
Comprehensive Collection and Prediction of ABC Transmembrane Protein Structures in the AI Era of Structural Biology

 , , , and
, , , and 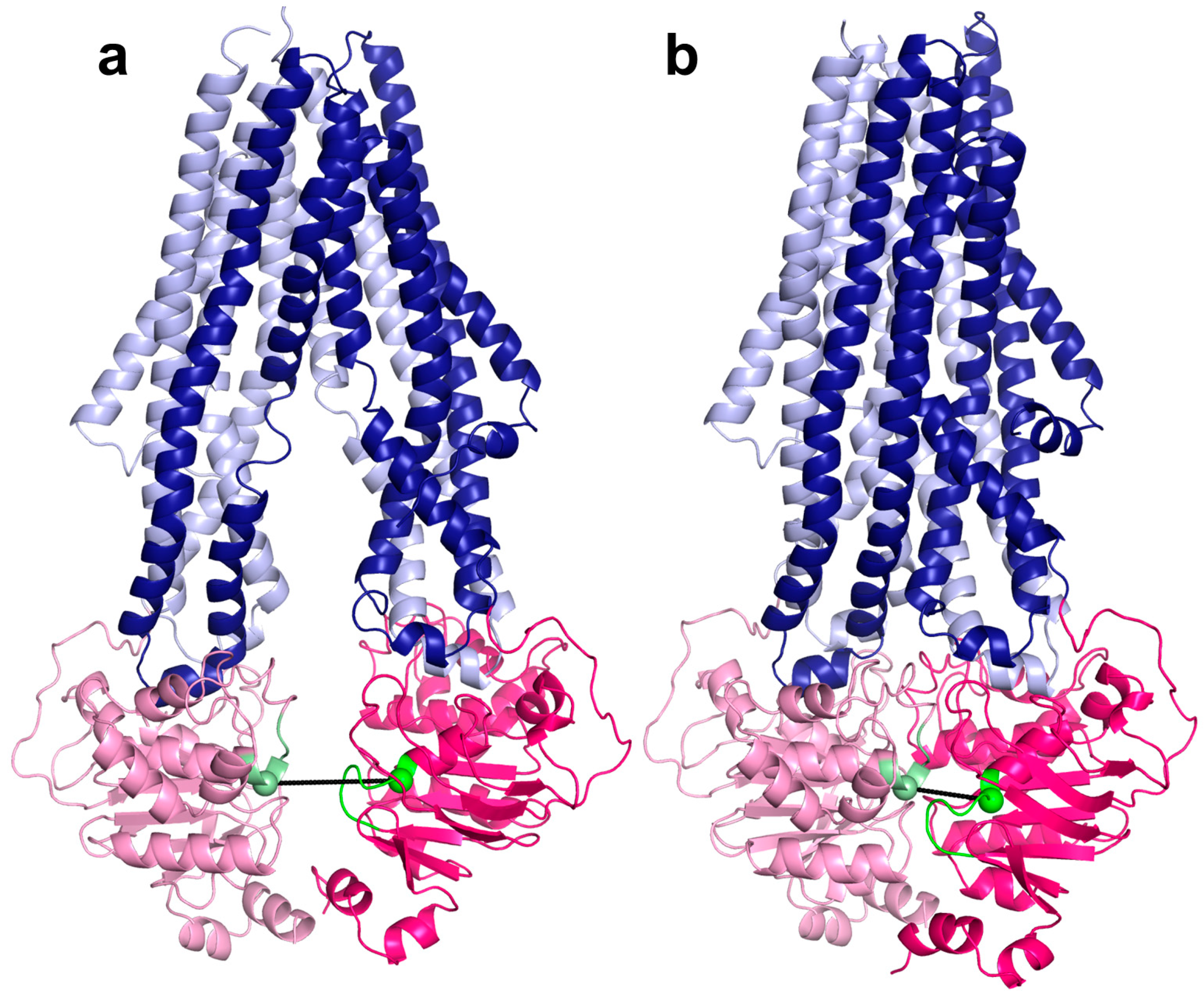{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
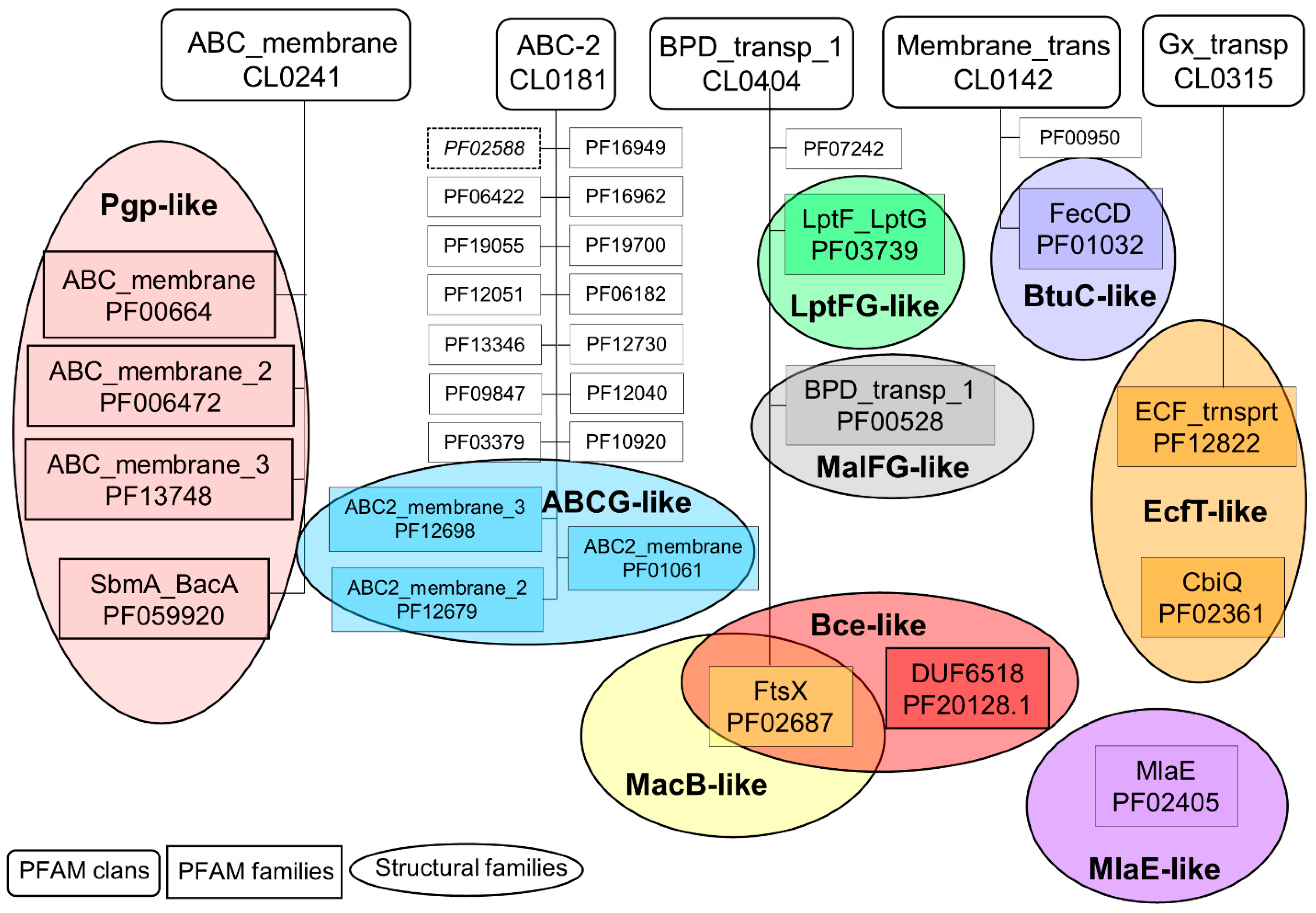
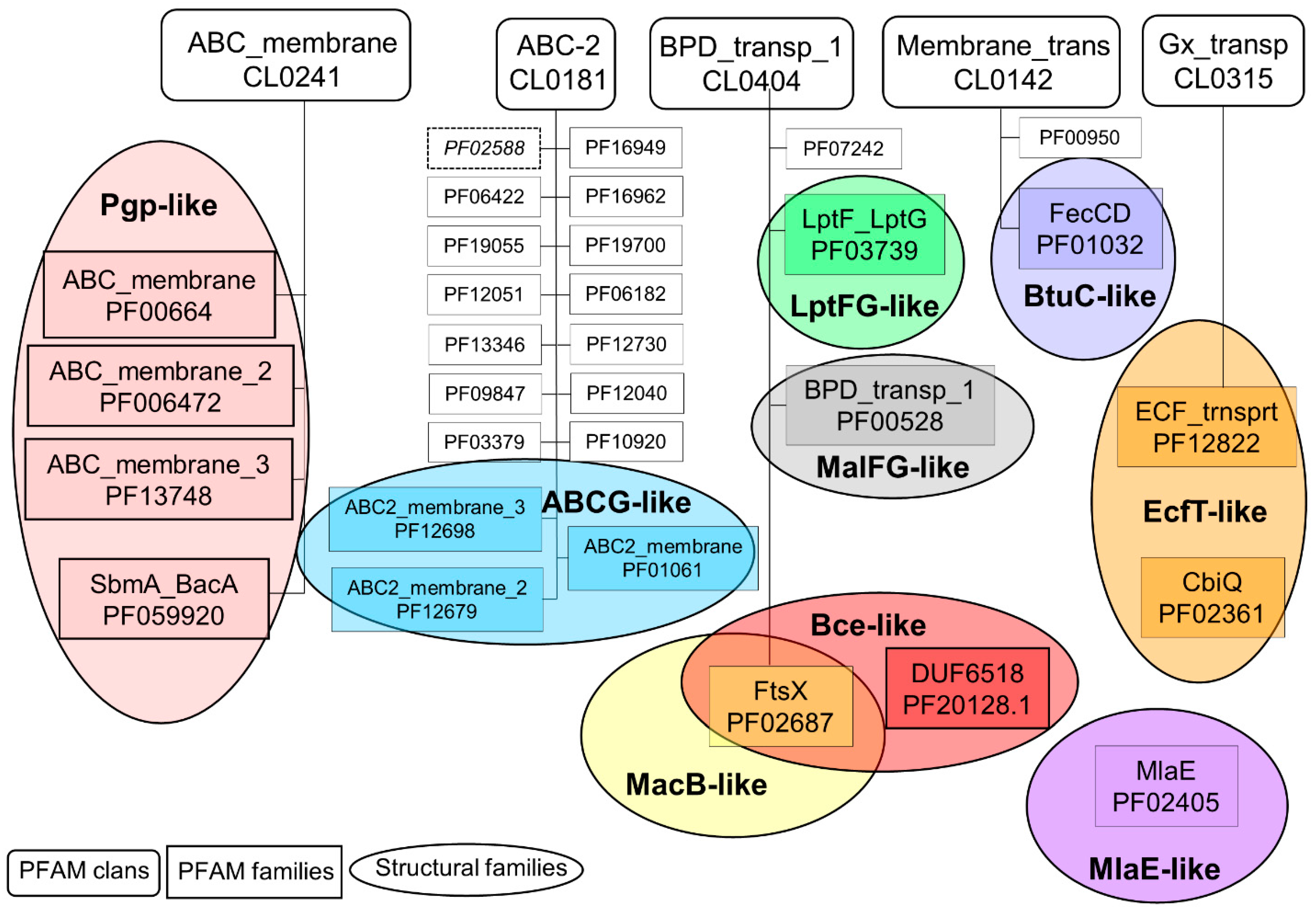
2.1. Connections between Transmembrane ABC Proteins Structures and Pfam Profiles
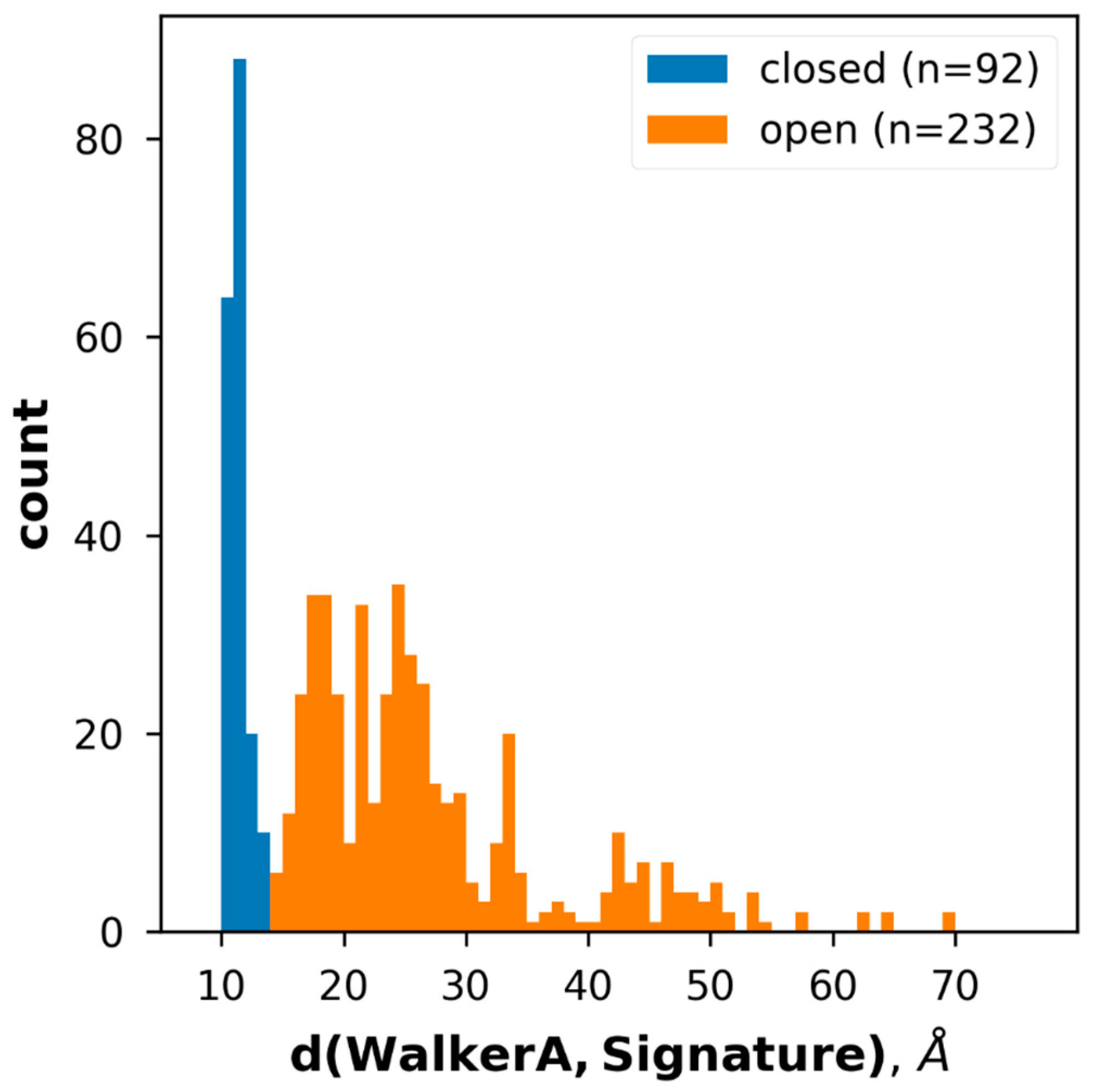
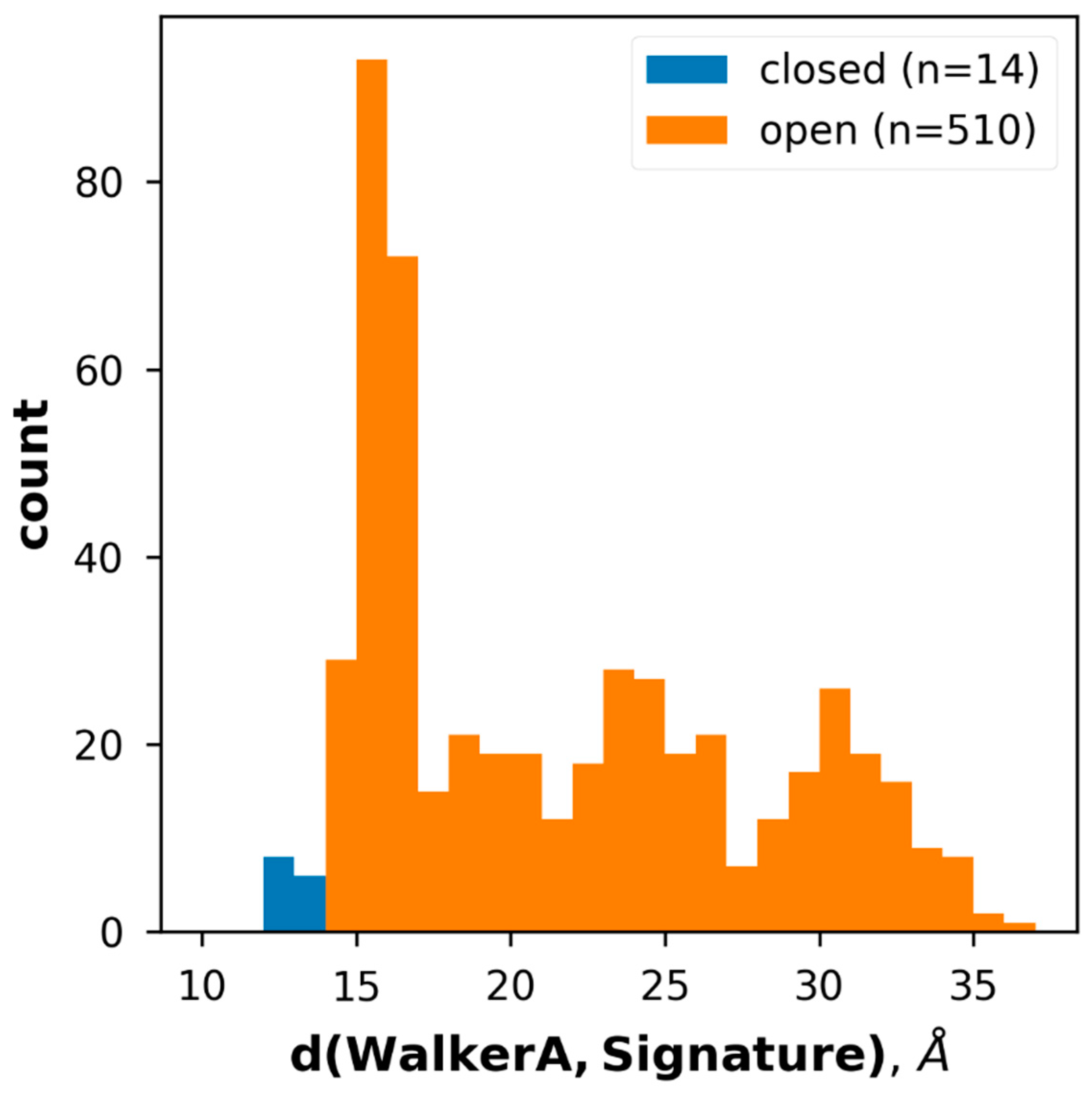
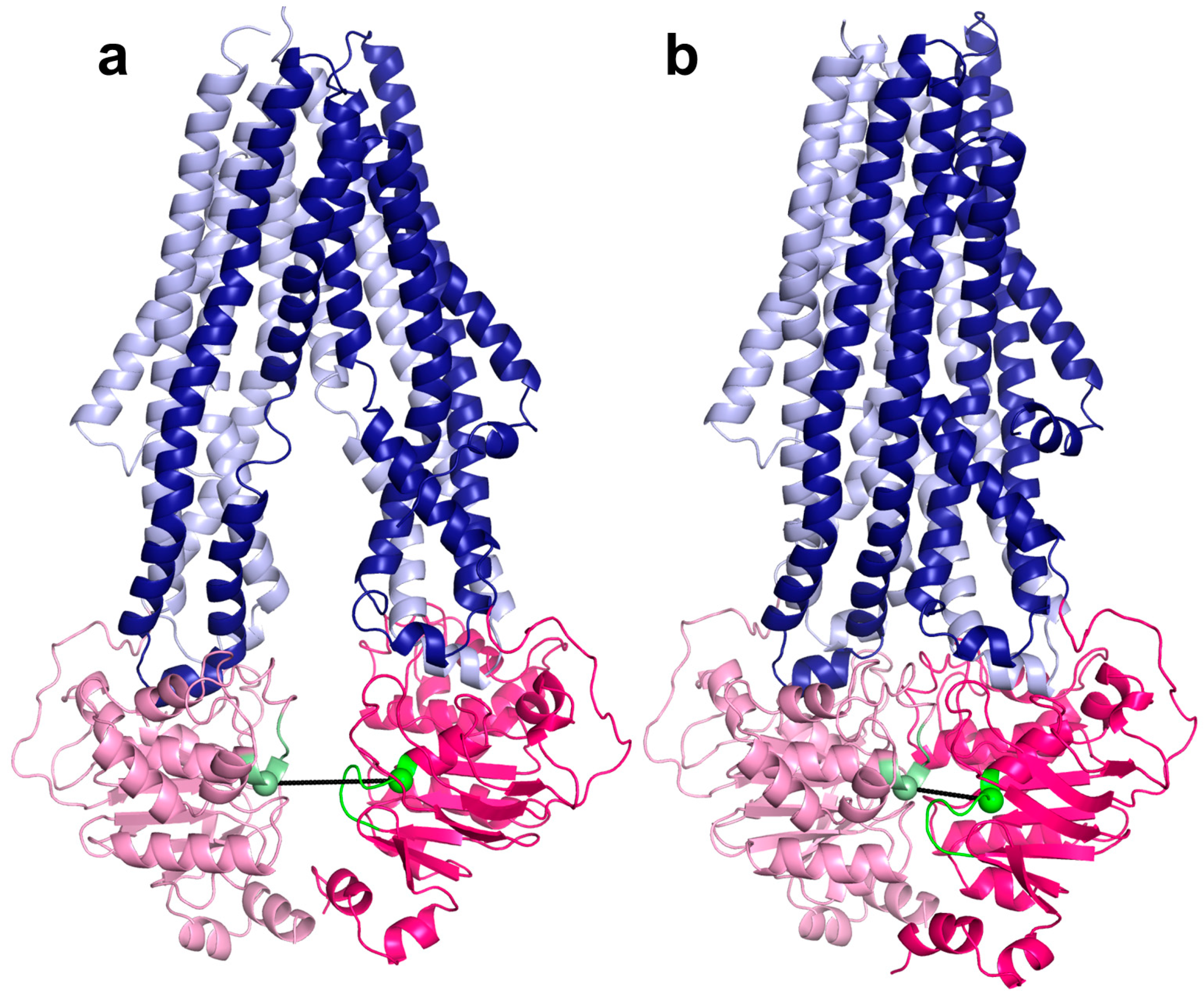
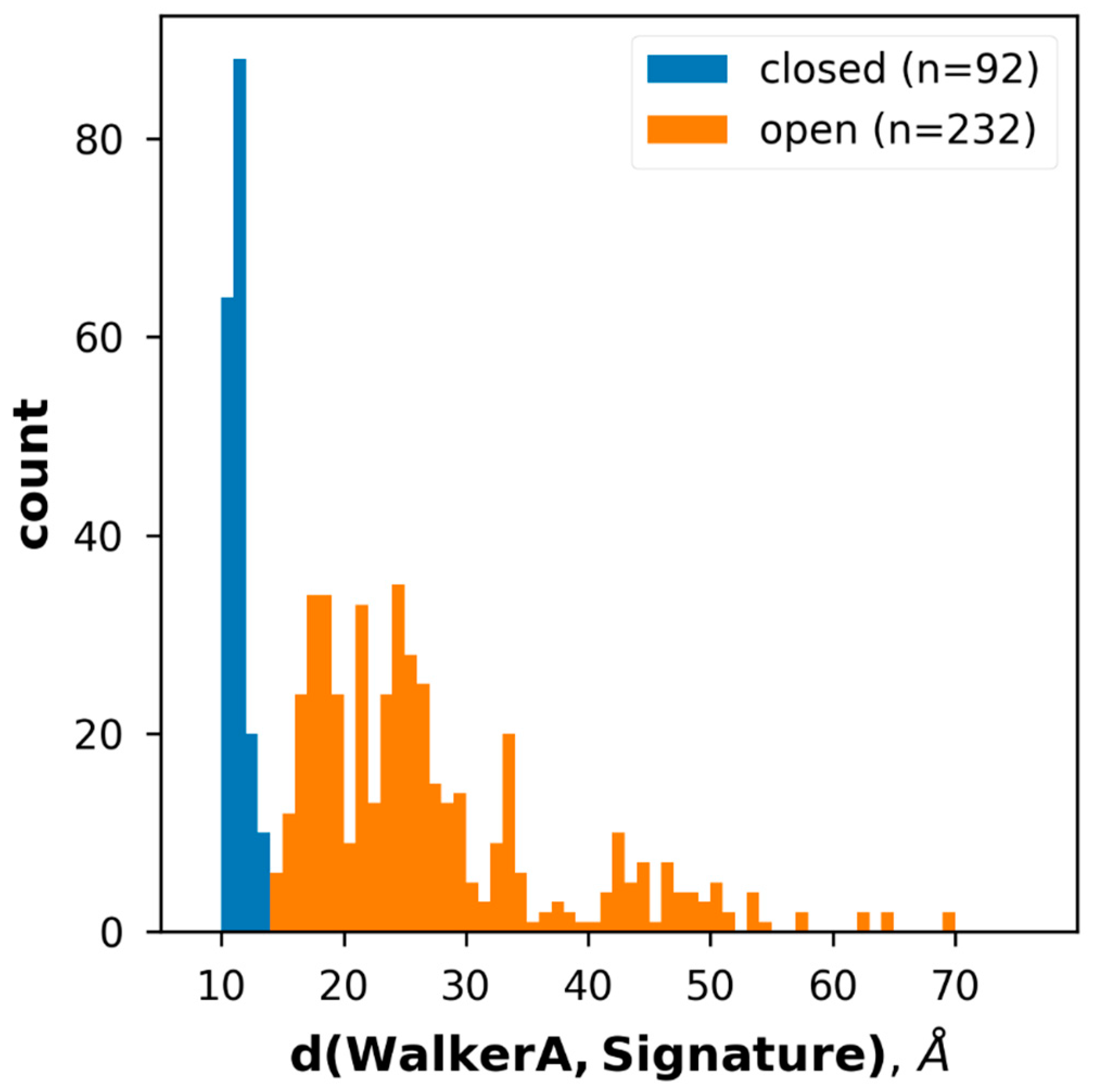
2.2. Conformational States of ABC Protein Structures
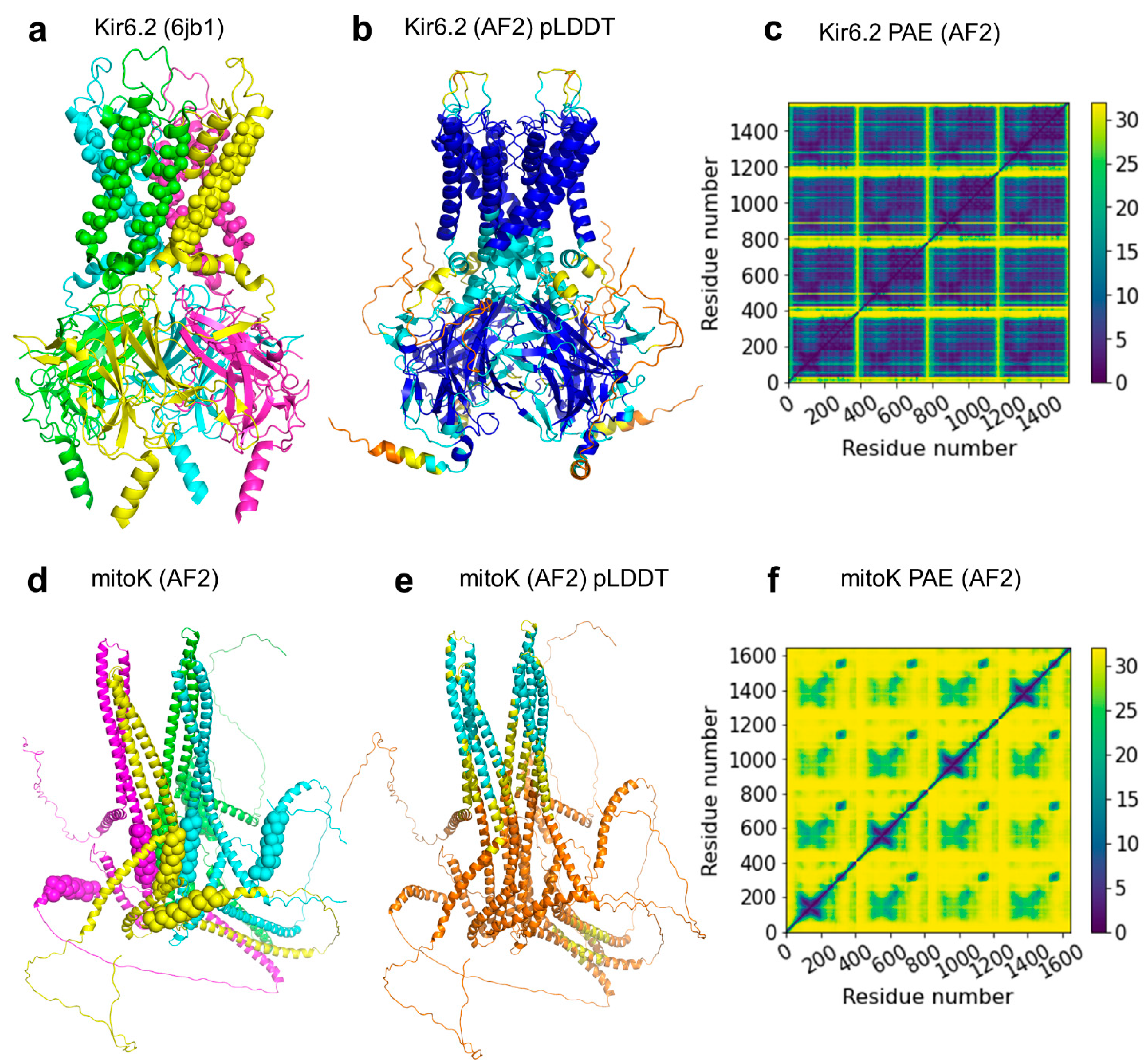
2.3. ABC Protein Structures Predicted by AlphaFold2
2.4. The ABC3D Database
2.5. Novel ABC Structures Accessible through the 3D-Beacons Network
3. Discussion
4. Methods and Materials
4.1. Databases and Associated Software
4.2. Identification and Classification of TM ABC Proteins
4.3. A Pipeline for Automatic Updates
4.4. Web Applications
4.5. Running AlphaFold2
