
The Human Cytomegalovirus β2.7 Long Non-Coding RNA Prevents Induction of Reactive Oxygen Species to Maintain Viral Gene Silencing during Latency


Abstract
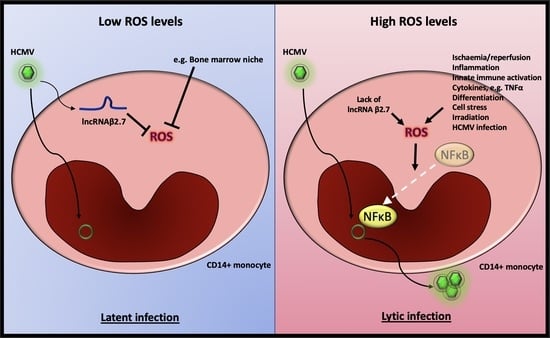
1. Introduction
2. Results
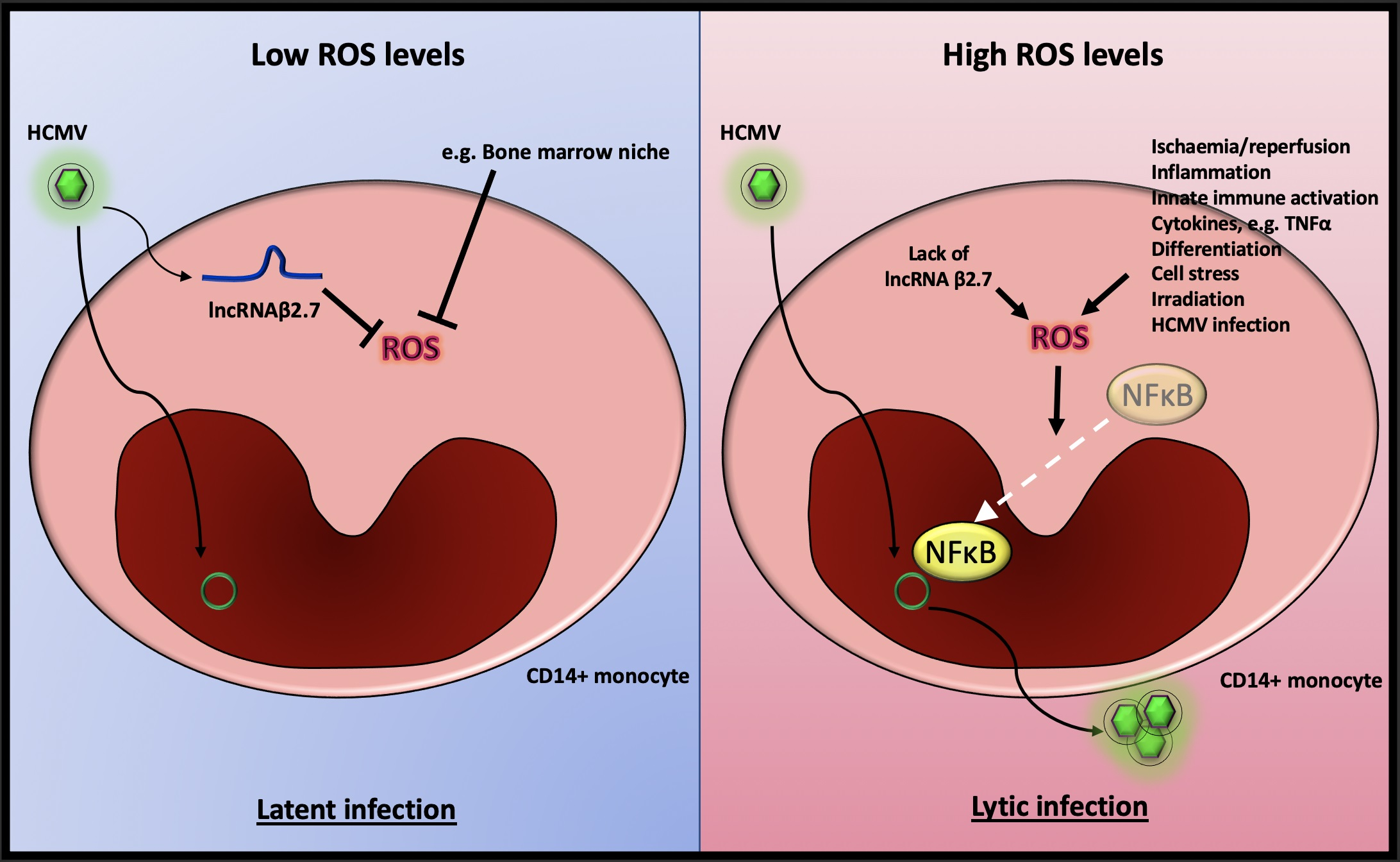
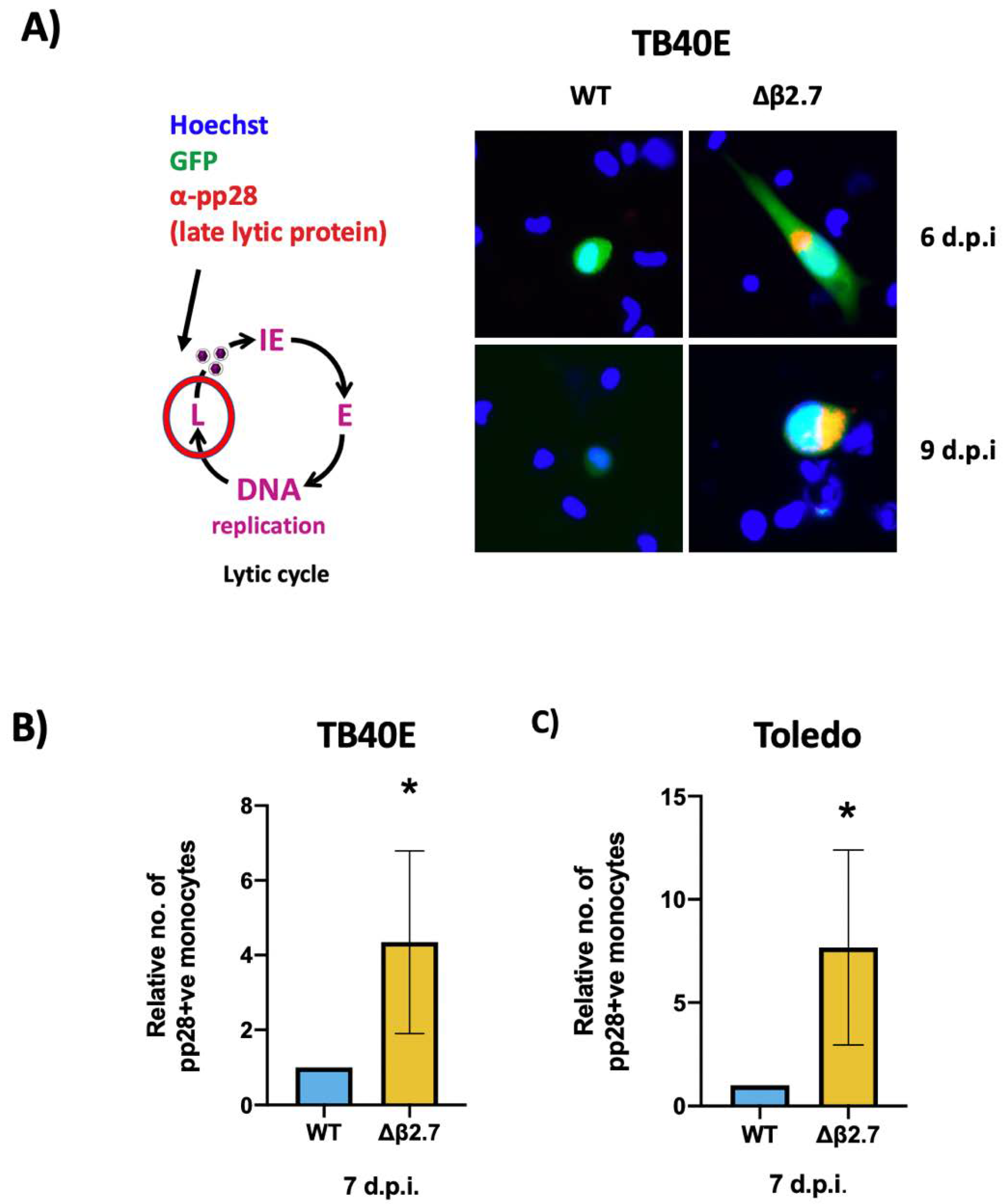
2.1. The Absence of β2.7 Results in Increased GFP Expression in a Sub-Population of Infected CD14+ Monocytes
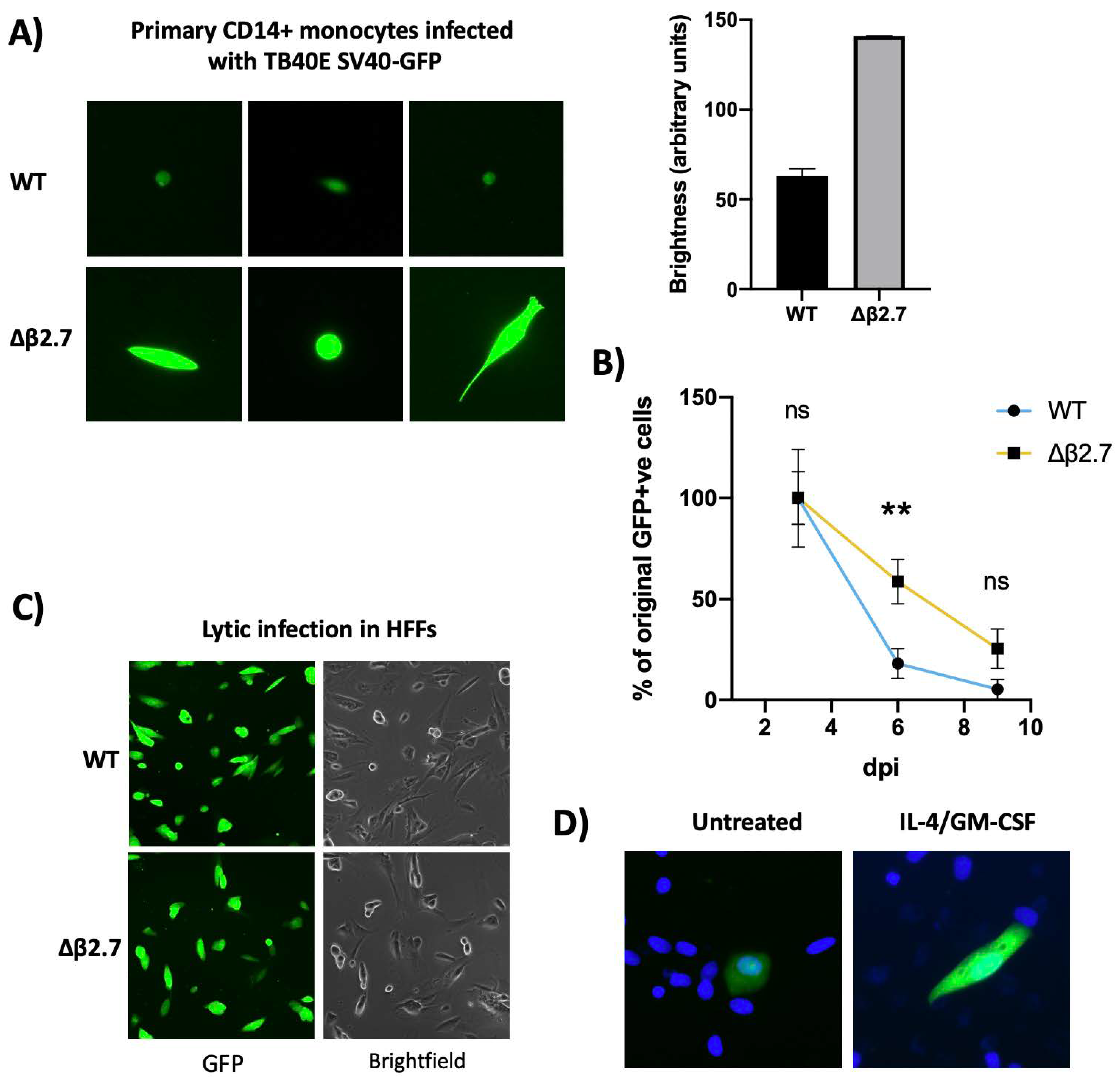
2.2. Viral Major Immediate Early Gene Expression Is Not Silenced in Δβ2.7 Infected Monocytes
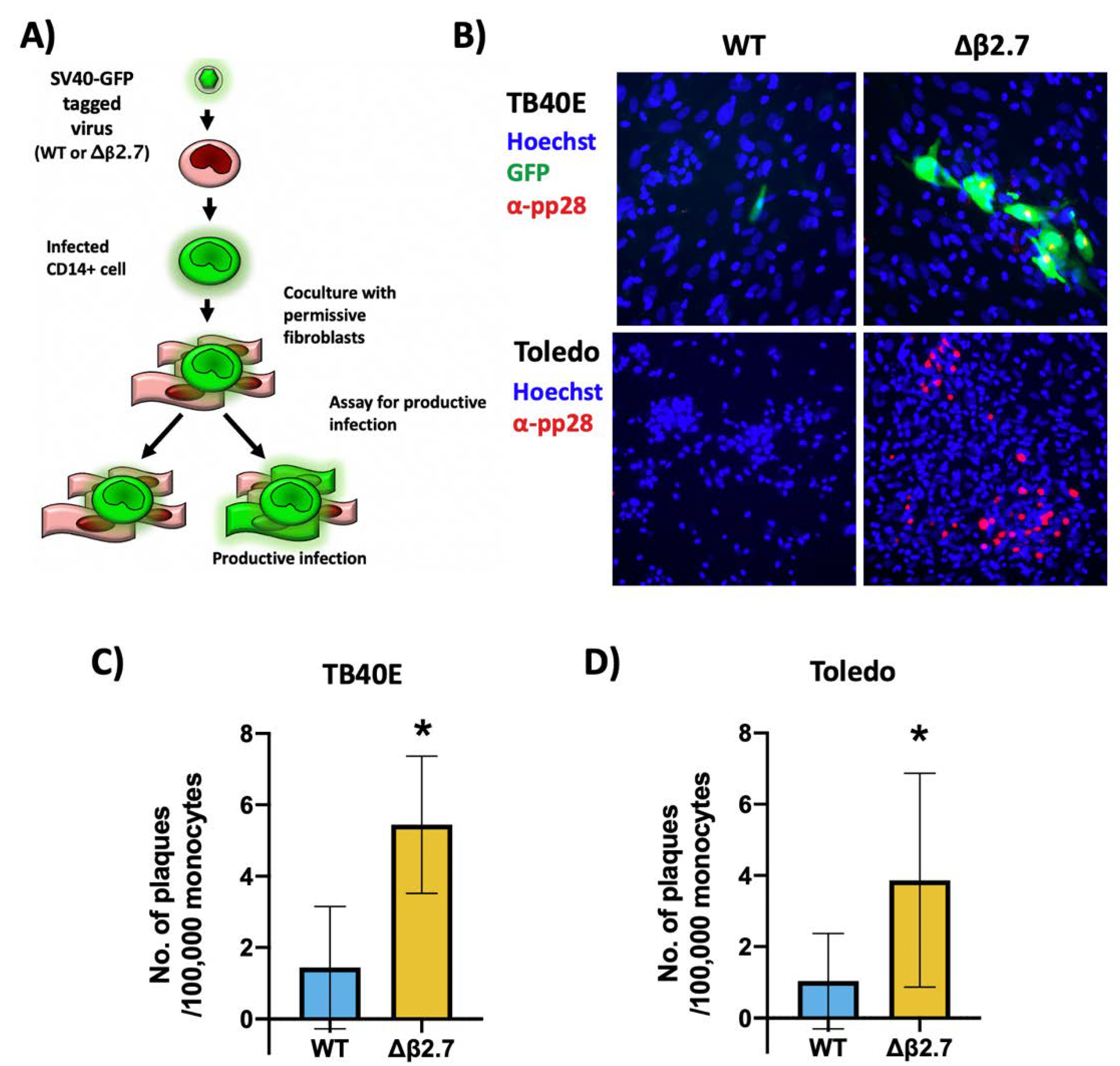
2.3. Δβ2.7 Infection Results in a Lytic Rather than Latent Infection in a Portion of Infected Monocytes
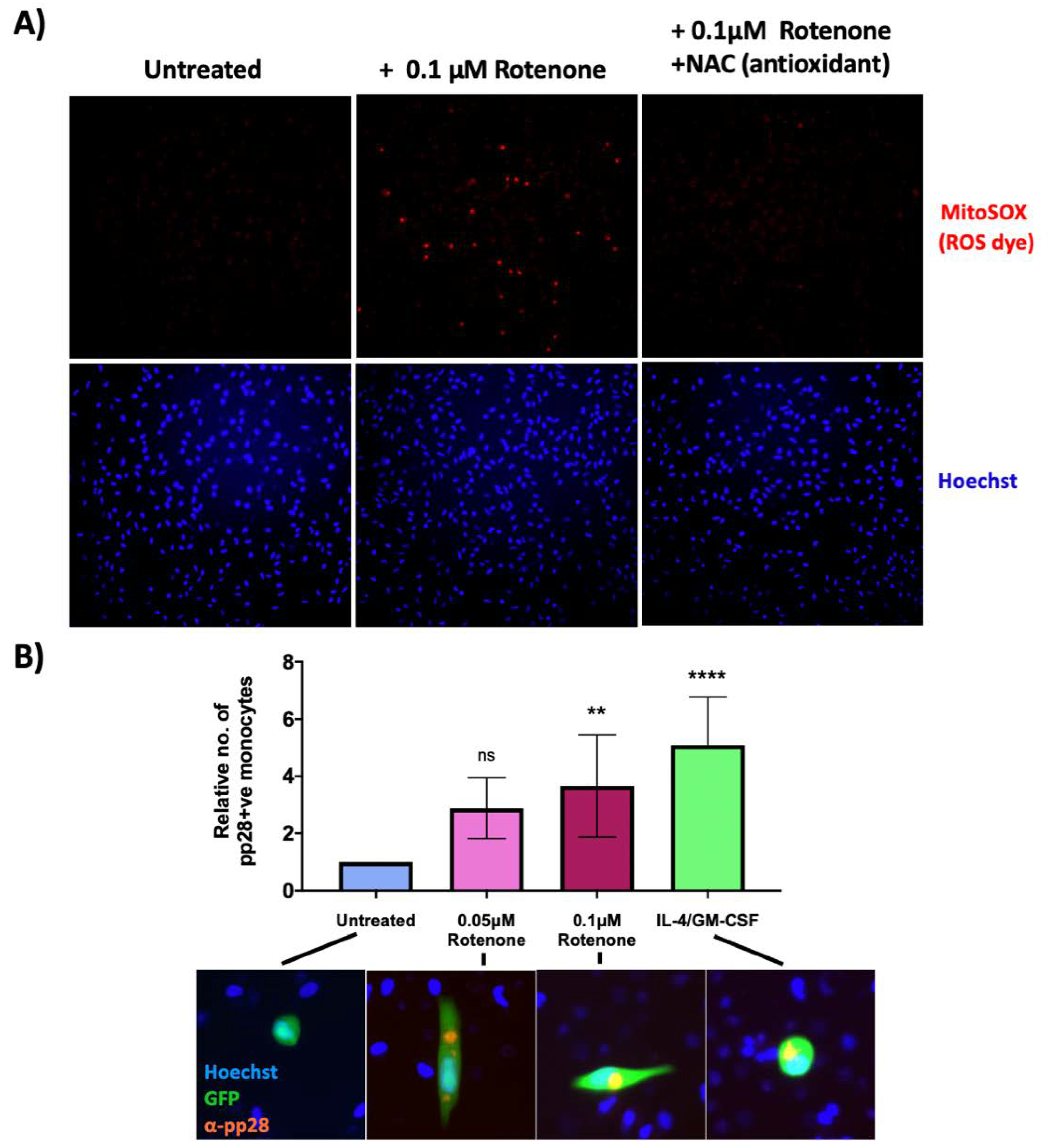
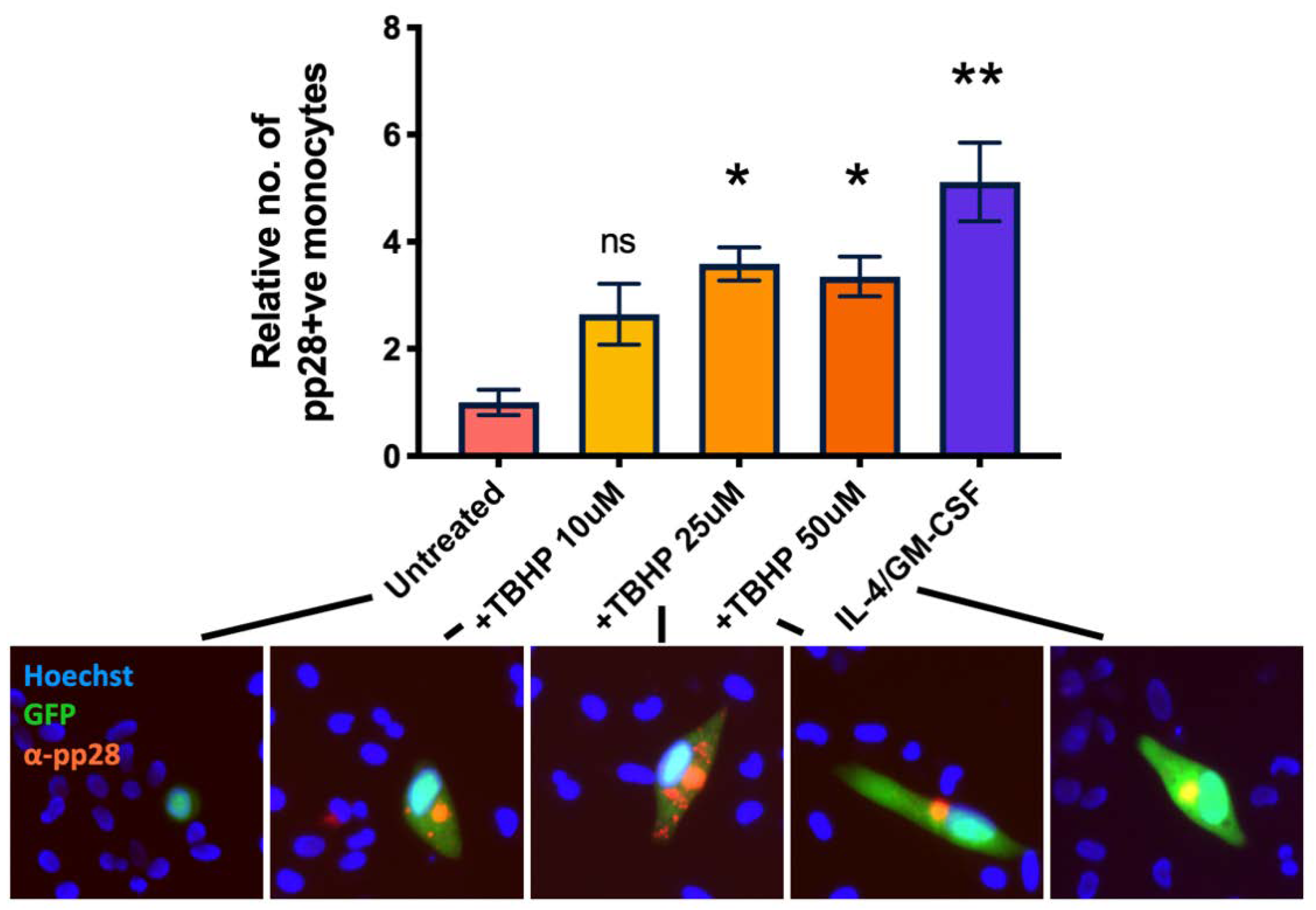
2.4. A Reactive Oxygen Species Inducer, Rotenone, Prevents HCMV Latency Establishment in a Sub-Population of Infected CD14+ Monocytes
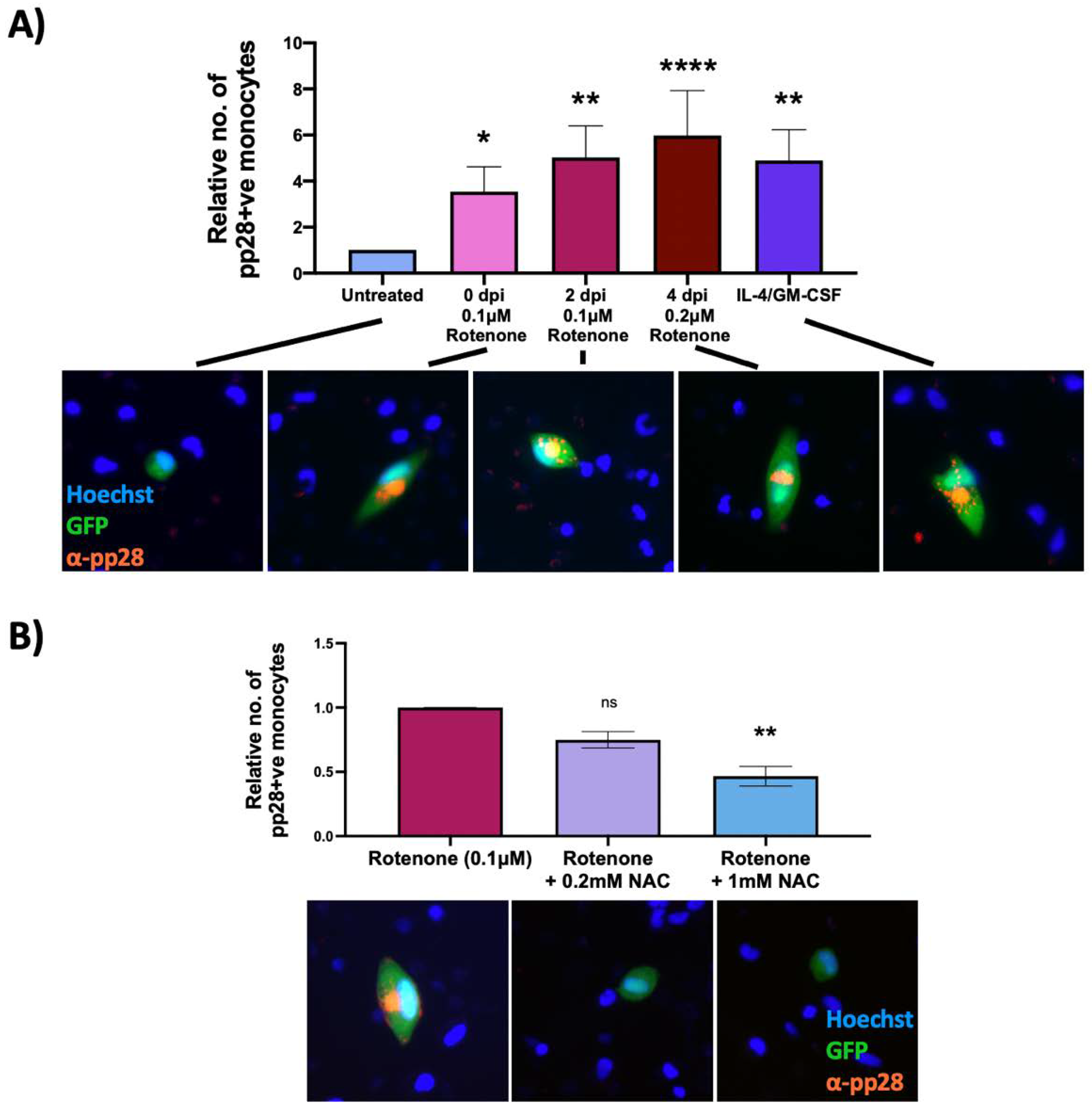
2.5. Reactive Oxygen Species Inducer, Rotenone, Reactivates Latent HCMV in CD14+ Monocytes
2.6. An Antioxidant Curbs Rotenone-Induced Lytic Gene Expression in Otherwise Latently Infected Monocytes
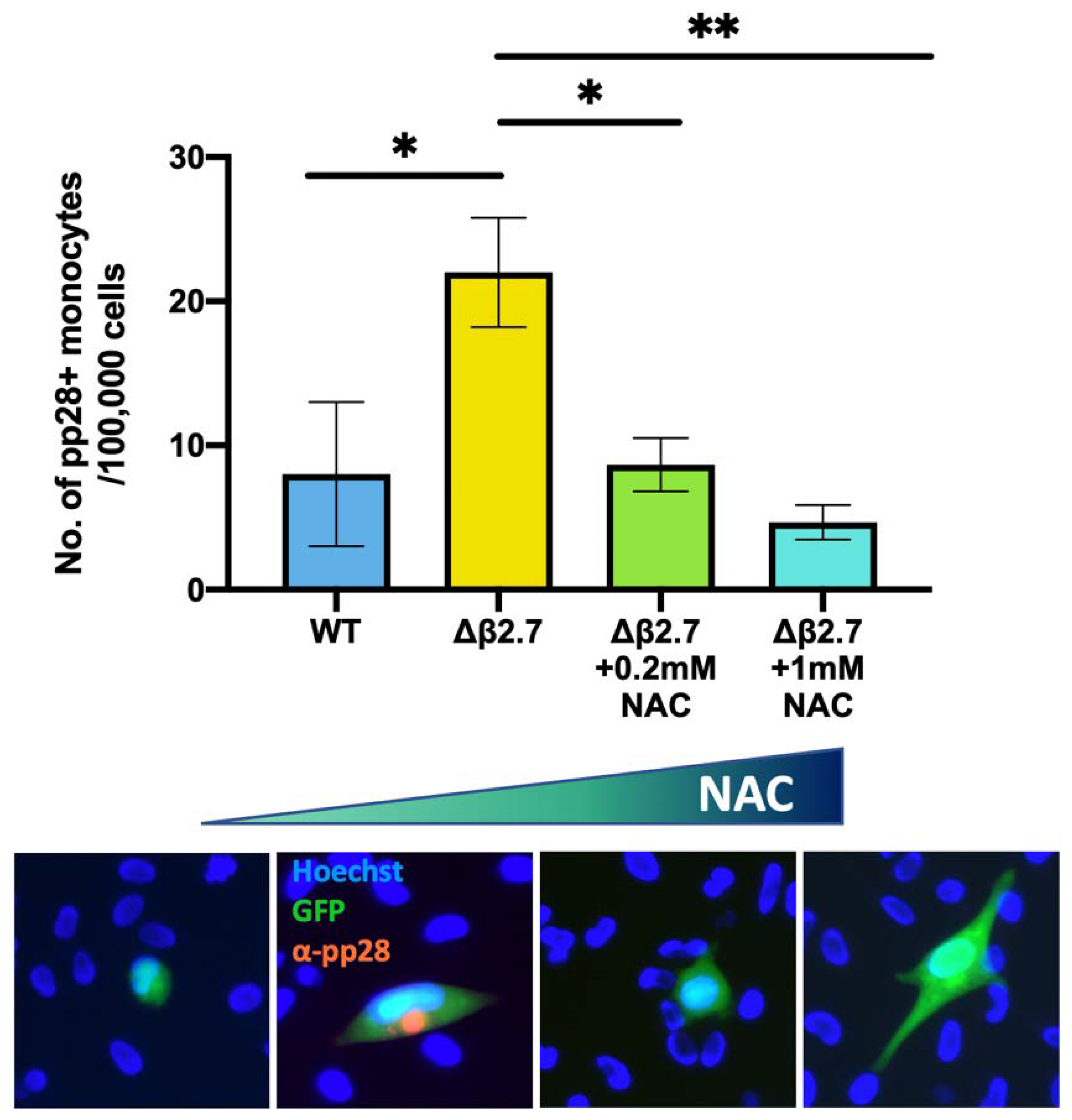
2.7. Direct Sources of ROS Can Also Prevent the Establishment/Maintenance of Latency in Primary Monocytes
2.8. An Antioxidant Reduces the Amount of Lytic Infection in Δβ2.7 Infection in Primary Monocytes
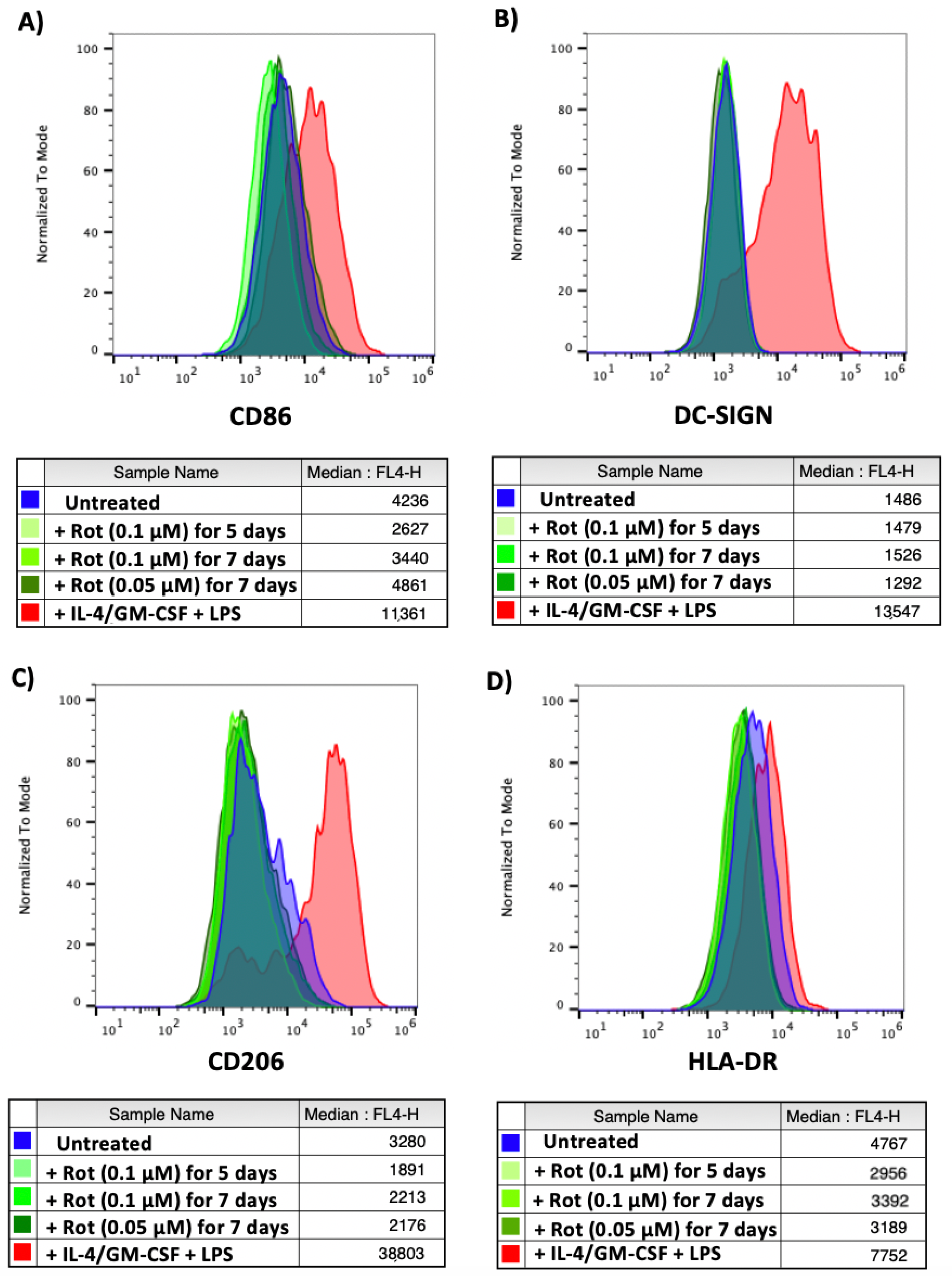
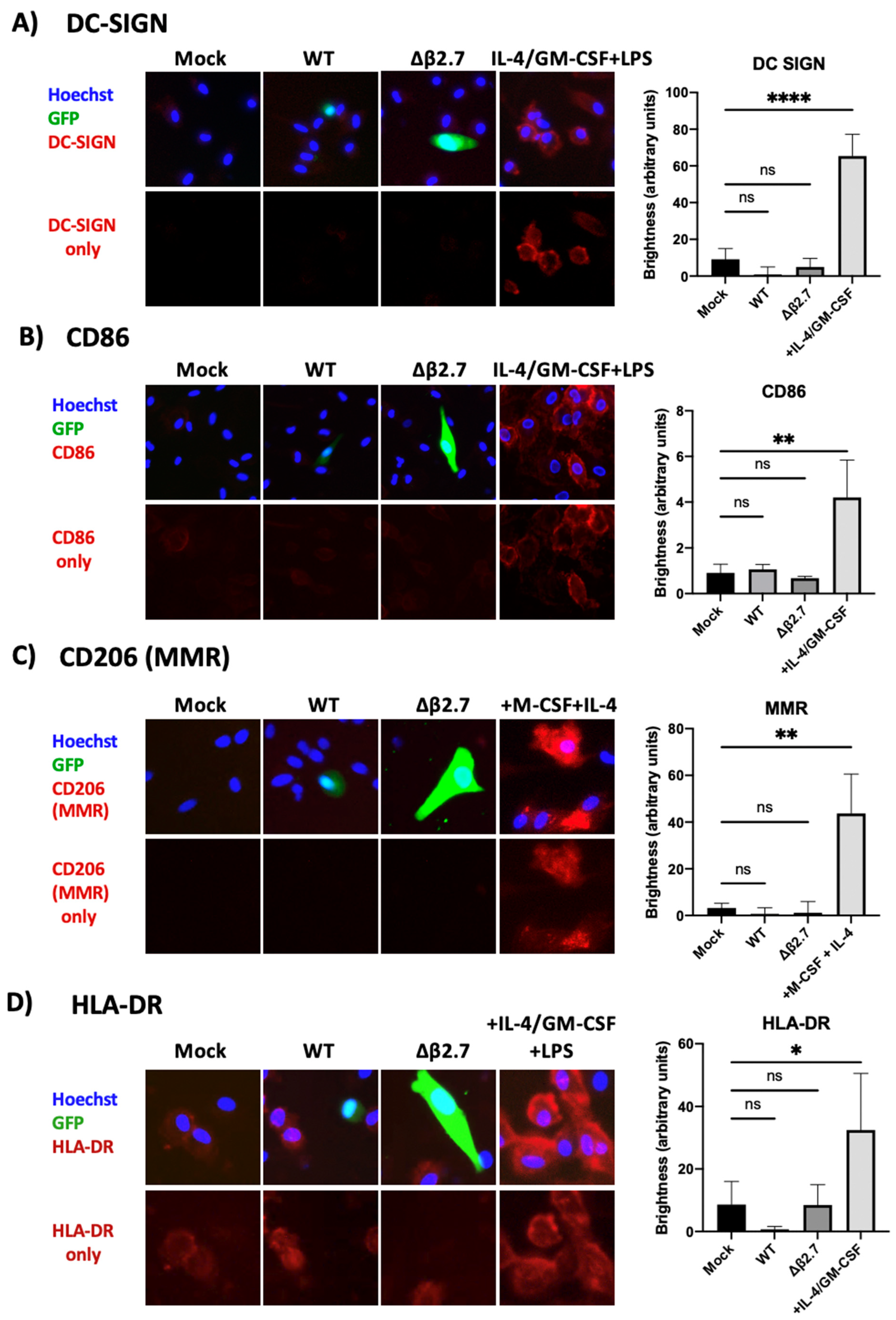
2.9. Higher ROS Levels Do Not Induce Differentiation of the Infected Monocytes
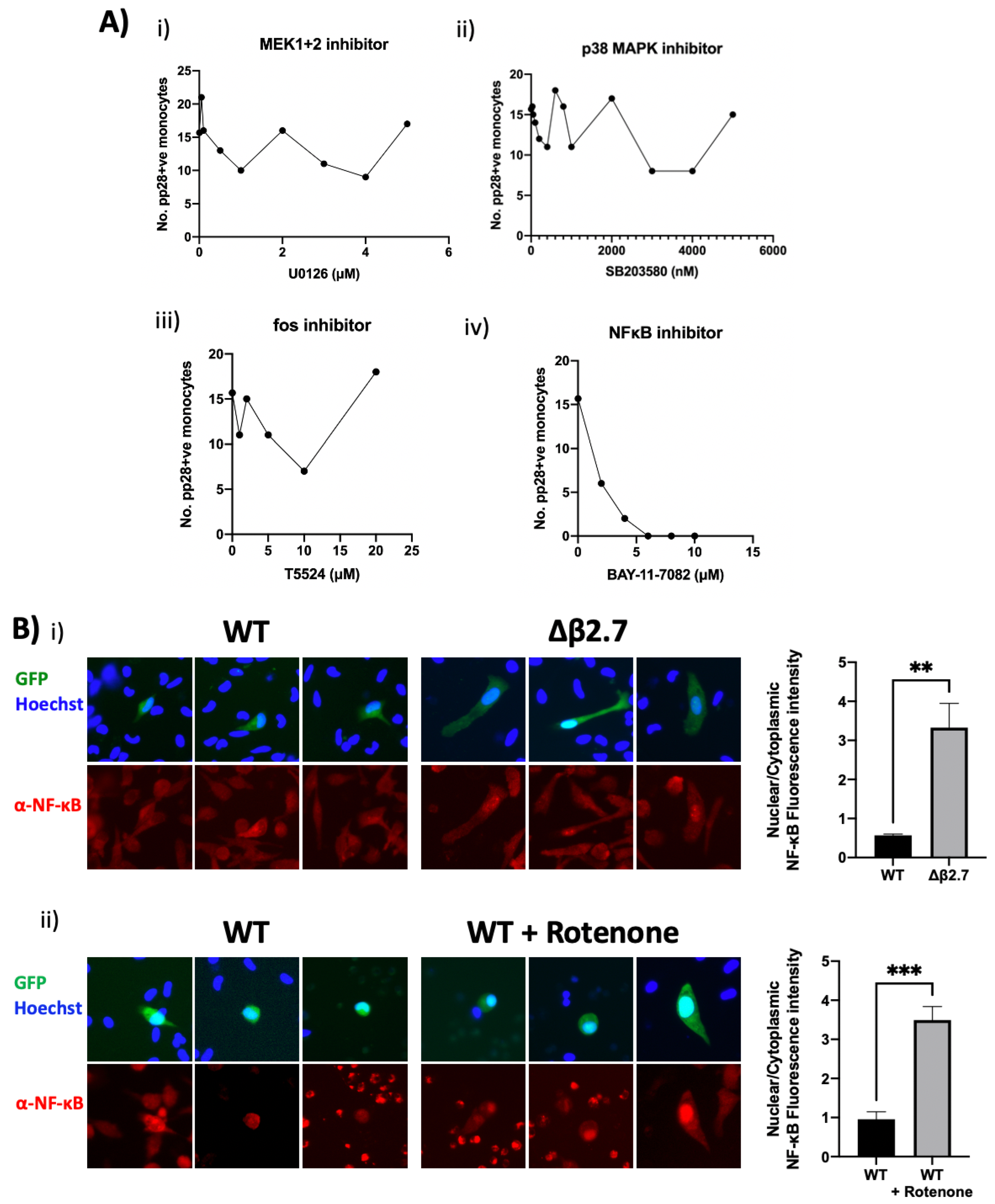
2.10. Δβ2.7 Infection and Rotenone Activate NF-κB
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Inhibitors
4.3. Human Cytomegaloviruses
4.4. Fluorescence Microscopy
4.5. DNA and RNA Extraction, Reverse Transcription and qPCR
4.6. Flow Cytometry
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of Cytomegalovirus Seroprevalence and Demographic Characteristics Associated with Infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Reeves, M. Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus Remains Latent in a Common Precursor of Dendritic and Myeloid Cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of Endogenous Human Cytomegalovirus in CD34+ Bone Marrow Progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef]
- Wills, M.R.; Poole, E.; Lau, B.; Krishna, B.; Sinclair, J.H. The Immunology of Human Cytomegalovirus Latency: Could Latent Infection Be Cleared by Novel Immunotherapeutic Strategies? Cell. Mol. Immunol. 2015, 12, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Dooley, A.L.; O’Connor, C.M. Regulation of the MIE Locus During HCMV Latency and Reactivation. Pathogens 2020, 9, 869. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B. Cell Signaling and Cytomegalovirus Reactivation: What Do Src Family Kinases Have to Do with It? Biochem. Soc. Trans. 2020, 48, 667–675. [Google Scholar] [CrossRef]
