
Identification of NHLRC1 as a Novel AKT Activator from a Lung Cancer Epigenome-Wide Association Study (EWAS)

 , , ,
, , ,  ,
,
Abstract
1. Background
2. Results
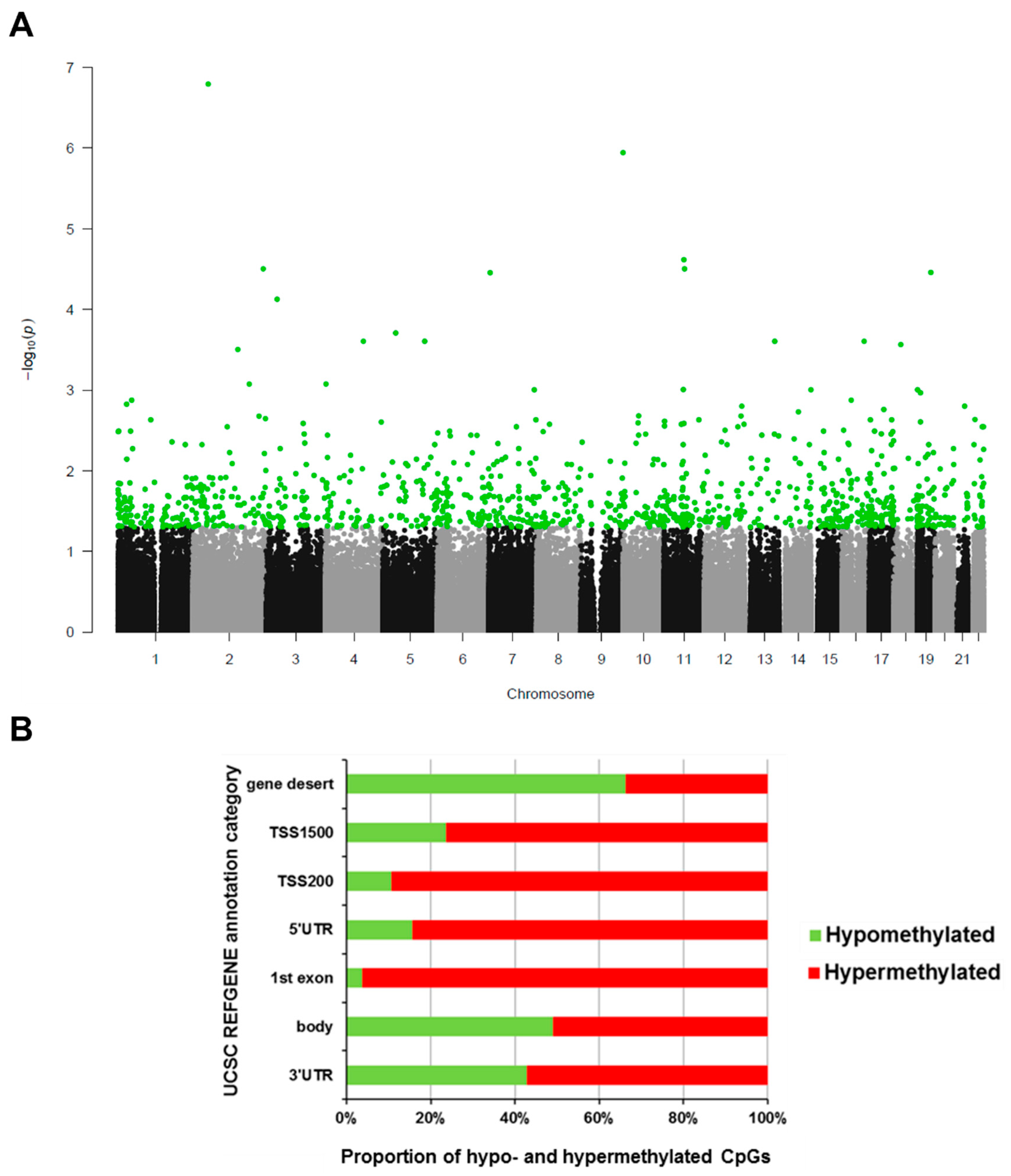
2.1. Differentially Methylated CpGs from EPIC HD EWAS
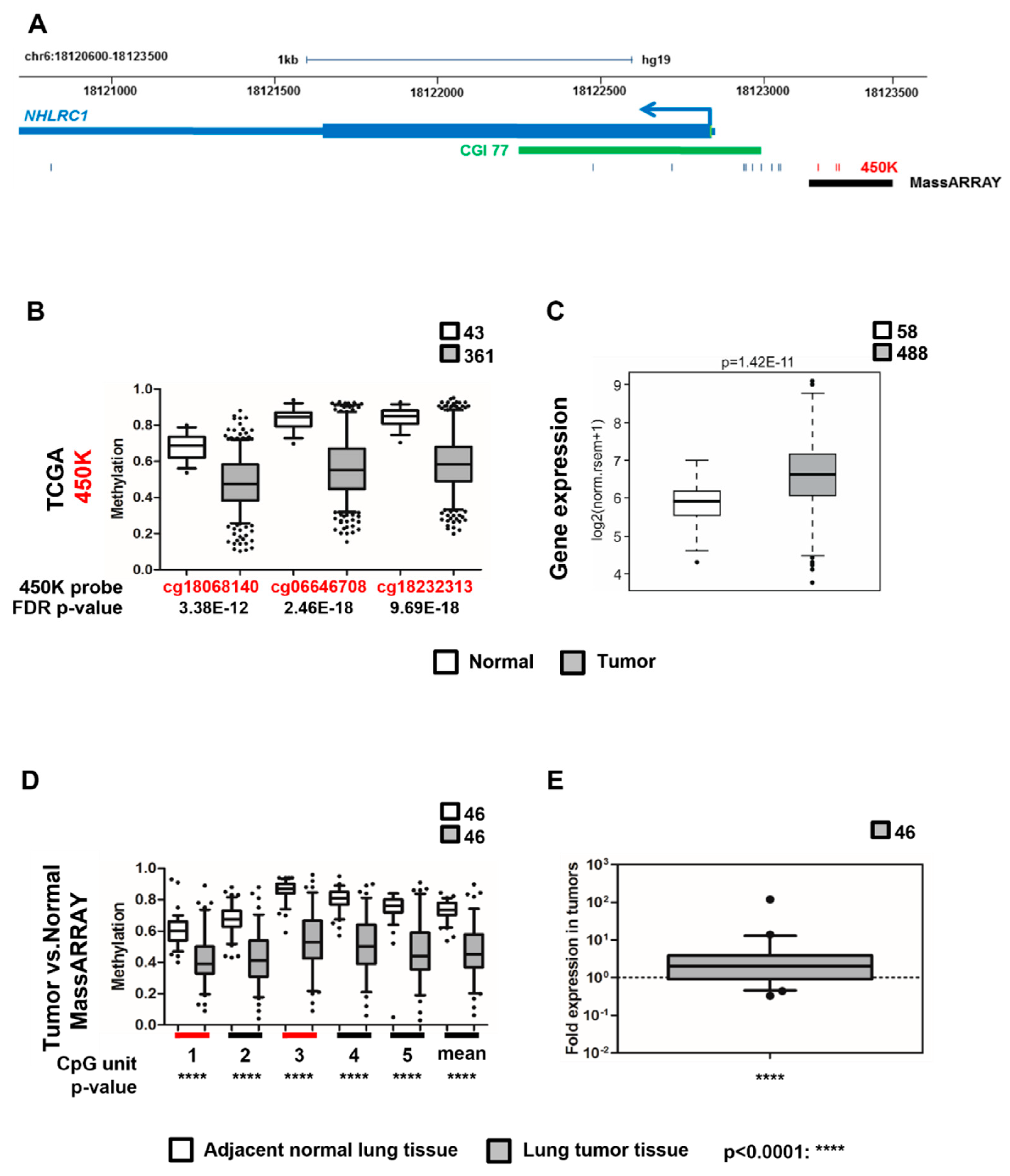
2.2. Top Hypermethylated EPIC HD EWAS DMPs Are Located within DMRs in Lung Tumor versus Adjacent Normal Tissue
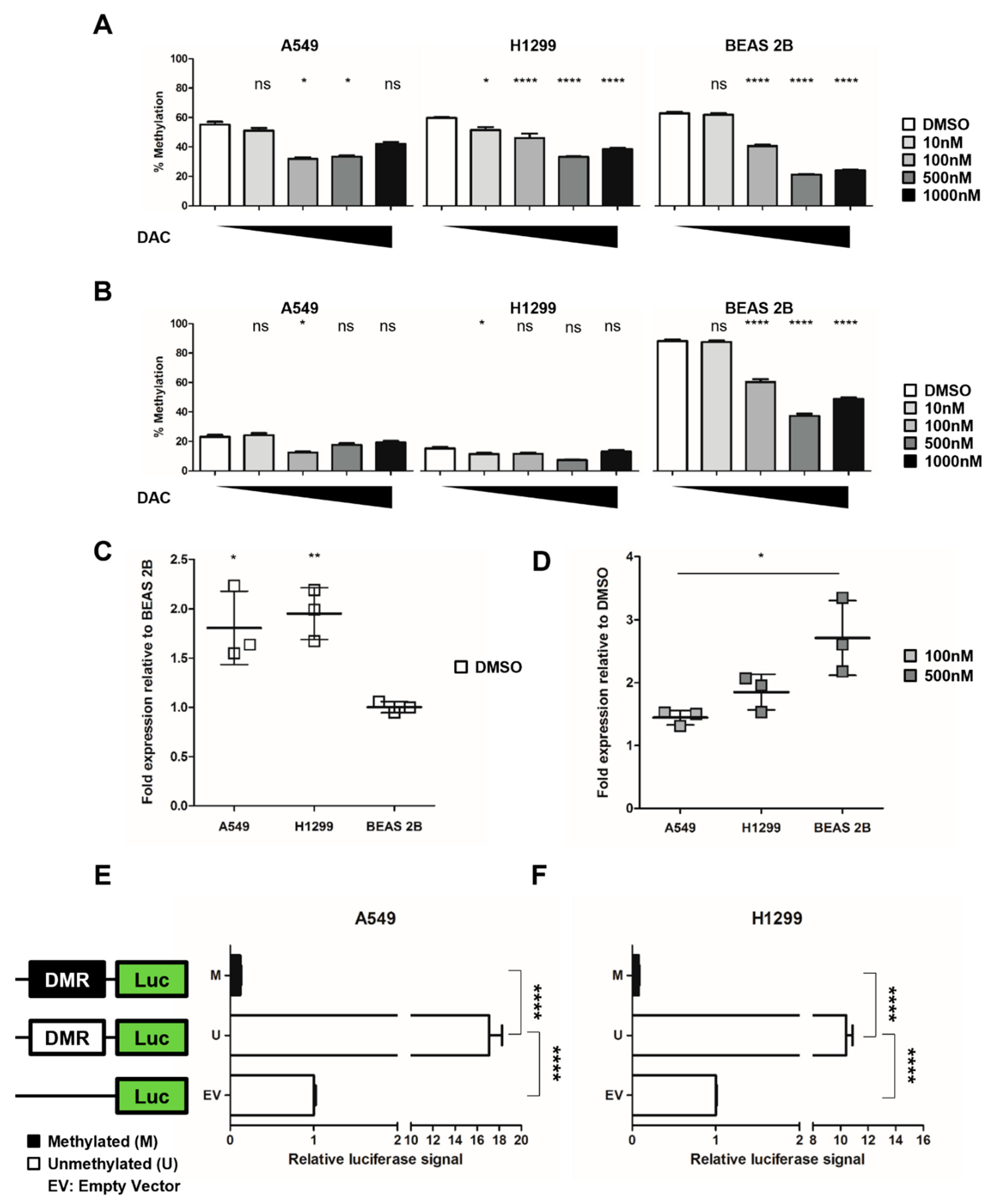