4.6. Data Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- George, A.M. ABC Transporters—40 Years on; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-23476-2. [Google Scholar]
- Szakács, G.; Váradi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The Role of ABC Transporters in Drug Absorption, Distribution, Metabolism, Excretion and Toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, Gating, and Regulation of the Cftr Anion Channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Váradi, A.; van de Wetering, K. PXE, a Mysterious Inborn Error Clarified. Trends Biochem. Sci. 2019, 44, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR Biology toward Combinatorial Pharmacotherapy: Expanded Classification of Cystic Fibrosis Mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkadi, B.; Homolya, L.; Hegedűs, T. The ABCG2/BCRP transporter and its variants—From structure to pathology. FEBS Lett. 2020, 594, 4012–4034. [Google Scholar] [CrossRef]
- Liu, X. Transporter-Mediated Drug-Drug Interactions and Their Significance. Adv. Exp. Med. Biol. 2019, 1141, 241–291. [Google Scholar] [CrossRef]
- Locher, K.P. Mechanistic Diversity in ATP-Binding Cassette (ABC) Transporters. Nat. Struct. Mol. Biol. 2016, 23, 487–493. [Google Scholar] [CrossRef] [Green Version]
- Srikant, S.; Gaudet, R. Mechanics and Pharmacology of Substrate Selection and Transport by Eukaryotic ABC Exporters. Nat. Struct. Mol. Biol. 2019, 26, 792–801. [Google Scholar] [CrossRef]
- Lewinson, O.; Orelle, C.; Seeger, M.A. Structures of ABC Transporters: Handle with Care. FEBS Lett. 2020, 594, 3799–3814. [Google Scholar] [CrossRef]
- Moraes, I.; Evans, G.; Sanchez-Weatherby, J.; Newstead, S.; Stewart, P.D.S. Membrane Protein Structure Determination—The next Generation. Biochim. Biophys. Acta 2014, 1838, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Varga, J.K.; E Tusnády, G. The TMCrys server for supporting crystallization of transmembrane proteins. Bioinformatics 2019, 35, 