- Kew, V.G.; Yuan, J.; Meier, J.; Reeves, M.B. Mitogen and Stress Activated Kinases Act Co-Operatively with CREB during the Induction of Human Cytomegalovirus Immediate-Early Gene Expression from Latency. PLoS Pathog. 2014, 10, e1004195. [Google Scholar] [CrossRef]
- Sinclair, J. Human Cytomegalovirus: Latency and Reactivation in the Myeloid Lineage. J. Clin. Virol. 2008, 41, 180–185. [Google Scholar] [CrossRef]
- Reeves, M.B.; Compton, T. Inhibition of Inflammatory Interleukin-6 Activity via Extracellular Signal-Regulated Kinase–Mitogen-Activated Protein Kinase Signaling Antagonizes Human Cytomegalovirus Reactivation from Dendritic Cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing. mBio 2018, 9, e00013-18. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and Trans Acting Factors Involved in Human Cytomegalovirus Experimental and Natural Latent Infection of CD14 (+) Monocytes and CD34 (+) Cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.R.; Roche, K.L.; Murphy, E.A.; Sinclair, J.H. A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes. Viruses 2022, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Carcy, R.; Cougnon, M.; Poet, M.; Durandy, M.; Sicard, A.; Counillon, L.; Blondeau, N.; Hauet, T.; Tauc, M.; Pisani, D.F. Targeting Oxidative Stress, a Crucial Challenge in Renal Transplantation Outcome. Free Radic. Biol. Med. 2021, 169, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Azevedo*, L.S.; Pierrotti, L.C.; Abdala, E.; Costa, S.F.; Strabelli, T.M.V.; Campos, S.V.; Ramos, J.F.; Latif, A.Z.A.; Litvinov, N.; Maluf, N.Z.; et al. Cytomegalovirus Infection in Transplant Recipients. Clinics 2015, 70, 515–523. [Google Scholar] [CrossRef]
- Reinke, P.; Prösch, S.; Kern, F.; Volk, H.D. Mechanisms of Human Cytomegalovirus (HCMV) (Re)Activation and Its Impact on Organ Transplant Patients. Transpl. Infect. Dis. 1999, 1, 157–164. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Murphy, E.A. A Myeloid Progenitor Cell Line Capable of Supporting Human Cytomegalovirus Latency and Reactivation, Resulting in Infectious Progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef]
- Elder, E.; Krishna, B.; Williamson, J.; Aslam, Y.; Farahi, N.; Wood, A.; Romashova, V.; Roche, K.; Murphy, E.; Chilvers, E.; et al. Monocytes Latently Infected with Human Cytomegalovirus Evade Neutrophil Killing. iScience 2019, 12, 13–26. [Google Scholar] [CrossRef]
- Krishna, B.A.; Spiess, K.; Poole, E.L.; Lau, B.; Voigt, S.; Kledal, T.N.; Rosenkilde, M.M.; Sinclair, J.H. Targeting the Latent Cytomegalovirus Reservoir with an Antiviral Fusion Toxin Protein. Nat. Commun. 2017, 8, 14321. [Google Scholar] [CrossRef]
