 0.2 | 0.3 | 0.3 | 0.6 |
| Chem. Pot | −4.7 | −5.2 | −5.0 | −5.2 |
| Electrophilicity | 4.0 | 6.9 | 7.8 | 16.5 |
| Electronegativity | 4.7 | 5.2 | 5.0 | 5.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferjani, H.; Bechaieb, R.; Alshammari, M.; Lemine, O.M.; Dege, N. New Organic–Inorganic Salt Based on Fluconazole Drug: TD-DFT Benchmark and Computational Insights into Halogen Substitution. Int. J. Mol. Sci. 2022, 23, 8765. https://doi.org/10.3390/ijms23158765
Ferjani H, Bechaieb R, Alshammari M, Lemine OM, Dege N. New Organic–Inorganic Salt Based on Fluconazole Drug: TD-DFT Benchmark and Computational Insights into Halogen Substitution. International Journal of Molecular Sciences. 2022; 23(15):8765. https://doi.org/10.3390/ijms23158765
Chicago/Turabian StyleFerjani, Hela, Rim Bechaieb, Marzook Alshammari, O. M. Lemine, and Necmi Dege. 2022. "New Organic–Inorganic Salt Based on Fluconazole Drug: TD-DFT Benchmark and Computational Insights into Halogen Substitution" International Journal of Molecular Sciences 23, no. 15: 8765. https://doi.org/10.3390/ijms23158765
APA StyleFerjani, H., Bechaieb, R., Alshammari, M., Lemine, O. M., & Dege, N. (2022). New Organic–Inorganic Salt Based on Fluconazole Drug: TD-DFT Benchmark and Computational Insights into Halogen Substitution. International Journal of Molecular Sciences, 23(15), 8765. https://doi.org/10.3390/ijms23158765