4203–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, S.C.; Ando, N. X-rays in the Cryo-Electron Microscopy Era: Structural Biology’s Dynamic Future. Biochemistry 2018, 57, 277–285. [Google Scholar] [CrossRef]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Gavira, J.A. Introduction to Protein Crystallization. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.J.; Trieber, C.; Overduin, M. Structural Biology of Endogenous Membrane Protein Assemblies in Native Nanodiscs. Curr. Opin. Struct. Biol. 2021, 69, 70–77. [Google Scholar] [CrossRef]
- Farkas, B.; Csizmadia, G.; Katona, E.; Tusnády, G.E.; Hegedűs, T. MemBlob Database and Server for Identifying Transmembrane Regions Using Cryo-EM Maps. Bioinformatics 2020, 36, 2595–2598. [Google Scholar] [CrossRef]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-accuracy protein structure prediction in CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1687–1699. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Hegedűs, T.; Geisler, M.; Lukács, G.L.; Farkas, B. Ins and Outs of AlphaFold2 Transmembrane Protein Structure Predictions. Cell. Mol. Life Sci. 2022, 79, 73. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, I.R.; Pei, J.; Baek, M.; Krishnakumar, A.; Anishchenko, I.; Ovchinnikov, S.; Zhang, J.; Ness, T.J.; Banjade, S.; Bagde, S.R.; et al. Computed structures of core eukaryotic protein complexes. Science 2021, 374, eabm4805. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The TrRosetta Server for Fast and Accurate Protein Structure Prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Duong Van Hoa, F.; Ekiert, D.; et al. Structural and Functional Diversity Calls for a New Classification of ABC Transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef]
- Decottignies, A.; Goffeau, A. Complete Inventory of the Yeast ABC Proteins. Nat. Genet. 1997, 15, 137–145. [Google Scholar] [CrossRef]
- Klein, I.; Sarkadi, B.; Váradi, A. An Inventory of the Human ABC Proteins. Biochim. Biophys. Acta BBA—Biomembr. 1999, 1461, 237–262. [Google Scholar] [CrossRef] [Green Version]
- Frank, G.A.; Shukla, S.; Rao, P.; Borgnia, M.J.; Bartesaghi, A.; Merk, A.; Mobin, A.; Esser, L.; Earl, L.A.; Gottesman, M.M.; et al. Cryo-EM Analysis of the Conformational Landscape of Human P-Glycoprotein (ABCB1) During Its Catalytic Cycle. Mol. Pharmacol. 2016, 90, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Beis, K. Structural Basis for the Mechanism of ABC Transporters. Biochem. Soc. Trans. 2015, 43, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Prescher, M.; Bonus, M.; Stindt, J.; Keitel-Anselmino, V.; Smits, S.H.; Gohlke, H.; Schmitt, L. Evidence for a credit-card-swipe mechanism in the human PC floppase ABCB4. Structure 2021, 29, 1144–1155. [Google Scholar] [CrossRef]
- Ford, R.C.; Beis, K. Learning the ABCs One at a Time: Structure and Mechanism of ABC Transporters. Biochem. Soc. Trans. 2019, 47, 23–36. [Google Scholar] [CrossRef]
- DALI and the Persistence of Protein Shape—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31606894/ (accessed on 2 June 2022).