- Silva, M.C.; Schröer, J.; Shenk, T. Human Cytomegalovirus Cell-to-Cell Spread in the Absence of an Essential Assembly Protein. Proc. Natl. Acad. Sci. USA 2005, 102, 2081–2086. [Google Scholar] [CrossRef]
- Cheung, A.K.L.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral Gene Expression during the Establishment of Human Cytomegalovirus Latent Infection in Myeloid Progenitor Cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Rak, M.; Buehler, J.C.; Igarashi-Hayes, S.; Kamil, J.P.; Moorman, N.J.; Goodrum, F. Alternative Promoters Drive Human Cytomegalovirus Reactivation from Latency. Proc. Natl. Acad. Sci. USA 2019, 116, 17492–17497. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.; Poole, E.; Van Damme, E.; Bunkens, L.; Sowash, M.; King, H.; Murphy, E.; Wills, M.; Van Loock, M.; Sinclair, J. Human Cytomegalovirus MiR-UL112-1 Promotes the down-Regulation of Viral Immediate Early-Gene Expression during Latency to Prevent T-Cell Recognition of Latently Infected Cells. J. Gen. Virol. 2016, 97, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain Proteins Regulate Human Cytomegalovirus Latency and Reactivation Allowing Epigenetic Therapeutic Intervention. Proc. Natl. Acad. Sci. USA 2021, 118, e2023025118. [Google Scholar] [CrossRef]
- Min, C.-K.; Shakya, A.K.; Lee, B.-J.; Streblow, D.N.; Caposio, P.; Yurochko, A.D. The Differentiation of Human Cytomegalovirus Infected-Monocytes Is Required for Viral Replication. Front. Cell. Infect. Microbiol. 2020, 10, 368. [Google Scholar] [CrossRef]
- Forte, E.; Swaminathan, S.; Schroeder, M.W.; Kim, J.Y.; Terhune, S.S.; Hummel, M. Tumor Necrosis Factor Alpha Induces Reactivation of Human Cytomegalovirus Independently of Myeloid Cell Differentiation Following Posttranscriptional Establishment of Latency. mBio 2018, 9, e01560-18. [Google Scholar] [CrossRef]
- Wang, L.; Duan, Q.; Wang, T.; Ahmed, M.; Zhang, N.; Li, Y.; Li, L.; Yao, X. Mitochondrial Respiratory Chain Inhibitors Involved in ROS Production Induced by Acute High Concentrations of Iodide and the Effects of SOD as a Protective Factor. Oxid. Med. Cell. Longev. 2015, 2015, 217670. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Mohammed, F.; Gorla, M.; Bisoyi, V.; Tammineni, P.; Sepuri, N.B.V. Rotenone-Induced Reactive Oxygen Species Signal the Recruitment of STAT3 to Mitochondria. FEBS Lett. 2020, 594, 1403–1412. [Google Scholar] [CrossRef]
- Heinz, S.; Freyberger, A.; Lawrenz, B.; Schladt, L.; Schmuck, G.; Ellinger-Ziegelbauer, H. Mechanistic Investigations of the Mitochondrial Complex I Inhibitor Rotenone in the Context of Pharmacological and Safety Evaluation. Sci. Rep. 2017, 7, 45465. