
5-Arylidenerhodanines as P-gp Modulators: An Interesting Effect of the Carboxyl Group on ABCB1 Function in Multidrug-Resistant Cancer Cells

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Biological Screening
2.2.1. The Rhodamine 123 Accumulation Assay
2.2.2. Cytotoxicity and Antiproliferative Assays
2.2.3. Drug Combination Assay
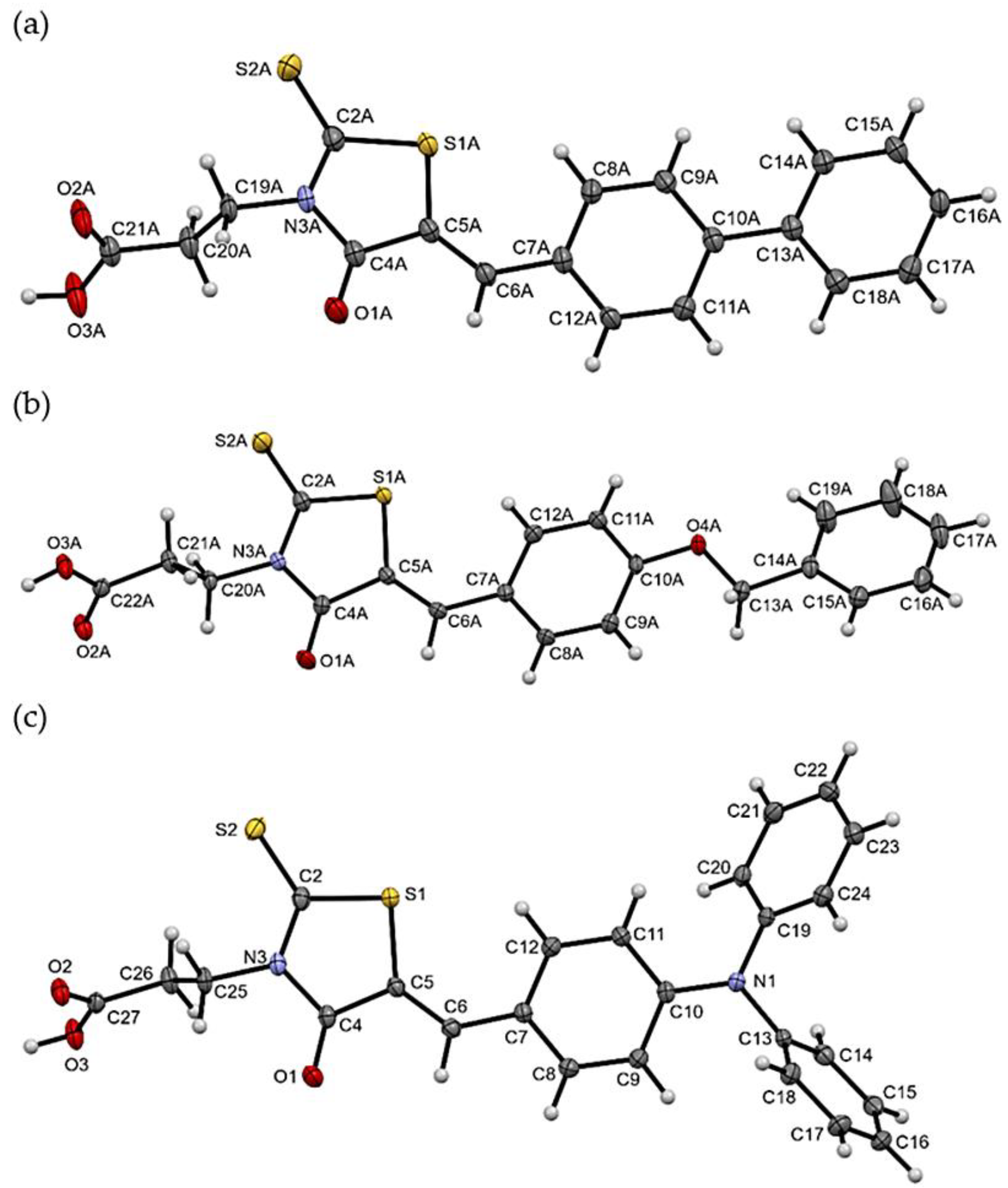
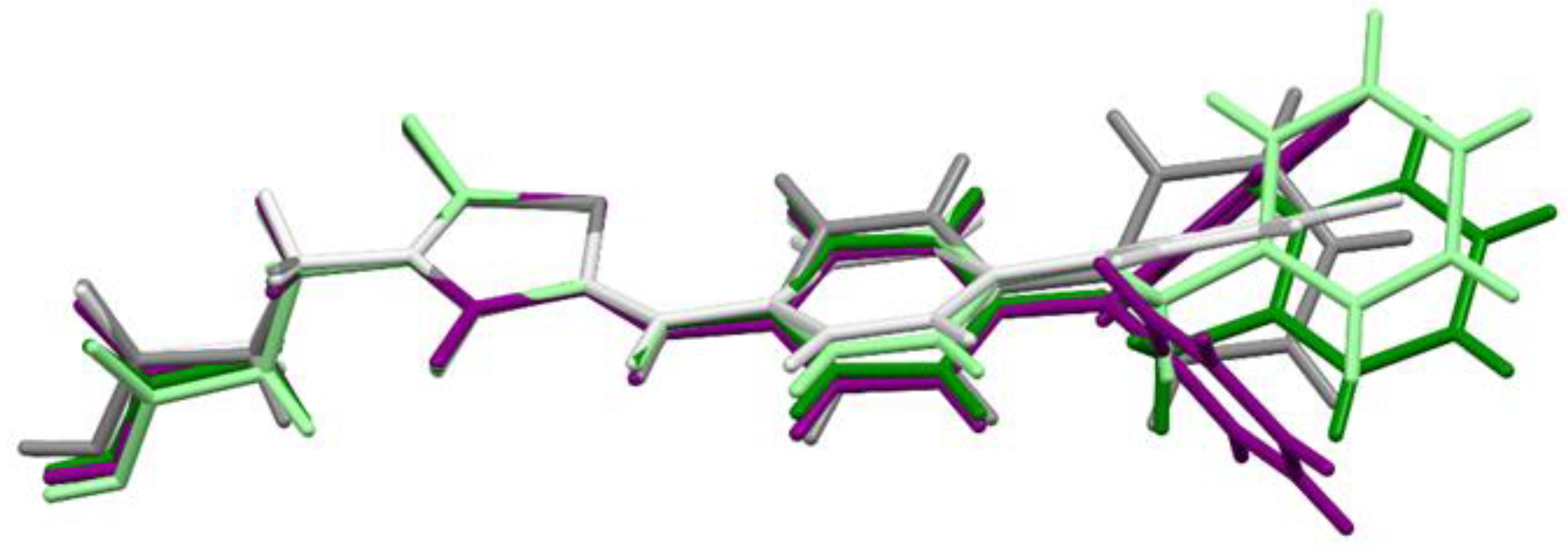
2.3. X-ray Studies of Compounds 3, 7, and 11
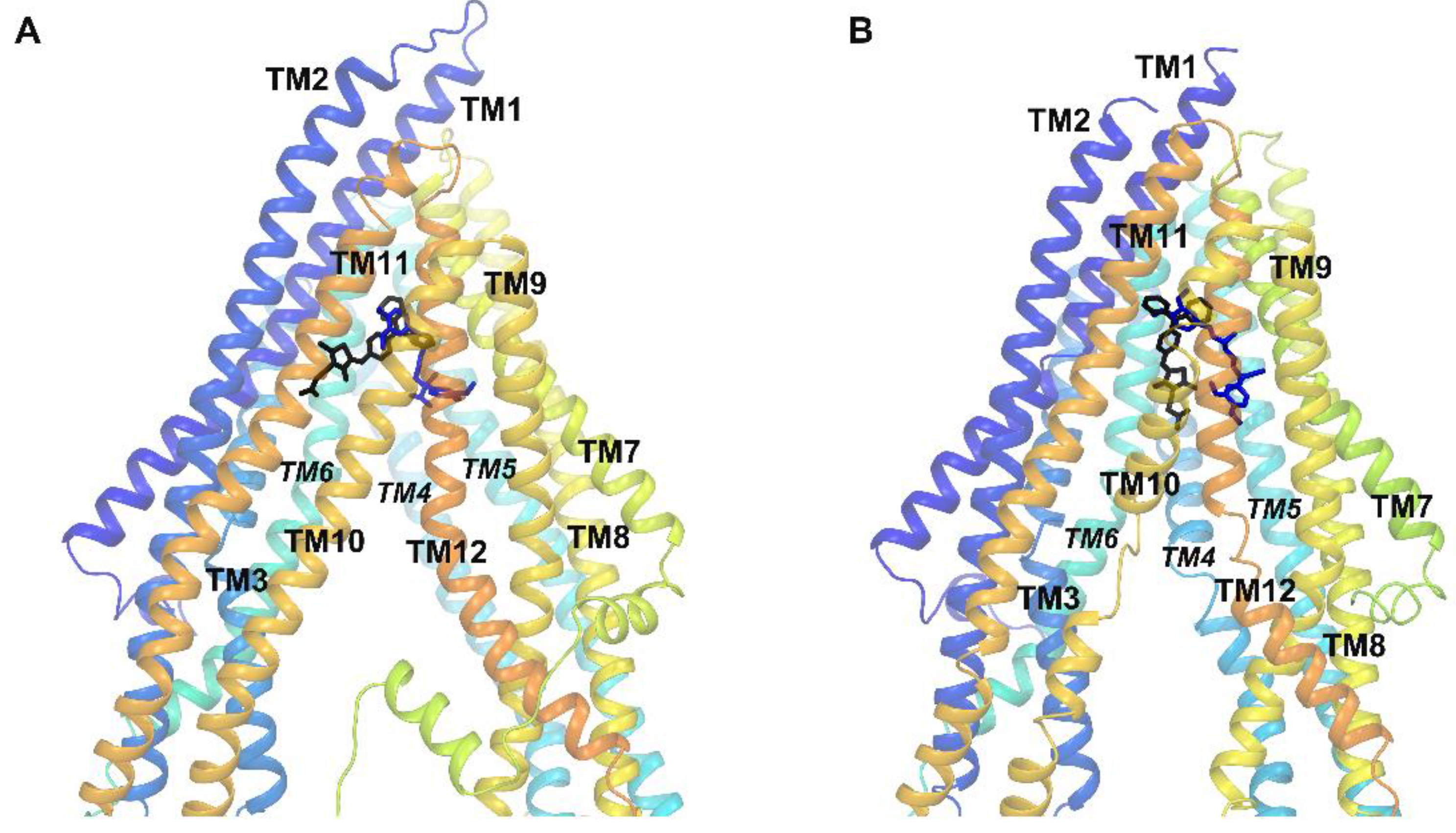
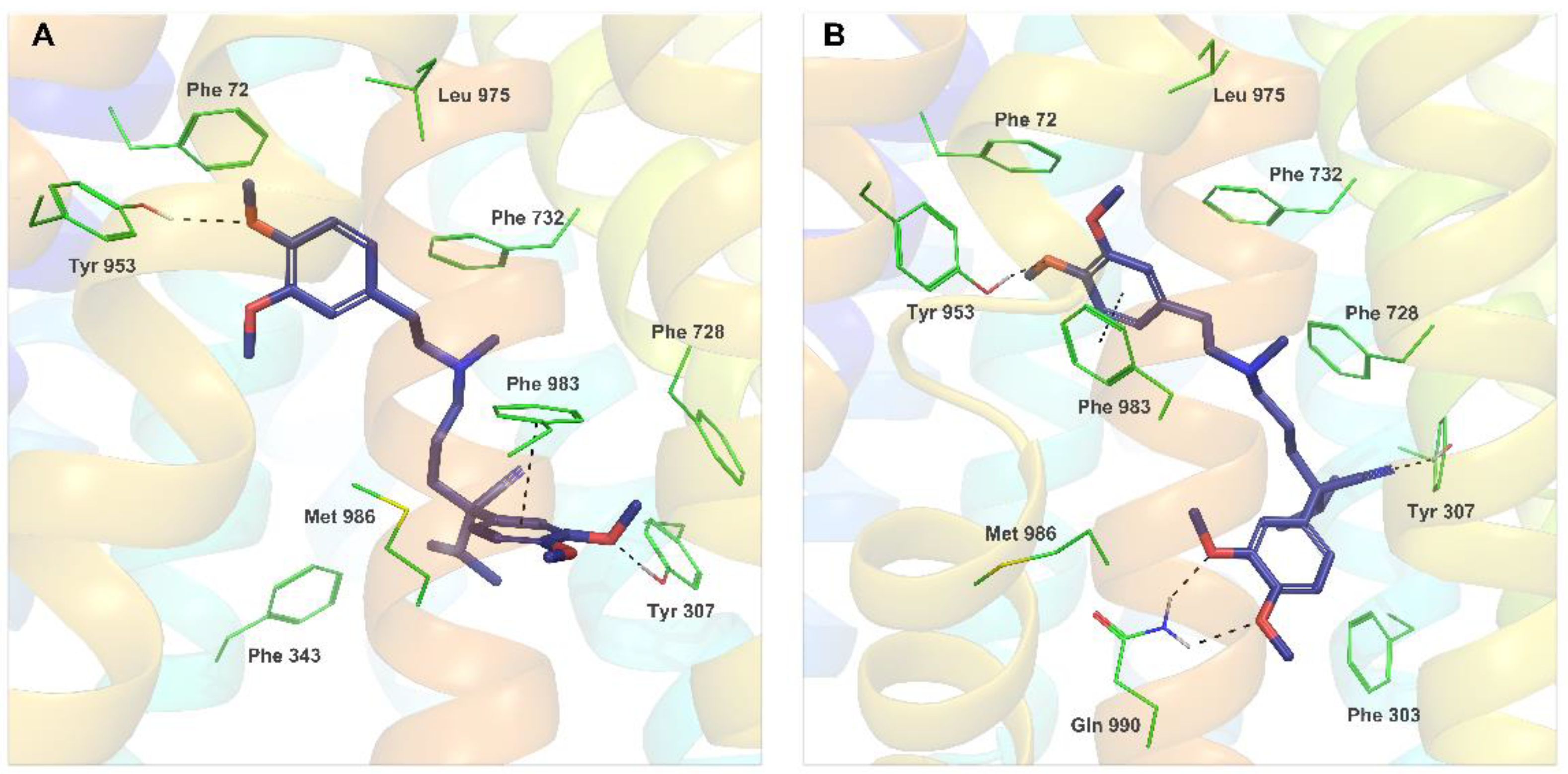
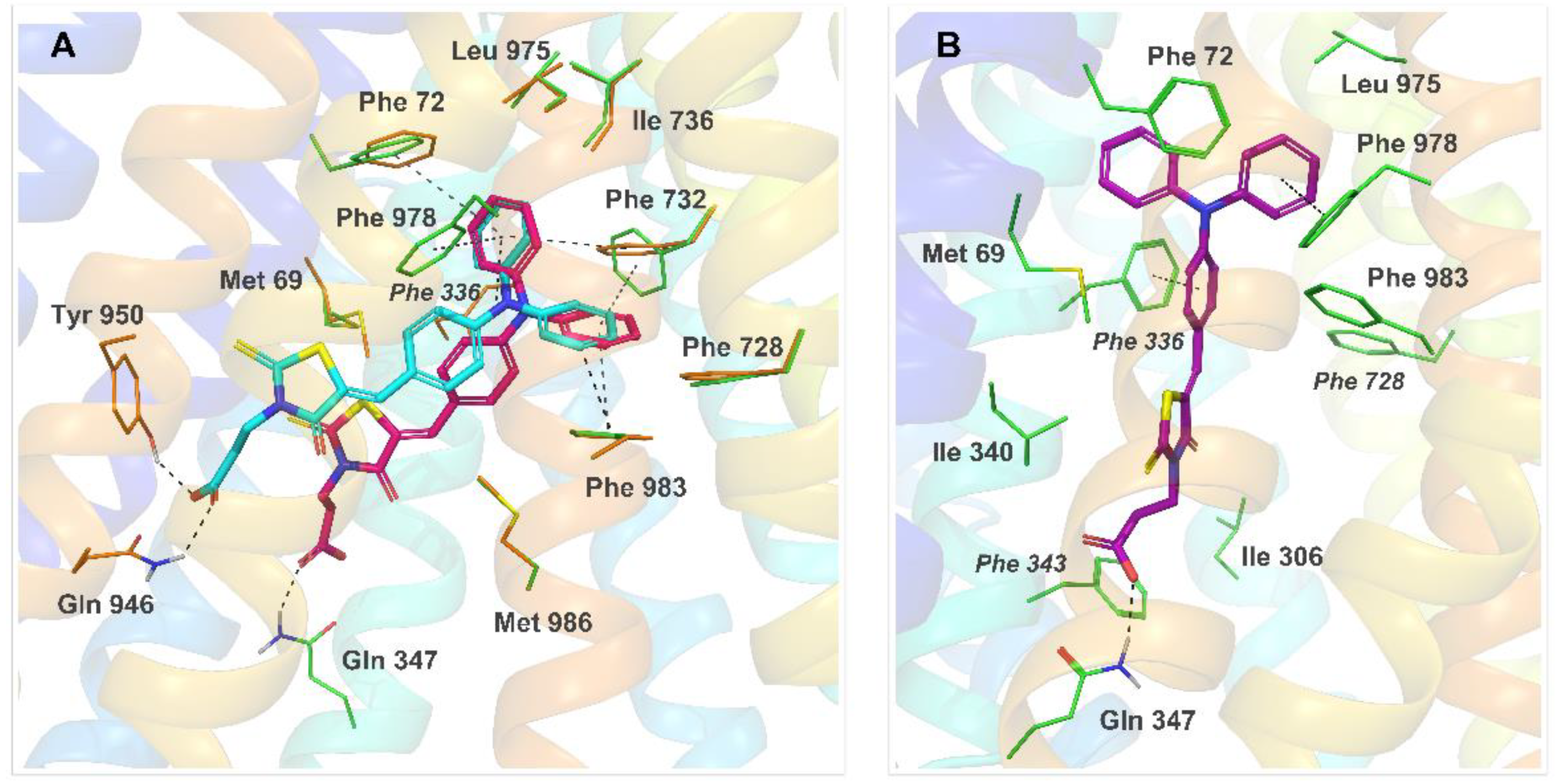
2.4. Molecular Modeling
2.5. Lipophilicity
3. Materials and Methods
3.1. Chemistry
3.1.1. General Method for the Synthesis of Compounds 1–8 and 10–12 [24]
3.1.2. Preparation of Compound 9 [44]
3.2. Biological Assays
3.2.1. Cell Lines
3.2.2. Modulation of P-gp: Rhodamine 123 Accumulation Assay
3.2.3. Cytotoxicity and Antiproliferative Assays
3.2.4. Drug Combination Assay
3.3. Crystallographic Study
Crystallographic Data