- Fox, N.K.; Brenner, S.E.; Chandonia, J.-M. SCOPe: Structural Classification of Proteins—Extended, Integrating SCOP and ASTRAL Data and Classification of New Structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.W.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xu, D.; Feng, Z.; Hou, W.-T.; Jiang, Y.-L.; Wang, L.; Sun, L.; Zhou, C.-Z.; Chen, Y. Cryo-EM Structure of Human Lysosomal Cobalamin Exporter ABCD4. Cell Res. 2019, 29, 1039–1041. [Google Scholar] [CrossRef]
- Wang, R.; Qin, Y.; Li, X. Structural Basis of Acyl-CoA Transport across the Peroxisomal Membrane by Human ABCD1. Cell Res. 2022, 32, 214–217. [Google Scholar] [CrossRef]
- Rempel, S.; Gati, C.; Nijland, M.; Thangaratnarajah, C.; Karyolaimos, A.; de Gier, J.W.; Guskov, A.; Slotboom, D.J. A Mycobacterial ABC Transporter Mediates the Uptake of Hydrophilic Compounds. Nature 2020, 580, 409–412. [Google Scholar] [CrossRef]
- Ghilarov, D.; Inaba-Inoue, S.; Stepien, P.; Qu, F.; Michalczyk, E.; Pakosz, Z.; Nomura, N.; Ogasawara, S.; Walker, G.C.; Rebuffat, S.; et al. Structural Insight into the Staphylococcus Aureus ATP-Driven Exporter of Virulent Peptide Toxins. Sci. Adv. 2021, 7, eabj5363. [Google Scholar] [CrossRef]
- George, N.L.; Schilmiller, A.L.; Orlando, B.J. Conformational Snapshots of the Bacitracin Sensing and Resistance Transporter BceAB. Proc. Natl. Acad. Sci. USA 2022, 119, e2123268119. [Google Scholar] [CrossRef]
- Mann, D.; Fan, J.; Somboon, K.; Farrell, D.P.; Muenks, A.; Tzokov, S.B.; DiMaio, F.; Khalid, S.; Miller, S.I.; Bergeron, J.R.C. Structure and Lipid Dynamics in the Maintenance of Lipid Asymmetry Inner Membrane Complex of A. Baumannii. Commun. Biol. 2021, 4, 817. [Google Scholar] [CrossRef]
- Yu, Q.; Ni, D.; Kowal, J.; Manolaridis, I.; Jackson, S.M.; Stahlberg, H.; Locher, K.P. Structures of ABCG2 under Turnover Conditions Reveal a Key Step in the Drug Transport Mechanism. Nat. Commun. 2021, 12, 4376. [Google Scholar] [CrossRef] [PubMed]
- Csizmadia, G.; Farkas, B.; Spagina, Z.; Tordai, H.; Hegedűs, T. Quantitative Comparison of ABC Membrane Protein Type I Exporter Structures in a Standardized Way. Comput. Struct. Biotechnol. J. 2018, 16, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Szabó, K.; Szakács, G.; Hegeds, T.; Sarkadi, B. Nucleotide Occlusion in the Human Cystic Fibrosis Transmembrane Conductance Regulator. Different Patterns in the Two Nucleotide Binding Domains. J. Biol. Chem. 1999, 274, 12209–12212. [Google Scholar] [CrossRef] [Green Version]
- Yeh, H.-I.; Yu, Y.-C.; Kuo, P.-L.; Tsai, C.-K.; Huang, H.-T.; Hwang, T.-C. Functional Stability of CFTR Depends on Tight Binding of ATP at Its Degenerate ATP-Binding Site. J. Physiol. 2021, 599, 4625–4642. [Google Scholar] [CrossRef] [PubMed]
- Nagy, T.; Tóth, Á.; Telbisz, Á.; Sarkadi, B.; Tordai, H.; Tordai, A.; Hegedűs, T. The transport pathway in the ABCG2 protein and its regulation revealed by molecular dynamics simulations. Cell. Mol. Life Sci. 2020, 78, 2329–2339. [Google Scholar] [CrossRef] [PubMed]
- Stockner, T.; de Vries, S.J.; Bonvin, A.M.J.J.; Ecker, G.F.; Chiba, P. Data-Driven Homology Modelling of P-Glycoprotein in the ATP-Bound State Indicates Flexibility of the Transmembrane Domains. FEBS J. 2009, 276, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Liao, M. ABCG2 Transports Anticancer Drugs via a Closed-to-Open Switch. Nat. Commun. 2020, 11, 2264. [Google Scholar] [CrossRef]
- Zhang, M.; Bao, Z.; Zhao, Q.; Guo, H.; Xu, K.; Wang, C.; Zhang, P. Structure of a Pantothenate Transporter and Implications for ECF Module Sharing and Energy Coupling of Group II ECF Transporters. Proc. Natl. Acad. Sci. USA 2014, 111, 18560–18565. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Qi, X.; Hong, S.; Xu, K.; He, F.; Zhang, M.; Chen, J.; Chao, D.; Zhao, W.; Li, D.; et al. Structure and Mechanism of a Group-I Cobalt Energy Coupling Factor Transporter. Cell Res. 2017, 27, 675–687. [Google Scholar] [CrossRef] [Green Version]
- Rempel, S.; Stanek, W.K.; Slotboom, D.J. ECF-Type ATP-Binding Cassette Transporters. Annu. Rev. Biochem. 2019, 88, 551–576. [Google Scholar] [CrossRef]