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.E.; Hill, G.E. An Assessment of Techniques to Manipulate Oxidative Stress in Animals. Funct. Ecol. 2017, 31, 9–21. [Google Scholar] [CrossRef]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive Oxygen Species in Haematopoiesis: Leukaemic Cells Take a Walk on the Wild Side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Zaccagnino, P.; Di Paola, M.; Saltarella, M.; Oliveros Celis, C.; Nico, B.; Santoro, G.; Lorusso, M. Role of Mitochondria and Reactive Oxygen Species in Dendritic Cell Differentiation and Functions. Free Radic. Biol. Med. 2008, 44, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, S.S.; Eligini, S.; Brambilla, M.; Tremoli, E.; Colli, S. Reactive Oxygen Species Mediate Cyclooxygenase-2 Induction during Monocyte to Macrophage Differentiation: Critical Role of NADPH Oxidase. Cardiovasc. Res. 2003, 60, 187–197. [Google Scholar] [CrossRef]
- Xu, Q.; Choksi, S.; Qu, J.; Jang, J.; Choe, M.; Banfi, B.; Engelhardt, J.F.; Liu, Z. NADPH Oxidases Are Essential for Macrophage Differentiation. J. Biol. Chem. 2016, 291, 20030–20041. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, M.; Shibutani, M.; Seike, S.; Yonezaki, M.; Takagishi, T.; Oda, M.; Kobayashi, K.; Sakurai, J. The P38 MAPK and JNK Pathways Protect Host Cells against Clostridium Perfringens Beta-Toxin. Infect. Immun. 2013, 81, 3703–3708. [Google Scholar] [CrossRef]
- Sattler, M.; Winkler, T.; Verma, S.; Byrne, C.H.; Shrikhande, G.; Salgia, R.; Griffin, J.D. Hematopoietic Growth Factors Signal through the Formation of Reactive Oxygen Species. Blood 1999, 93, 2928–2935. [Google Scholar] [CrossRef]
- Sinha, A.; Singh, A.; Satchidanandam, V.; Natarajan, K. Impaired Generation of Reactive Oxygen Species during Differentiation of Dendritic Cells (DCs) by Mycobacterium Tuberculosis Secretory Antigen (MTSA) and Subsequent Activation of MTSA-DCs by Mycobacteria Results in Increased Intracellular Survival. J. Immunol. Baltim. 2006, 177, 468–478. [Google Scholar] [CrossRef]
- Nishi, K.; Oda, T.; Takabuchi, S.; Oda, S.; Fukuda, K.; Adachi, T.; Semenza, G.L.; Shingu, K.; Hirota, K. LPS Induces Hypoxia-Inducible Factor 1 Activation in Macrophage-Differentiated Cells in a Reactive Oxygen Species-Dependent Manner. Antioxid. Redox Signal. 2008, 10, 983–995. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.-G. ROS Play a Critical Role in the Differentiation of Alternatively Activated Macrophages and the Occurrence of Tumor-Associated Macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.F.; Yeung, H.T.; Lam, Y.M.; Ng, R.K. Reactive Oxygen Species Activate Differentiation Gene Transcription of Acute Myeloid Leukemia Cells via the JNK/c-JUN Signaling Pathway. Leuk. Res. 2018, 68, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage Activation and Polarization. Front. Biosci. J. Virtual Libr. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Bertani, F.R.; Mozetic, P.; Fioramonti, M.; Iuliani, M.; Ribelli, G.; Pantano, F.; Santini, D.; Tonini, G.; Trombetta, M.; Businaro, L.; et al. Classification of M1/M2-Polarized Human Macrophages by Label-Free Hyperspectral Reflectance Confocal Microscopy and Multivariate Analysis. Sci. Rep. 2017, 7, 8965. [Google Scholar] [CrossRef]