3.4. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nobili, S.; Landini, I.; Giglioni, B.; Mini, E. Pharmacological strategies for overcoming multidrug resistance. Curr. Drug Targets 2006, 7, 861–879. [Google Scholar] [CrossRef]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef]
- Park, S.H.; Park, C.J.; Kim, D.Y.; Lee, B.R.; Kim, Y.J.; Cho, Y.U.; Jang, S. MRP1 and P-glycoprotein expression assays would be useful in the additional detection of treatment non-responders in CML patients without ABL1 mutation. Leuk. Res. 2015, 39, 1109–1116. [Google Scholar] [CrossRef]
- Slot, A.J.; Molinski, S.V.; Cole, S.P. Mammalian multidrug-resistance proteins (MRPs). Essays Biochem. 2011, 50, 179–207. [Google Scholar] [CrossRef]
- Gatti, L.; Beretta, G.L.; Cossa, G.; Zunino, F.; Perego, P. ABC transporters as potential targets for modulation of drug resistance. Mini Rev. Med. Chem. 2009, 9, 1102–1112. [Google Scholar] [CrossRef]
- Szakacs, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles heel of multidrug-resistant cancer by exploiting the fitness cost of resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef]
- Lai, J.-I.; Tseng, Y.-J.; Chen, M.-H.; Huang, C.-Y.F.; Chang, P.M.-H. Clinical perspective of FDA approved drugs with P-glycoprotein inhibition activities for potential cancer therapeutics. Front. Oncol. 2020, 10, 561936. [Google Scholar] [CrossRef]
- Seelig, A. P-glycoprotein: One mechanism, many tasks and the consequences for pharmacotherapy of cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011–2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef]
- Dong, J.; Qin, Z.; Zhang, W.-D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.-S.; Cheng, X.-D.; Qin, J.-J. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: An update. Drug Resist. Updat. 2020, 49, 100681. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef]
- Maddila, S.; Gorle, S.; Jonnalagadda, S.B. Drug screening of rhodanine derivatives for antibacterial activity. Expert Opin. Drug Discov. 2020, 15, 203–229. [Google Scholar] [CrossRef]
- Yin, L.J.; bin Ahmad Kamar, A.K.D.; Fung, G.T.; Liang, C.T.; Avupati, V.R. Review of anticancer potentials and structure-activity relationships (SAR) of rhodanine derivatives. Biomed. Pharmacother. 2022, 145, 112406. [Google Scholar] [CrossRef]
