


Nutritional Interventions for Patients with Mitochondrial POLG-Related Diseases: A Systematic Review on Efficacy and Safety

Abstract
:1. Introduction
2. Results
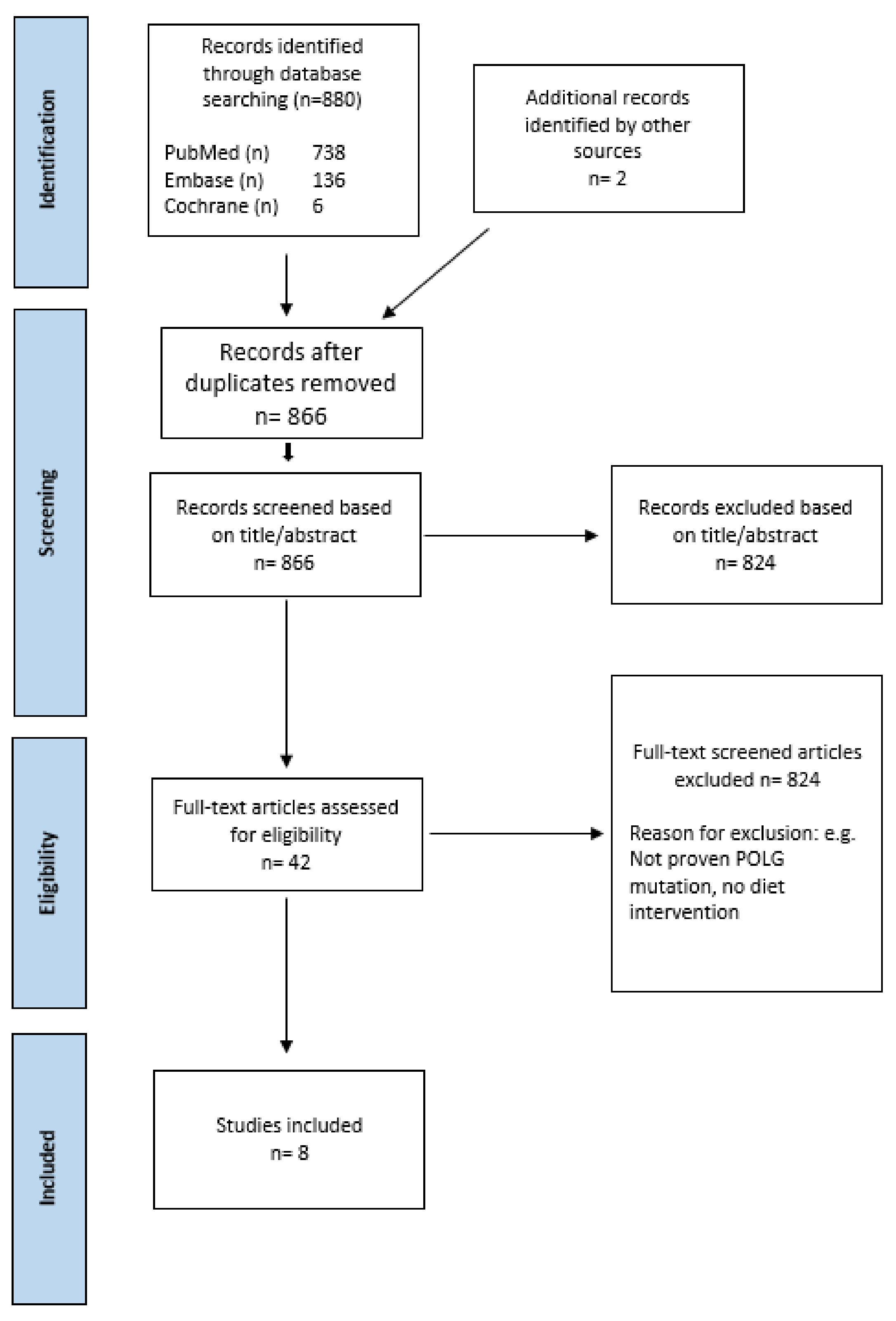
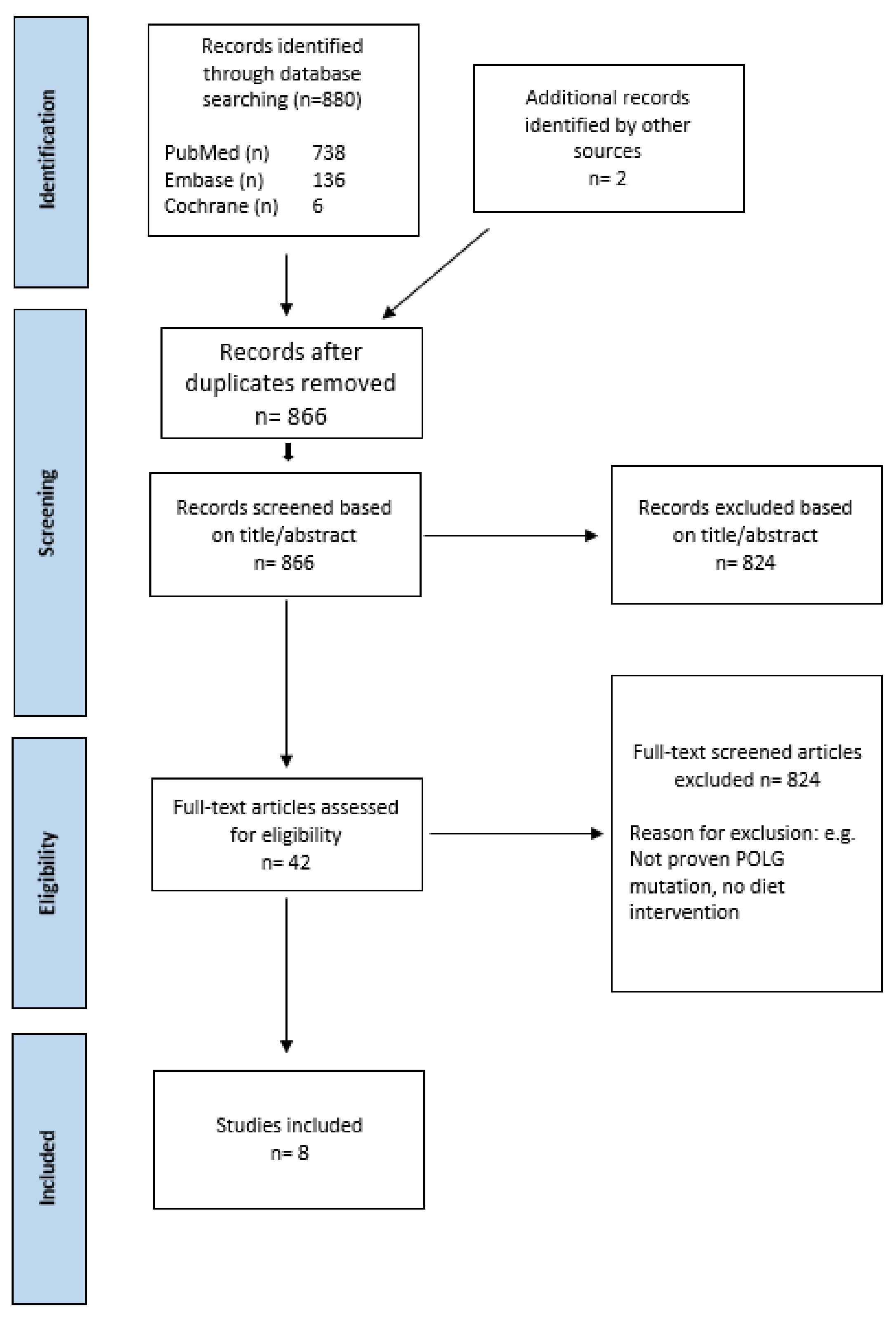
2.1. Study Selection
2.2. Study Characteristics
2.3. Study Quality
2.4. Primary Outcomes
2.4.1. Epileptic Seizures
2.4.2. Liver Impairment
2.4.3. Clinical Symptom Improvements
2.5. Secondary Outcomes
2.5.1. Compliance
2.5.2. Macronutrient Composition and Effect
2.5.3. Nutritional Supplements
2.5.4. Effect on AEDs
3. Discussion
3.1. Seizures
3.2. Liver Impairment
3.3. Mitochondrial Respiratory Chain (MRC)
3.4. Macronutrient Composition and Effect
3.5. Compliance
3.6. AEDs
3.7. Nutritional Supplements
4. Methods
4.1. Search Strategy
4.2. Study Selection
4.3. Eligibility Criteria
4.4. Exclusion Criteria
4.5. Data Extraction
4.6. Quality Appraisal and Interrater Reliability
4.7. Outcome Measures
5. Conclusions
6. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Search Strategy