- Bio-Rad Macrophage Polarization—Mini-Review. Available online: https://www.bio-rad-antibodies.com/macrophage-polarization-minireview.html (accessed on 5 August 2021).
- Engering, A.; Geijtenbeek, T.B.H.; van Vliet, S.J.; Wijers, M.; van Liempt, E.; Demaurex, N.; Lanzavecchia, A.; Fransen, J.; Figdor, C.G.; Piguet, V.; et al. The Dendritic Cell-Specific Adhesion Receptor DC-SIGN Internalizes Antigen for Presentation to T Cells. J. Immunol. 2002, 168, 2118–2126. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-J.; Gu, Y.; Wang, C.-Z.; Jin, Y.; Wen, X.-M.; Ma, J.-C.; Tang, L.-J.; Mao, Z.-W.; Qian, J.; Lin, J. The M2 Macrophage Marker CD206: A Novel Prognostic Indicator for Acute Myeloid Leukemia. Oncoimmunology 2019, 9, 1683347. [Google Scholar] [CrossRef]
- Amatruda, T.T.; Bohman, R.; Ranyard, J.; Koeffler, H.P. Pattern of Expression of HLA-DR and HLA-DQ Antigens and MRNA in Myeloid Differentiation. Blood 1987, 69, 1225–1236. [Google Scholar]
- Casasola-LaMacchia, A.; Ritorto, M.S.; Seward, R.J.; Ahyi-Amendah, N.; Ciarla, A.; Hickling, T.P.; Neubert, H. Human Leukocyte Antigen Class II Quantification by Targeted Mass Spectrometry in Dendritic-like Cell Lines and Monocyte-Derived Dendritic Cells. Sci. Rep. 2021, 11, 1028. [Google Scholar] [CrossRef]
- Buchmeier, N.A.; Cooper, N.R. Suppression of Monocyte Functions by Human Cytomegalovirus. Immunology 1989, 66, 278–283. [Google Scholar] [PubMed]
- Slobedman, B.; Mocarski, E.S.; Arvin, A.M.; Mellins, E.D.; Abendroth, A. Latent Cytomegalovirus Down-Regulates Major Histocompatibility Complex Class II Expression on Myeloid Progenitors. Blood 2002, 100, 2867–2873. [Google Scholar] [CrossRef]
- Elder, E.G.; Krishna, B.A.; Williamson, J.; Lim, E.Y.; Poole, E.; Sedikides, G.X.; Wills, M.; O’Connor, C.M.; Lehner, P.J.; Sinclair, J. Interferon-Responsive Genes Are Targeted during the Establishment of Human Cytomegalovirus Latency. mBio 2019, 10, e02574-19. [Google Scholar] [CrossRef] [PubMed]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-KappaB Activation by Reactive Oxygen Species: Fifteen Years Later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, e792639. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and Macrophage Differentiation: Circulation Inflammatory Monocyte as Biomarker for Inflammatory Diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, e2795090. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte Recruitment during Infection and Inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef]
- Pasha, A.; Calvani, M.; Favre, C. Β3-Adrenoreceptors as ROS Balancer in Hematopoietic Stem Cell Transplantation. Int. J. Mol. Sci. 2021, 22, 2835. [Google Scholar] [CrossRef] [PubMed]