- Trotsko, N. Antitubercular properties of thiazolidin-4-ones-A review. Eur. J. Med. Chem. 2021, 215, 113266. [Google Scholar] [CrossRef]
- Mousavi, S.M.; Zarei, M.; Hashemi, S.A.; Babapoor, A.; Amani, A.M. A conceptual review of rhodanine: Current applications of antiviral drugs, anticancer and antimicrobial activities. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1132–1148. [Google Scholar] [CrossRef]
- Spengler, G.; Evaristo, M.; Handzlik, J.; Serly, J.; Molnar, J.; Viveiros, M.; Kiec-Kononowicz, K.; Amaral, L. Biological activity of hydantoin derivatives on P-glycoprotein (ABCB1) of mouse lymphoma cells. Anticancer. Res. 2010, 30, 4867–4871. [Google Scholar]
- Spengler, G.; Handzlik, J.; Ocsovszki, I.; Viveiros, M.; Kiec-Kononowicz, K.; Molnar, J.; Amaral, L. Modulation of multidrug efflux pump activity by new hydantoin derivatives on colon adenocarcinoma cells without inducing apoptosis. Anticancer. Res. 2011, 31, 3285–3288. [Google Scholar]
- Martins, A.; Dymek, A.; Handzlik, J.; Spengler, G.; Armada, A.; Molnar, J.; Kiec-Kononowicz, K.; Amaral, L. Activity of fourteen new hydantoin compounds on the human ABCB1 efflux pump. Vivo 2012, 26, 293–297. [Google Scholar]
- Zeslawska, E.; Kincses, A.; Spengler, G.; Nitek, W.; Wyrzuc, K.; Kiec-Kononowicz, K.; Handzlik, J. The 5-aromatic hydantoin-3-acetate derivatives as inhibitors of the tumour multidrug resistance efflux pump P-glycoprotein (ABCB1): Synthesis, crystallographic and biological studies. Bioorg. Med. Chem. 2016, 24, 2815–2822. [Google Scholar] [CrossRef]
- Zeslawska, E.; Kincses, A.; Spengler, G.; Nitek, W.; Tejchman, W.; Handzlik, J. Pharmacophoric features for a very potent 5-spirofluorenehydantoin inhibitor of cancer efflux pump ABCB1, based on X-ray analysis. Chem. Biol. Drug Des. 2019, 93, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Zeslawska, E.; Kucwaj-Brysz, K.; Kincses, A.; Spengler, G.; Szymanska, E.; Czopek, A.; Marc, M.A.; Kaczor, A.; Nitek, W.; Dominguez-Alvarez, E.; et al. An insight into the structure of 5-spiro aromatic derivatives of imidazolidine-2,4-dione, a new group of very potent inhibitors of tumor multidrug resistance in T-lymphoma cells. Bioorg. Chem. 2021, 109, 104735. [Google Scholar] [CrossRef] [PubMed]
- Zeslawska, E.; Szymanska, E.; Nitek, W.; Handzlik, J. Crystallographic studies of piperazine derivatives of 3-methyl-5-spirofluorenehydantoin in search of structural features of P-gp inhibitors. Acta Crystallogr. C Struct. Chem. 2021, 77, 467–478. [Google Scholar] [CrossRef]
- Tejchman, W.; Korona-Glowniak, I.; Malm, A.; Zylewski, M.; Suder, P. Antibacterial properties of 5-substituted derivatives of rhodanine-3-carboxyalkyl acids. Med. Chem. Res. 2017, 26, 1316–1324. [Google Scholar] [CrossRef]
- Nencki, M. Ueber die Einwirkung der Monochloressigsäure auf Sulfocyansäure und ihre Salze. J. Prakt. Chem. Chem. 1877, 16, 1–17. [Google Scholar] [CrossRef]
- Körner, H. Über einige Derivate der Dithiocarbamino-essigsäure. Ber. Dtsch. Chem. Ges. 1908, 41, 1901–1905. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-4: LigPrep; Epik; Protein Preparation Wizard. Macromodel; Glide; Prime; MM-GBSA; Schrödinger, LLC: New York, NY, USA, 2021.