- Setyawati, I.; Stanek, W.K.; Majsnerowska, M.; Swier, L.J.Y.M.; Pardon, E.; Steyaert, J.; Guskov, A.; Slotboom, D.J. In Vitro Reconstitution of Dynamically Interacting Integral Membrane Subunits of Energy-Coupling Factor Transporters. eLife 2020, 9, e64389. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.; Muñoz, A.; Zhang, X.; Düfer, M.; Drews, G.; Krippeit-Drews, P.; Aguilar-Bryan, L. ABCC8 and ABCC9: ABC Transporters That Regulate K+ Channels. Pflügers Arch. -Eur. J. Physiol. 2007, 453, 703–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.-X.; Ding, D.; Wang, M.; Kang, Y.; Zeng, X.; Chen, L. Ligand Binding and Conformational Changes of SUR1 Subunit in Pancreatic ATP-Sensitive Potassium Channels. Protein Cell 2018, 9, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-Sensitive Potassium Channel in Mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Heginbotham, L.; Lu, Z.; Abramson, T.; MacKinnon, R. Mutations in the K+ Channel Signature Sequence. Biophys. J. 1994, 66, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Doyle, D.A.; Morais Cabral, J.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. Is the Emperor Wearing Shorts? The Published Structures of ABC Transporters. FEBS Lett. 2020, 594, 3790–3798. [Google Scholar] [CrossRef]
- Neumann, J.; Rose-Sperling, D.; Hellmich, U.A. Diverse Relations between ABC Transporters and Lipids: An Overview. Biochim. Biophys. Acta BBA—Biomembr. 2017, 1859, 605–618. [Google Scholar] [CrossRef]
- Farkas, B.; Tordai, H.; Padányi, R.; Tordai, A.; Gera, J.; Paragi, G.; Hegedűs, T. Discovering the chloride pathway in the CFTR channel. Cell. Mol. Life Sci. 2019, 77, 765–778. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Grudinin, S. NOLB: Nonlinear Rigid Block Normal-Mode Analysis Method. J. Chem. Theory Comput. 2017, 13, 2123–2134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased scale and scope after 10 years of protein dynamics modelling with Python. Bioinformatics 2021, 37, 3657–3659. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhou, X.; Wuyun, Q.; Pearce, R.; Li, Y.; Zhang, Y. FUpred: Detecting Protein Domains through Deep-Learning-Based Contact Map Prediction. Bioinformatics 2020, 36, 3749–3757. [Google Scholar] [CrossRef] [PubMed]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching the AlphaFold Models with Ligands and Co-Factors. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bagdonas, H.; Fogarty, C.A.; Fadda, E.; Agirre, J. The Case for Post-Predictional Modifications in the AlphaFold Protein Structure Database. Nat. Struct. Mol. Biol. 2021, 28, 869–870. [Google Scholar] [CrossRef]
- Gyimesi, G.; Borsodi, D.; Sarankó, H.; Tordai, H.; Sarkadi, B.; Hegedűs, T. ABCMdb: A Database for the Comparative Analysis of Protein Mutations in ABC Transporters, and a Potential Framework for a General Application. Hum. Mutat. 2012, 33, 1547–1556. [Google Scholar] [CrossRef]
- Tordai, H.; Jakab, K.; Gyimesi, G.; András, K.; Brózik, A.; Sarkadi, B.; Hegedus, T. ABCMdb Reloaded: Updates on Mutations in ATP Binding Cassette Proteins. Database 2017, 2017, bax023. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-Align: A Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- Wojdyr, M. GEMMI: A Library for Structural Biology. J. Open Source Softw. 2022, 7, 4200. [Google Scholar] [CrossRef]
- Bayer, M. SQLAlchemy. In The Architecture of Open Source Applications Volume II: Structure, Scale, and a Few More Fearless Hacks; Brown, A., Wilson, G., Eds.; 2012; Available online: https://aosabook.org (accessed on 10 March 2022).
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tordai, H.; Suhajda, E.; Sillitoe, I.; Nair, S.; Varadi, M.; Hegedus, T. Comprehensive Collection and Prediction of ABC Transmembrane Protein Structures in the AI Era of Structural Biology. Int. J. Mol. Sci. 2022, 23, 8877. https://doi.org/10.3390/ijms23168877
Tordai H, Suhajda E, Sillitoe I, Nair S, Varadi M, Hegedus T. Comprehensive Collection and Prediction of ABC Transmembrane Protein Structures in the AI Era of Structural Biology. International Journal of Molecular Sciences. 2022; 23(16):8877. https://doi.org/10.3390/ijms23168877
Chicago/Turabian StyleTordai, Hedvig, Erzsebet Suhajda, Ian Sillitoe, Sreenath Nair, Mihaly Varadi, and Tamas Hegedus. 2022. "Comprehensive Collection and Prediction of ABC Transmembrane Protein Structures in the AI Era of Structural Biology" International Journal of Molecular Sciences 23, no. 16: 8877. https://doi.org/10.3390/ijms23168877
APA StyleTordai, H., Suhajda, E., Sillitoe, I., Nair, S., Varadi, M., & Hegedus, T. (2022). Comprehensive Collection and Prediction of ABC Transmembrane Protein Structures in the AI Era of Structural Biology. International Journal of Molecular Sciences, 23(16), 8877. https://doi.org/10.3390/ijms23168877