- Reboredo, M.; Greaves, R.F.; Hahn, G. Human Cytomegalovirus Proteins Encoded by UL37 Exon 1 Protect Infected Fibroblasts against Virus-Induced Apoptosis and Are Required for Efficient Virus Replication. J. Gen. Virol. 2004, 85, 3555–3567. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of Mitochondrial Networks by the Human Cytomegalovirus UL37 Gene Product Viral Mitochondrion-Localized Inhibitor of Apoptosis. J. Virol. 2003, 77, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Skaletskaya, A.; Bartle, L.M.; Chittenden, T.; McCormick, A.L.; Mocarski, E.S.; Goldmacher, V.S. A Cytomegalovirus-Encoded Inhibitor of Apoptosis That Suppresses Caspase-8 Activation. Proc. Natl. Acad. Sci. USA 2001, 98, 7829–7834. [Google Scholar] [CrossRef]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.-J. Reactive Oxygen Species Hydrogen Peroxide Mediates Kaposi’s Sarcoma-Associated Herpesvirus Reactivation from Latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, J.; Sun, R. Oxidative Stress Induces Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus and Death of Primary Effusion Lymphoma Cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Li, T.; Tan, B.; Ramos da Silva, S.; Jung, J.U.; Feng, P.; Gao, S.-J. FoxO1 Suppresses Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication and Controls Viral Latency. J. Virol. 2019, 93, e01681-18. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Fang, C.-Y.; Wu, C.-C.; Tsai, C.-H.; Lin, S.-F.; Chen, J.-Y. Reactive Oxygen Species Mediate Epstein-Barr Virus Reactivation by N-Methyl-N’-Nitro-N-Nitrosoguanidine. PLoS ONE 2013, 8, e84919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, J.; Zhong, J. Chaetocin Reactivates the Lytic Replication of Epstein-Barr Virus from Latency via Reactive Oxygen Species. Sci. China Life Sci. 2017, 60, 66–71. [Google Scholar] [CrossRef]
- Speir, E.; Shibutani, T.; Yu, Z.X.; Ferrans, V.; Epstein, S.E. Role of Reactive Oxygen Intermediates in Cytomegalovirus Gene Expression and in the Response of Human Smooth Muscle Cells to Viral Infection. Circ. Res. 1996, 79, 1143–1152. [Google Scholar] [CrossRef]
- Speir, E. Cytomegalovirus Gene Regulation by Reactive Oxygen Species: Agents in Atherosclerosis. Ann. N. Y. Acad. Sci. 2000, 899, 363–374. [Google Scholar] [CrossRef]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Ponath, V.; Kaina, B. Death of Monocytes through Oxidative Burst of Macrophages and Neutrophils: Killing in Trans. PLoS ONE 2017, 12, e0170347. [Google Scholar] [CrossRef]
- Kuwabara, W.M.T.; Zhang, L.; Schuiki, I.; Curi, R.; Volchuk, A.; Alba-Loureiro, T.C. NADPH Oxidase-Dependent Production of Reactive Oxygen Species Induces Endoplasmatic Reticulum Stress in Neutrophil-Like HL60 Cells. PLoS ONE 2015, 10, e0116410. [Google Scholar] [CrossRef]