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Cryst. D Biol. Cryst. 2015, 71, 732–741. [Google Scholar] [CrossRef]
- Nicklisch, S.C.; Rees, S.D.; McGrath, A.P.; Gökirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Robey, R.; Gottesman, M.; Ambudkar, S. Multidrug transporters: Recent insights from cryo-electron microscopy-derived atomic structures and animal models. F1000Research 2020, 9, 17. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.N.; Smolinski, M.P. Discovery and characterization of potent dual P-glycoprotein and CYP3A4 inhibitors: Design, synthesis, cryo-EM analysis, and biological evaluations. J. Med. Chem. 2022, 65, 191–216. [Google Scholar] [CrossRef]
- Kaczor, A.; Nove, M.; Kincses, A.; Spengler, G.; Szymanska, E.; Latacz, G.; Handzlik, J. Search for ABCB1 modulators among 2-amine-5-arylideneimidazolones as a new perspective to overcome cancer multidrug resistance. Molecules 2020, 25, 2258. [Google Scholar] [CrossRef]
- Pimthon, J.; Dechaanontasup, R.; Ratanapiphop, C.; Phromprasert, C. Homology modeling and substrate binding studies of human P-glycoprotein. Pharm. Sci. Asia 2017, 44, 96–107. [Google Scholar] [CrossRef]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef]
- Prajapati, R.; Singh, U.; Patil, A.; Khomane, K.S.; Bagul, P.; Bansal, A.K.; Sangamwar, A.T. In silico model for P-glycoprotein substrate prediction: Insights from molecular dynamics and in vitro studies. J. Comput. Aided. Mol. Des. 2013, 27, 347–363. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J. Biol. Chem. 2000, 275, 39272–39278. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem. 2001, 276, 14972–14979. [Google Scholar] [CrossRef]
- Wang, R.B.; Kuo, C.L.; Lien, L.L.; Lien, E.J. Structure-activity relationship: Analyses of p-glycoprotein substrates and inhibitors. J. Clin. Pharm. Ther. 2003, 28, 203–228. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef]
- Esswein, A.; Schaefer, W.; Tsaklakidis, C.; Honold, K.; Kaluza, K. Thiazolidine derivatives for the treatment and prevention of metabolic bone disorders. Patent Application No. 63310/99, 30 January 1999. [Google Scholar]
- Smith, T.K.; Young, B.L.; Denton, H.; Hughes, D.L.; Wagner, G.K. First small molecular inhibitors of T. brucei dolicholphosphate mannose synthase (DPMS), a validated drug target in African sleeping sickness. Bioorg. Med. Chem. Lett. 2009, 19, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, M.M.; Pastan, I.; Gottesman, M.M. Certain calcium channel blockers bind specifically to multidrug-resistant human KB carcinoma membrane vesicles and inhibit drug binding to P-glycoprotein. J. Biol. Chem. 1987, 262, 2166–2170. [Google Scholar] [CrossRef]
- Domínguez-Álvarez, E.; Gajdács, M.; Spengler, G.; Palop, J.A.; Marć, M.A.; Kieć-Kononowicz, K.; Amaral, L.; Molnár, J.; Jacob, C.; Handzlik, J.; et al. Identification of selenocompounds with promising properties to reverse cancer multidrug resistance. Bioorg. Med. Chem. Lett. 2016, 26, 2821–2824. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, D.S.P.; Szemerédi, N.; Spengler, G.; Mulhovo, S.; dos Santos, D.J.V.A.; Ferreira, M.-J.U. Exploring the monoterpene indole alkaloid scaffold for reversing P-glycoprotein-mediated multidrug resistance in cancer. Pharmaceuticals 2021, 14, 862. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar]