2.3. NHLRC1 Expression Is Epigenetically Regulated
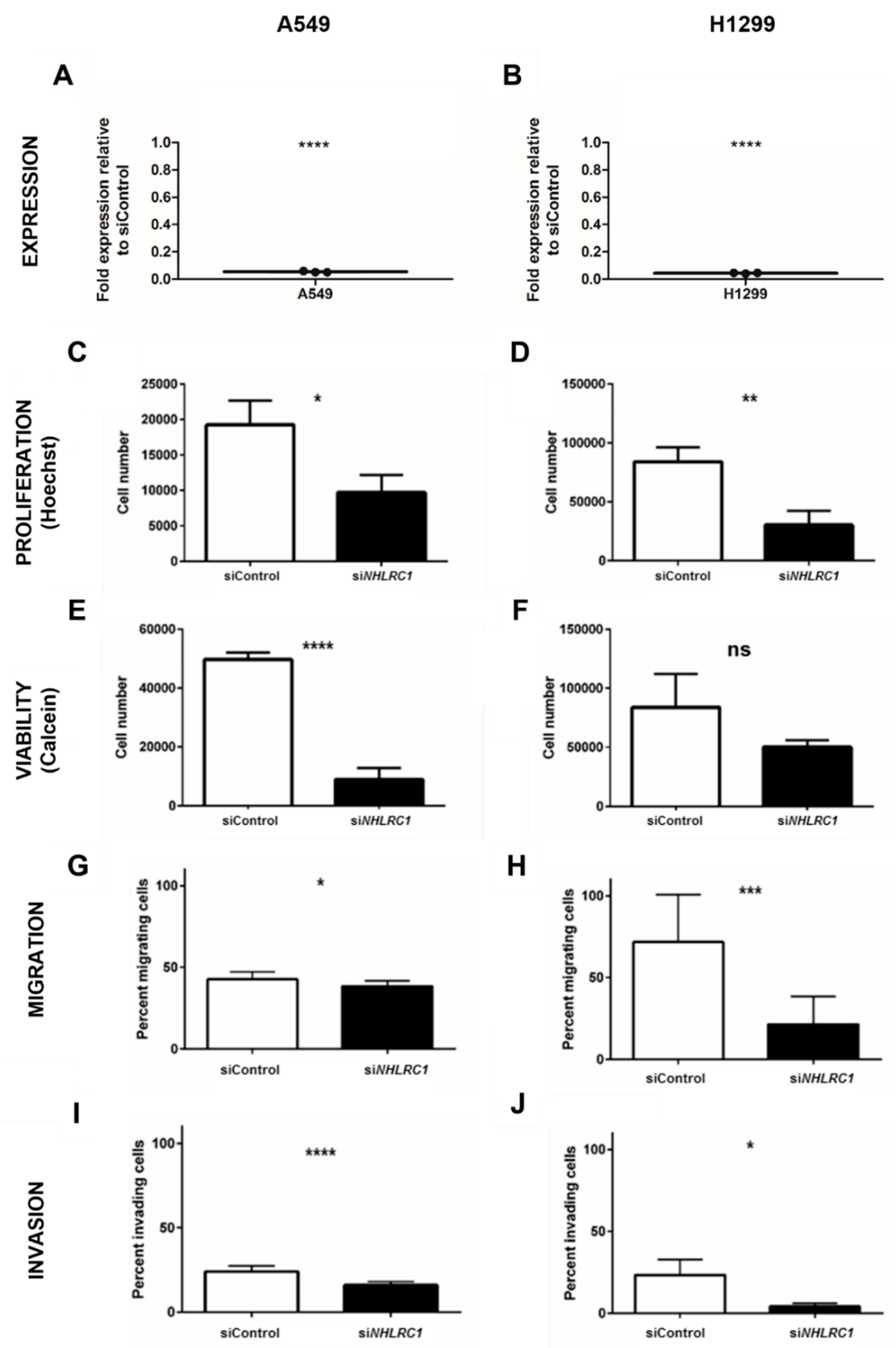
2.4. NHLRC1 Stimulates Proliferation, Viability, Migration and Invasion
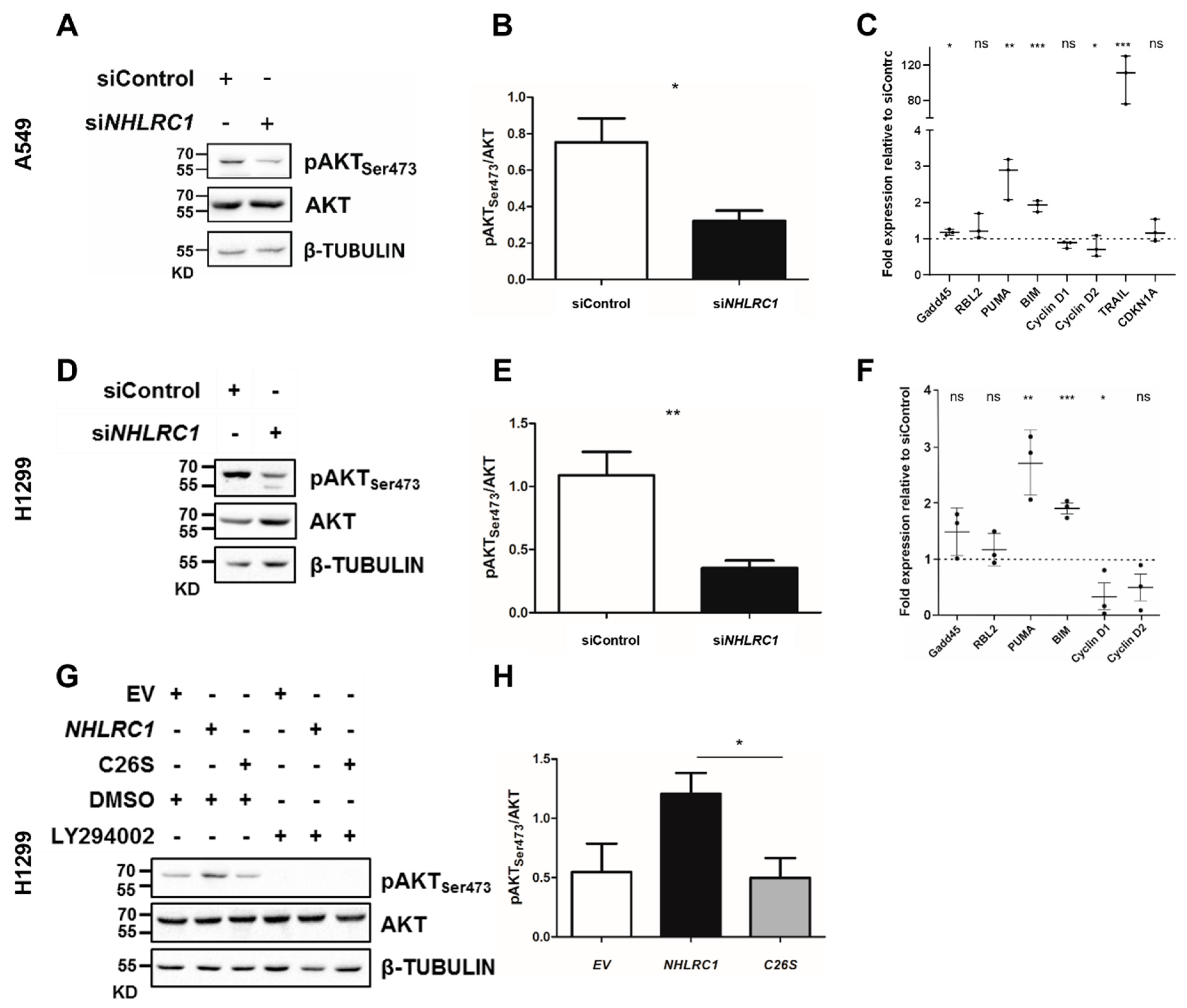
2.5. NHLRC1 Regulates AKT Activation and Modulates Expression of AKT Regulated Genes in Lung Cancer Cells
3. Discussion
4. Methods
4.1. Study Population
4.2. DNA Isolation
4.3. HumanMethylation450K BeadChip Array-Based Analyses of EPIC HD Samples
4.4. HumanMethylation450K DNA Methylation Data Analyses of TCGA Data for Tumor versus Adjacent Normal Tissue
4.5. Sequence-Specific Mass Spectrometry DNA Methylation Analysis (MassARRAY, Agena Bioscience, Hamburg, Germany)
4.6. DNA Methylation Analysis by Pyrosequencing
4.7. RNA Isolation, DNase I Treatment, Reverse Transcription and Expression Analyses
4.8. Routine Cell Culture and Treatments
4.8.1. 5-Aza-2′-Deoxycytidine Cell Treatment
4.8.2. NHLRC1 Knock-Down and Overexpression
4.9. Functional Assays
4.9.1. Proliferation–Viability Assay
4.9.2. Dual Luciferase Promoter Assay
4.9.3. Transwell Migration–Invasion Assays
4.9.4. Western Blot
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- IARC Lung Cancer Statistics. Available online: https://gco.iarc.fr/today/data/pdf/fact-sheets/cancers/cancer-fact-sheets-11.pdf (accessed on 17 August 2022).