- Swindle, E.J.; Hunt, J.A.; Coleman, J.W. A Comparison of Reactive Oxygen Species Generation by Rat Peritoneal Macrophages and Mast Cells Using the Highly Sensitive Real-Time Chemiluminescent Probe Pholasin: Inhibition of Antigen-Induced Mast Cell Degranulation by Macrophage-Derived Hydrogen Peroxide. J. Immunol. 2002, 169, 5866–5873. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.A.; Chan, G.C.; O’Connor, C.M. Modulation of Host Cell Signaling during Cytomegalovirus Latency and Reactivation. Virol. J. 2021, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Wass, A.B.; O’Connor, C.M. Activator Protein-1 Transactivation of the Major Immediate Early Locus Is a Determinant of Cytomegalovirus Reactivation from Latency. Proc. Natl. Acad. Sci. USA 2020, 117, 20860–20867. [Google Scholar] [CrossRef] [PubMed]
- Dupont, L.; Du, L.; Poulter, M.; Choi, S.; McIntosh, M.; Reeves, M.B. Src Family Kinase Activity Drives Cytomegalovirus Reactivation by Recruiting MOZ Histone Acetyltransferase Activity to the Viral Promoter. J. Biol. Chem. 2019, 294, 12901–12910. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, D. Reactive Oxygen Species (ROS)-Responsive Nanomedicine for Solving Ischemia-Reperfusion Injury. Front. Chem. 2020, 8, 732. [Google Scholar] [CrossRef] [PubMed]
- Minami, K.; Bae, S.; Uehara, H.; Zhao, C.; Lee, D.; Iske, J.; Fanger, M.W.; Reder, J.; Morrison, I.; Azuma, H.; et al. Targeting of Intragraft Reactive Oxygen Species by APP-103, a Novel Polymer Product, Mitigates Ischemia/Reperfusion Injury and Promotes the Survival of Renal Transplants. Am. J. Transplant. 2020, 20, 1527–1537. [Google Scholar] [CrossRef]
- Ávila, C.; Líbano, L.; Rojas, I.; Rodrigo, R. Role of Ischemia-Reperfusion in Oxidative Stress-Mediated Injury during Kidney Transplantation. Clin. Res. Trials 2019, 5, 1–4. [Google Scholar] [CrossRef]
- Shi, S.; Xue, F. Current Antioxidant Treatments in Organ Transplantation. Oxid. Med. Cell. Longev. 2016, 2016, 8678510. [Google Scholar] [CrossRef]
- Zhang, Z.; Qiu, L.; Yan, S.; Wang, J.-J.; Thomas, P.M.; Kandpal, M.; Zhao, L.; Iovane, A.; Liu, X.; Thorp, E.B.; et al. A Clinically Relevant Murine Model Unmasks a “Two-Hit” Mechanism for Reactivation and Dissemination of Cytomegalovirus after Kidney Transplant. Am. J. Transplant. 2019, 19, 2421–2433. [Google Scholar] [CrossRef]
- Poole, E.; Reeves, M.; Sinclair, J.H. The Use of Primary Human Cells (Fibroblasts, Monocytes, and Others) to Assess Human Cytomegalovirus Function. In Human Cytomegaloviruses Methods and Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1119, pp. 81–98. [Google Scholar]
- Rostam, H.M.; Reynolds, P.M.; Alexander, M.R.; Gadegaard, N.; Ghaemmaghami, A.M. Image Based Machine Learning for Identification of Macrophage Subsets. Sci. Rep. 2017, 7, 3521. [Google Scholar] [CrossRef]
- McSharry, B.P.; Tomasec, P.; Neale, M.L.; Wilkinson, G.W.G. The Most Abundantly Transcribed Human Cytomegalovirus Gene (Beta 2.7) Is Non-Essential for Growth In Vitro. J. Gen. Virol. 2003, 84, 2511–2516. [Google Scholar] [CrossRef]