- The PyMOL Molecular Graphics System; Version 2.5.2; Schrödinger, LLC: New York, NY, USA, 2021.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
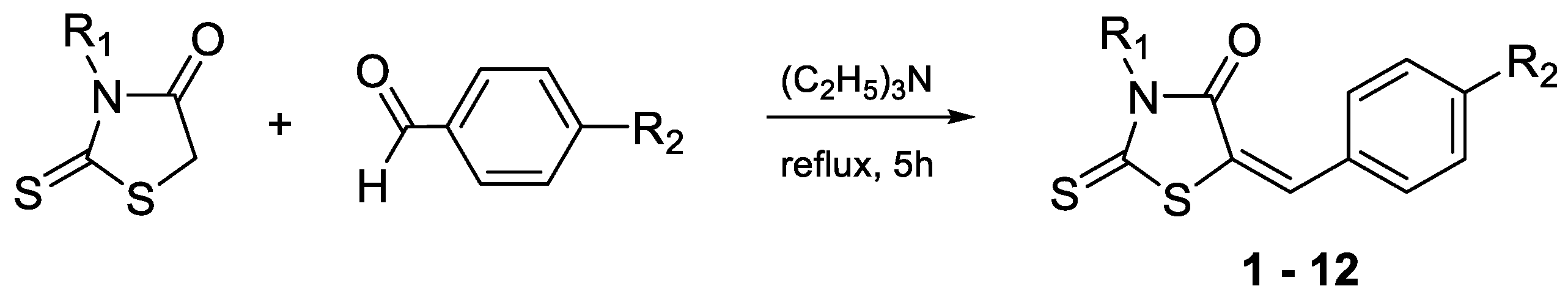{kind=link}

| Cmpd. | R1 | R2 |
|---|---|---|
| 1 | -H | -Ph |
| 2 | -CH2COOH | -Ph |
| 3 | -(CH2)2COOH | -Ph |
| 4 | -(CH2)3COOH | -Ph |
| 5 | -H | -OCH2Ph |
| 6 | -CH2COOH | -OCH2Ph |
| 7 | -(CH2)2COOH | -OCH2Ph |
| 8 | -(CH2)3COOH | -OCH2Ph |
| 9 | -H | -N(Ph)2 |
| 10 | -CH2COOH | -N(Ph)2 |
| 11 | -(CH2)2COOH | -N(Ph)2 |
| 12 | -(CH2)3COOH | -N(Ph)2 |
| Cmpd. | FAR 1 2 μM | FAR 1 20 μM |
|---|---|---|
| 1 | <1 | 12.704 |
| 2 | <1 | <1 |
| 3 | <1 | <1 |
| 4 | <1 | <1 |
| 5 | <1 | 15.366 |
| 6 | <1 | <1 |
| 7 | <1 | <1 |
| 8 | <1 | <1 |
| 9 | 21.658 | 32.184 |
| 10 | 27.501 | 75.107 |
| 11 | 35.245 | 76.226 |
| 12 | 35.547 | 78.491 |
| Verapamil | nd | 4.380 |
| IC50 ± SD (μM) | ||||
|---|---|---|---|---|
| Cmpd. | Cytotoxic Effect | Antiproliferative Effect | ||
| PAR | MDR | PAR | MDR | |
| 1 | 48.28 ± 1.54 | 56.09 ± 1.72 | 35.84 ± 0 | 39.26 ± 1.05 |
| 2 | >100 | >100 | 43.10 ± 0.66 | 55.17 ± 1.89 |
| 3 | 13.88 ± 0.49 | 17.02 ± 1.05 | 28.60 ± 0.72 | 36.75 ± 1.72 |
| 4 | >100 | >100 | 8.12 ± 0.89 | 4.30 ± 0.21 |
| 5 | 45.31 ± 0.94 | 49.57 ± 0.29 | 28.13 ± 0.43 | 50.08 ± 1.44 |
| 6 | >100 | >100 | 36.14 ± 1.49 | 63.48 ± 2.80 |
| 7 | 63.26 ± 2.61 | >100 | 36.12 ± 1.11 | 47.16 ± 0.54 |
| 8 | 47.01 ± 1.99 | 20.99 ± 0.95 | 7.45 ± 0.57 | 8.37 ± 0.61 |
| 9 * | nd | nd | nd | nd |
| 10 | >25 | >25 | 7.31 ± 0.70 | 9.28 ± 0.22 |
| 11 | >25 | >25 | 8.92 ± 1.35 | 42.10 ± 0.73 |
| 12 | >25 | >25 | 15.30 ± 2.24 | 11.16 ± 1.48 |
| Cmpd. | Ratio 1 | CI | Interaction |
|---|---|---|---|
| 1 | 69.6:1 | 1.50 (18) | Antagonism |
| 3 | 11.6:1 | 2.33 (83) | Antagonism |
| 5 | 34.8:1 | 1.60 (18) | Antagonism |
| 8 | 27.8:1 | 0.92 (8) | Nearly additive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żesławska, E.; Tejchman, W.; Kincses, A.; Spengler, G.; Nitek, W.; Żuchowski, G.; Szymańska, E. 5-Arylidenerhodanines as P-gp Modulators: An Interesting Effect of the Carboxyl Group on ABCB1 Function in Multidrug-Resistant Cancer Cells. Int. J. Mol. Sci. 2022, 23, 10812. https://doi.org/10.3390/ijms231810812
Żesławska E, Tejchman W, Kincses A, Spengler G, Nitek W, Żuchowski G, Szymańska E. 5-Arylidenerhodanines as P-gp Modulators: An Interesting Effect of the Carboxyl Group on ABCB1 Function in Multidrug-Resistant Cancer Cells. International Journal of Molecular Sciences. 2022; 23(18):10812. https://doi.org/10.3390/ijms231810812
Chicago/Turabian StyleŻesławska, Ewa, Waldemar Tejchman, Annamária Kincses, Gabriella Spengler, Wojciech Nitek, Grzegorz Żuchowski, and Ewa Szymańska. 2022. "5-Arylidenerhodanines as P-gp Modulators: An Interesting Effect of the Carboxyl Group on ABCB1 Function in Multidrug-Resistant Cancer Cells" International Journal of Molecular Sciences 23, no. 18: 10812. https://doi.org/10.3390/ijms231810812
APA StyleŻesławska, E., Tejchman, W., Kincses, A., Spengler, G., Nitek, W., Żuchowski, G., & Szymańska, E. (2022). 5-Arylidenerhodanines as P-gp Modulators: An Interesting Effect of the Carboxyl Group on ABCB1 Function in Multidrug-Resistant Cancer Cells. International Journal of Molecular Sciences, 23(18), 10812. https://doi.org/10.3390/ijms231810812