- Cancer Research UK Lung Cancer Statistics. Available online: http://www.cancerresearchuk.org/cancer-info/cancerstats/keyfacts/lung-cancer/#incidence.30.11.2014 (accessed on 30 November 2021).
- WHO. Global Health Estimates 2016: Disease Burden by Cause, Age, Sex, by Country and by Region, 2000–2016; World Health Organization: Geneva, Switzerland, 2018; Available online: https://www.Who.Int/healthinfo/global_burden_disease/estimates/en/index1.html (accessed on 9 July 2019).
- Schneider, B.J. Non-small cell lung cancer staging: Proposed revisions to the TNM system. Cancer Imaging 2008, 8, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Bruder, C.; Bulliard, J.-L.; Germann, S.; Konzelmann, I.; Bochud, M.; Leyvraz, M.; Chiolero, A. Estimating lifetime and 10-year risk of lung cancer. Prev. Med. Rep. 2018, 11, 125–130. [Google Scholar] [CrossRef]
- Sasieni, P.D.; Shelton, J.; Ormiston-Smith, N.; Thomson, C.S.; Silcocks, P.B. What is the lifetime risk of developing cancer? The effect of adjusting for multiple primaries. Br. J. Cancer 2011, 105, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Chatterjee, N.; Yu, K.; Goldin, L.R.; Goldstein, A.M.; Rotunno, M.; Mirabello, L.; Jacobs, K.; Wheeler, W.; Yeager, M.; et al. A Genome-wide Association Study of Lung Cancer Identifies a Region of Chromosome 5p15 Associated with Risk for Adenocarcinoma. Am. J. Hum. Genet. 2009, 85, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Liu, X.; Li, J.; Ouyang, R.; Chen, P. The role of cigarette smoke-induced epigenetic alterations in inflammation. Epigenetics Chromatin 2019, 12, 1–25. [Google Scholar] [CrossRef]
- Fasanelli, F.; Baglietto, L.; Ponzi, E.; Guida, F.; Campanella, G.; Johansson, M.; Grankvist, K.; Johansson, M.; Assumma, M.B.; Naccarati, A.; et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat. Commun. 2015, 6, 10192. [Google Scholar] [CrossRef]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-Smoking-Related Differential DNA Methylation: 27K Discovery and Replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef]
- Shenker, N.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2012, 22, 843–851. [Google Scholar] [CrossRef]
- Orozco, L.D.; Morselli, M.; Rubbi, L.; Guo, W.; Go, J.; Shi, H.; Lopez, D.; Furlotte, N.A.; Bennett, B.J.; Farber, C.R.; et al. Epigenome-Wide Association of Liver Methylation Patterns and Complex Metabolic Traits in Mice. Cell Metab. 2015, 21, 905–917. [Google Scholar] [CrossRef]
- Van Veldhoven, K.; Polidoro, S.; Baglietto, L.; Severi, G.; Sacerdote, C.; Panico, S.; Mattiello, A.; Palli, D.; Masala, G.; Krogh, V.; et al. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin. Epigenetics 2015, 7, 1–12. [Google Scholar] [CrossRef]
- Baglietto, L.; Ponzi, E.; Haycock, P.; Hodge, A.; Assumma, M.B.; Jung, C.-H.; Chung, J.; Fasanelli, F.; Guida, F.; Campanella, G.; et al. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int. J. Cancer 2016, 140, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Arab, K.; Park, Y.J.; Lindroth, A.M.; Schäfer, A.; Oakes, C.; Weichenhan, D.; Lukanova, A.; Lundin, E.; Risch, A.; Meister, M.; et al. Long Noncoding RNA TARID Directs Demethylation and Activation of the Tumor Suppressor TCF21 via GADD45A. Mol. Cell 2014, 55, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Arab, K.; Karaulanov, E.; Musheev, M.; Trnka, P.; Schäfer, A.; Grummt, I.; Niehrs, C. GADD45A binds R-loops and recruits TET1 to CpG island promoters. Nat. Genet. 2019, 51, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Ohtaki, S.; Wanibuchi, M.; Kataoka-Sasaki, Y.; Sasaki, M.; Oka, S.; Noshiro, S.; Akiyama, Y.; Mikami, T.; Mikuni, N.; Kocsis, J.D.; et al. ACTC1 as an invasion and prognosis marker in glioma. J. Neurosurg. 2017, 126, 467–475. [Google Scholar] [CrossRef]
- The ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Lübbert, M.; Suciu, S.; Baila, L.; Rüter, B.H.; Platzbecker, U.; Giagounidis, A.; Selleslag, D.; LaBar, B.; Germing, U.; Salih, H.R.; et al. Low-Dose Decitabine Versus Best Supportive Care in Elderly Patients With Intermediate- or High-Risk Myelodysplastic Syndrome (MDS) Ineligible for Intensive Chemotherapy: Final Results of the Randomized Phase III Study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J. Clin. Oncol. 2011, 29, 1987–1996. [Google Scholar] [CrossRef]
- Michalowsky, L.A.; Jones, P.A. Differential nuclear protein binding to 5-azacytosine-containing DNA as a potential mechanism for 5-aza-2′-deoxycytidine resistance. Mol. Cell. Biol. 1987, 7, 3076–3083. [Google Scholar] [CrossRef]
- Wasserman, W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004, 5, 276–287. [Google Scholar] [CrossRef]
- Sandelin, A.; Carninci, P.; Lenhard, B.; Ponjavic, J.; Hayashizaki, Y.; Hume, D.A. Mammalian RNA polymerase II core promoters: Insights from genome-wide studies. Nat. Rev. Genet. 2007, 8, 424–436. [Google Scholar] [CrossRef]