- Roback, J.D.; Hillyer, C.D.; Drew, W.L.; Laycock, M.E.; Luka, J.; Mocarski, E.S.; Slobedman, B.; Smith, J.W.; Soderberg-Naucler, C.; Todd, D.S.; et al. Multicenter Evaluation of PCR Methods Fordetecting CMV DNA in Blood Donors. Transfusion 2001, 41, 1249–1257. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company and Catalogue No. | Dilution |
|---|---|---|
| anti-IE | Argene, 11-003 | 1/1000 |
| anti-pp28 | Abcam, ab6502 | 1/1000 |
| anti-NF-κB | Abcam, ab16502 | 1/500 |
| anti-HLA-DR-APC | Biolegend, #307609 | 1/200 |
| anti-DC-SIGN-APC | Biolegend, #330107 | 1/200 |
| anti-CD86-APC | Biolegend, #305412 | 1/200 |
| anti-MMR-Alexa Fluor 647 | Biolegend, #321116 | 1/200 |
| Mouse IgG2a κ APC | Biolegend, #400219 | Equivalent concentration to test antibody |
| Mouse IgG1 κ APC | Biolegend, #400119 | Equivalent concentration to test antibody |
| Target | Primer Sequence |
|---|---|
| IE F | GTC CTG ACA GAA CTC GTC AAA |
| IE R | TAA AGG CGC CAG TGA ATT TTT CTT C |
| GAPDH F | TGC ACC ACC AAC TGC TTA GC |
| GAPDH R | GGC ATG GAC TGT GGT CAT GAG |
| GAPDH promoter F | CGG CTA CTA GCG GTT TTA CG |
| GAPDH promoter R | AAG AAG ATG CGG CTG ACT GT |
| UL44 promoter F | AAC CTG AGC GTG TTT GTG |
| UL44 promoter R | CGT GCA AGT CTC GAC TAA G |
| Antibody | Source |
|---|---|
| anti-HLA-DR-APC | Biolegend, #307609 |
| anti-DC-SIGN-APC | Biolegend, #330107 |
| anti-MMR Alexa Fluor 647 | Biolegend, #321116 |
| anti-CD86-APC | Biolegend, #305412 |
| Mouse IgG2a κ APC | Biolegend, #400219 |
| Mouse IgG1 κ APC | Biolegend, #400119 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perera, M.R.; Sinclair, J.H. The Human Cytomegalovirus β2.7 Long Non-Coding RNA Prevents Induction of Reactive Oxygen Species to Maintain Viral Gene Silencing during Latency. Int. J. Mol. Sci. 2022, 23, 11017. https://doi.org/10.3390/ijms231911017
Perera MR, Sinclair JH. The Human Cytomegalovirus β2.7 Long Non-Coding RNA Prevents Induction of Reactive Oxygen Species to Maintain Viral Gene Silencing during Latency. International Journal of Molecular Sciences. 2022; 23(19):11017. https://doi.org/10.3390/ijms231911017
Chicago/Turabian StylePerera, Marianne R., and John H. Sinclair. 2022. "The Human Cytomegalovirus β2.7 Long Non-Coding RNA Prevents Induction of Reactive Oxygen Species to Maintain Viral Gene Silencing during Latency" International Journal of Molecular Sciences 23, no. 19: 11017. https://doi.org/10.3390/ijms231911017
APA StylePerera, M. R., & Sinclair, J. H. (2022). The Human Cytomegalovirus β2.7 Long Non-Coding RNA Prevents Induction of Reactive Oxygen Species to Maintain Viral Gene Silencing during Latency. International Journal of Molecular Sciences, 23(19), 11017. https://doi.org/10.3390/ijms231911017