- Hannus, M.; Beitzinger, M.; Engelmann, J.C.; Weickert, M.-T.; Spang, R.; Hannus, S.; Meister, G. siPools: Highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014, 42, 8049–8061. [Google Scholar] [CrossRef]
- Singh, S.; Singh, P.K.; Bhadauriya, P.; Ganesh, S. Lafora disease E3 ubiquitin ligase malin is recruited to the processing bodies and regulates the microRNA-mediated gene silencing process via the decapping enzyme Dcp1a. RNA Biol. 2012, 9, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Nada, S.; Kimura, H.; Tajima, S.; Takahashi, Y.; Kitamura, A.; Oneyama, C.; Okada, M. The mTOR Pathway Controls Cell Proliferation by Regulating the FoxO3a Transcription Factor via SGK1 Kinase. PLoS ONE 2014, 9, e88891. [Google Scholar] [CrossRef] [PubMed]
- de Brachène, A.C.; Demoulin, J.-B. FOXO transcription factors in cancer development and therapy. Exp. 2015, 73, 1159–1172. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Ausserlechner, M.J.; Salvador, C.; Deutschmann, A.; Bodner, M.; Viola, G.; Bortolozzi, R.; Basso, G.; Hagenbuchner, J.; Obexer, P. Therapy-resistant acute lymphoblastic leukemia (ALL) cells inactivate FOXO3 to escape apoptosis induction by TRAIL and Noxa. Oncotarget 2013, 4, 995–1007. [Google Scholar] [CrossRef]
- Gilley, J.; Coffer, P.J.; Ham, J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 2003, 162, 613–622. [Google Scholar] [CrossRef]
- You, H.; Pellegrini, M.; Tsuchihara, K.; Yamamoto, K.; Hacker, G.; Erlacher, M.; Villunger, A.; Mak, T.W. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 2006, 203, 1657–1663. [Google Scholar] [CrossRef]
- Schmidt, M.; de Mattos, S.F.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell Cycle Inhibition by FoxO Forkhead Transcription Factors Involves Downregulation of Cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Nakamura, N.; Sansal, I.; Bergeron, L.; Sellers, W.R. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell 2002, 2, 81–91. [Google Scholar] [CrossRef]
- Kops, G.J.P.L.; Medema, R.; Glassford, J.; Essers, M.; Dijkers, P.F.; Coffer, P.J.; Lam, E.; Burgering, B.M.T. Control of Cell Cycle Exit and Entry by Protein Kinase B-Regulated Forkhead Transcription Factors. Mol. Cell. Biol. 2002, 22, 2025–2036. [Google Scholar] [CrossRef]
- Chen, J.; Yusuf, I.; Andersen, H.-M.; Fruman, D.A. Foxo transcription factors cooperate with delta ef1 to activate growth suppressive genes in b lymphocytes. J. Immunol. 2006, 176, 2711–2721. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA Repair Pathway Stimulated by the Forkhead Transcription Factor FOXO3a Through the Gadd45 Protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, J.; Xu, F.; Zhu, Q.; Ge, D.; Lu, C. Novel methylation-driven genes identified as prognostic indicators for lung squamous cell carcinoma. Am. J. Transl. Res. 2019, 11, 1997–2012. [Google Scholar]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; et al. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011, 43, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Young, E.J.; Ianzano, L.; Munteanu, I.; Zhao, X.; Christopoulos, C.C.; Avanzini, G.; Elia, M.; Ackerley, C.A.; Jovic, N.J.; et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003, 35, 125–127. [Google Scholar] [CrossRef]
- Kecmanovic, M.; Keckarevic-Markovic, M.; Keckarevic, D.; Stevanovic, G.; Jovic, N. Genetics of Lafora progressive myoclonic epilepsy: Current perspectives. Appl. Clin. Genet. 2016, 9, 49–53. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Upadhyay, M.; Gupta, S.; Bhadauriya, P.; Ganesh, S. Lafora disease proteins laforin and malin negatively regulate the HIPK2-p53 cell death pathway. Biochem. Biophys. Res. Commun. 2015, 464, 106–111. [Google Scholar] [CrossRef]
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; VanderBeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2003, 25, 157–167. [Google Scholar] [CrossRef]
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite Motif Protein 32 Facilitates Cell Growth and Migration via Degradation of Abl-Interactor 2. Cancer Res. 2008, 68, 5572–5580. [Google Scholar] [CrossRef]
- Romá-Mateo, C.; Moreno, D.; Vernia, S.; Rubio, T.; Bridges, T.M.; Gentry, M.S.; Sanz, P. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol. Biol. 2011, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xumonette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2016, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Ström, A.; Gustafsson, J.Å. Estrogen receptor β upregulates FOXO3a and causes induction of apoptosis through PUMA in prostate cancer. Oncogene 2014, 33, 4213–4225. [Google Scholar] [CrossRef]
- Fennell, D.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef]
- Liu, H.; Yin, J.; Wang, C.; Gu, Y.; Deng, M.; He, Z. FOXO3a mediates the cytotoxic effects of cisplatin in lung cancer cells. Anti-Cancer Drugs 2014, 25, 898–907. [Google Scholar] [CrossRef]
- Team R. Rstudio: Integrated Development for R; Rstudio PBC: Boston, MA, USA, 2020; Available online: http://www.Rstudio.Com (accessed on 17 August 2022).
- Assenov, Y.; Müller, F.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 2014, 11, 1138–1140. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Marabita, F.; Lechner, M.; Bartlett, T.; Tegner, J.; Gomez-Cabrero, D.; Beck, S. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013, 29, 189–196. [Google Scholar] [CrossRef]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Díez-Villanueva, A.; Mallona, I.; Peinado, M.A. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin 2015, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, M.; Nelson, M.R.; Stanssens, P.; Zabeau, M.; Liloglou, T.; Xinarianos, G.; Cantor, C.R.; Field, J.K.; Boom, D.V.D. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc. Natl. Acad. Sci. USA 2005, 102, 15785–15790. [Google Scholar] [CrossRef] [PubMed]
- Bean, G.R.; Ganesan, Y.T.; Dong, Y.; Takeda, S.; Liu, H.; Chan, P.M.; Huang, Y.; Chodosh, L.A.; Zambetti, G.P.; Hsieh, J.J.-D.; et al. PUMA and BIM Are Required for Oncogene Inactivation–Induced Apoptosis. Sci. Signal. 2013, 6, ra20. [Google Scholar] [CrossRef] [PubMed]
- Hülsmann, H.J.; Rolff, J.; Bender, C.; Jarahian, M.; Korf, U.; Herwig, R.; Fröhlich, H.; Thomas, M.; Merk, J.; Fichtner, I.; et al. Activation of AMP-activated protein kinase sensitizes lung cancer cells and H1299 xenografts to erlotinib. Lung Cancer 2014, 86, 151–157. [Google Scholar] [CrossRef][Green Version]
- Gilbert, D.F.; Erdmann, G.; Zhang, X.; Fritzsche, A.; Demir, K.; Jaedicke, A.; Muehlenberg, K.; Wanker, E.E.; Boutros, M. A Novel Multiplex Cell Viability Assay for High-Throughput RNAi Screening. PLoS ONE 2011, 6, e28338. [Google Scholar] [CrossRef]
- Scherf, D.B.; Sarkisyan, N.; Jacobsson, H.; Claus, R.; Bermejo, J.L.; Peil, B.; Gu, L.; Muley, T.; Meister, M.; Dienemann, H.; et al. Epigenetic screen identifies genotype-specific promoter DNA methylation and oncogenic potential of CHRNB4. Oncogene 2012, 32, 3329–3338. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EPIC HD 450K Discovery Sample Set (Blood Samples) | |
|---|---|
| Total sample number cases and controls (n) | 132 |
| Mean age at baseline (years) | 56 (range: 39–65) |
| Mean time from blood draw to diagnosis (years) | 4.6 (range: 1.1–8.6) |
| Incident cases of lung cancer (n) | 66 |
| Males (n) | 55 cases + 55 controls |
| Females (n) | 11 cases + 11 controls |
| Current smokers (n) | 48 cases + 48 controls |
| Former smokers (n) | 16 cases + 16 controls |
| Mean pack years (n) | 31.4 (range: 0.6–66.3) |
| Number of pairs considered in the analysis (n) | 63 * |
| EPIC HD Top Hypomethylated CpGs | ||||||||||
| Eank | 450K Probe | Genomic Location (hg19) | Closest Coding Element | Distance to Closest TSS [bp] | EPIC HD Mean Methylation Difference [%] | EPIC HD BH p-Value | TCGA SCC Mean Methylation Difference [%] | TCGA SCC FDR p-Value | TCGA ADC Mean Methylation Difference [%] | TCGA ADC FDR p-Value |
| 1 | cg22586603 | chr8:129985596 | LINC00976 | 7350 | −8.9 | 2.7 × 10−2 | −22.1 | 1.9 × 10−12 | −11.9 | 2.0 × 10−4 |
| 2 | cg13291208 | chr8:70475388 | SULF1 | 29,757 | −6.6 | 3.6 × 10−2 | −18.2 | 1.3 × 10−6 | −23.4 | 1.2 × 10−8 |
| 3 | cg06126421 | chr6:30720080 | IER3 | 7753 | −6.3 | 2.4 × 10−2 | −17.3 | 1.5 × 10−8 | −4.4 | 3.7 × 10−1 |
| 4 | cg13204432 | chr2:51074939 | NRXN1 | 184,735 | −4.1 | 1.6 × 10−7 | −9.1 | 4.5 × 10−12 | −8.5 | 4.9 × 10−6 |
| 5 | cg05951044 | chr18:23597141 | SS18 | 73,470 | −4.1 | 2.7 × 10−4 | −8.7 | 1.2 × 10−6 | −3.6 | 4.2 × 10−2 |
| 6 | cg04955914 | chr2:220040571 | CNPPD1 | 1144 | −3.9 | 2.1 × 10−3 | −6.0 | 5.0 × 10−3 | −7.1 | 3.3 × 10−2 |
| EPIC HD Top Hypermethylated CpGs | ||||||||||
| Rank | 450K Probe | Genomic Location (hg19) | Closest Coding Element | Distance to Closest TSS [bp] | EPIC HD Mean Methylation Difference [%] | EPIC HD BH p-Value | TCGA SCC Mean Methylation Difference [%] | TCGA SCC FDR p-Value | TCGA ADC Mean Methylation Difference [%] | TCGA ADC FDR p-Value |
| 1 | cg06646708 | chr6:18123224 | NHLRC1 | 374 | 6.7 | 3.2 × 10−2 | −29.3 | 6.3 × 10−25 | −23.9 | 2.5 × 10−18 |
| 2 | cg17242351 | chr15:35086890 | ACTC1 | 1037 | 6.6 | 1.7 × 10−3 | 22.9 | 1.0 × 10−8 | 22.0 | 1.7 × 10−10 |
| 3 | cg07714266 | chr7:28075371 | JAZF1 | 145,066 | 5.5 | 7.7 × 10−3 | −14.4 | 4.5 × 10−11 | −2.1 | 1.9 × 10−1 |
| 4 | cg26484090 | chr6:99960583 | USP45 | 2669 | 5.4 | 2.0 × 10-2 | −3.2 | 2.7 × 10−3 | −1.2 | 3.2 × 10−1 |
| 5 | cg00554165 | chr5:172536348 | CREBRF | 52,993 | 5.2 | 4.7 × 10−3 | 4.6 | 1.7 × 10−5 | 0.2 | 5.8 × 10−1 |
| 6 | cg14187813 | chr2:97651611 | FAM178B | 690 | 4.9 | 3.6 × 10−3 | −15.2 | 9.9 × 10−9 | 3.0 | 5.3 × 10−1 |
| Lung Tumor versus Adjacent Normal Lung Tissue (Replication Set I) | |
|---|---|
| Total lung tumour-adjacent normal lung samples (n) | 200 |
| Adenocarcinoma (n) | 50 (pairs) |
| Squamous cell carcinoma (n) | 50 (pairs) |
| Mean age at diagnosis (years) | 64 (range: 43–81) |
| Males (n) | 68 |
| Females (n) | 32 |
| Current smokers (n) | 45 |
| Former smokers (n) | 54 |
| Unknown smoking status (n) | 1 |
| Mean pack years (py) | 46 (range: 1–150) |
| Number of patients considered in the analysis (n) | 94 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faltus, C.; Lahnsteiner, A.; Barrdahl, M.; Assenov, Y.; Hüsing, A.; Bogatyrova, O.; Laplana, M.; Johnson, T.; Muley, T.; Meister, M.; et al. Identification of NHLRC1 as a Novel AKT Activator from a Lung Cancer Epigenome-Wide Association Study (EWAS). Int. J. Mol. Sci. 2022, 23, 10699. https://doi.org/10.3390/ijms231810699
Faltus C, Lahnsteiner A, Barrdahl M, Assenov Y, Hüsing A, Bogatyrova O, Laplana M, Johnson T, Muley T, Meister M, et al. Identification of NHLRC1 as a Novel AKT Activator from a Lung Cancer Epigenome-Wide Association Study (EWAS). International Journal of Molecular Sciences. 2022; 23(18):10699. https://doi.org/10.3390/ijms231810699
Chicago/Turabian StyleFaltus, Christian, Angelika Lahnsteiner, Myrto Barrdahl, Yassen Assenov, Anika Hüsing, Olga Bogatyrova, Marina Laplana, Theron Johnson, Thomas Muley, Michael Meister, and et al. 2022. "Identification of NHLRC1 as a Novel AKT Activator from a Lung Cancer Epigenome-Wide Association Study (EWAS)" International Journal of Molecular Sciences 23, no. 18: 10699. https://doi.org/10.3390/ijms231810699
APA StyleFaltus, C., Lahnsteiner, A., Barrdahl, M., Assenov, Y., Hüsing, A., Bogatyrova, O., Laplana, M., Johnson, T., Muley, T., Meister, M., Warth, A., Thomas, M., Plass, C., Kaaks, R., & Risch, A. (2022). Identification of NHLRC1 as a Novel AKT Activator from a Lung Cancer Epigenome-Wide Association Study (EWAS). International Journal of Molecular Sciences, 23(18), 10699. https://doi.org/10.3390/ijms231810699