{kind=link}
| Search | Query |
|---|---|
| #1 | DNA Polymerase gamma [MESH] |
| #2 | DNA-Directed DNA Polymerase/genetics [MESH] |
| #3 | Mitochondrial Diseases/therapy [MESH] |
| #4 | DNA-Directed DNA Polymerase [MESH] |
| #5 | POLG protein: human [MESH] |
| #6 | Status Epilepticus/pathology [MESH] |
| #7 | POLG1 [Title/Abstract] |
| #8 | POLG mutation [Title/Abstract] |
| #9 | nDNA mutation [Title/Abstract] |
| #10 | POLG [Title/Abstract] |
| #11 | Alpers huttenlocher syndrom * [Title/Abstract] |
| #12 | MEMSA [Title/Abstract] |
| #13 | Myoclonic epilepsy myopathy sensory ataxia [Title/Abstract] |
| #14 | SCAE [Title/Abstract] |
| #15 | Spinocerebellar ataxia with epilepsy [Title/Abstract] |
| #16 | ataxia neuropathy spectrum [Title/Abstract] |
| #17 | MIRAS [Title/Abstract] |
| #18 | SANDO [Title/Abstract] |
| #19 | #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 [Hits:51,680]. |
| #20 | Diet: Ketogenic * [MESH] |
| #21 | Glycemic Index [MESH] |
| #22 | Diet: Ketogenic/methods * [MESH] |
| #23 | Diet: High-Fat [MESH] |
| #24 | Diet * [MESH] |
| #25 | Epilepsy/diet therapy [MESH] |
| #26 | Ketogenic diet [Title/Abstract] |
| #27 | low glycaemic index diet [Title/Abstract] |
| #28 | High fat diet [Title/Abstract] |
| #29 | Modified atkins diet [Title/Abstract] |
| #30 | #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 OR #28 OR #29 [Hits:1,343,284]. #19 AND #30 [Hits:738]. |
| Search | Query |
|---|---|
| #1 | DNA Polymerase gamma [MESH] |
| #2 | DNA-Directed DNA Polymerase [MESH] |
| #3 | POLG1 [Title:Abstract:Keyword] |
| #4 | POLG mutation [Title:Abstract:Keyword] |
| #5 | POLG [Title:Abstract:Keyword] |
| #6 | Alpers huttenlocher syndrome [Title:Abstract:Keyword] |
| #7 | MEMSA [Title:Abstract:Keyword] |
| #8 | SCAE [Title:Abstract:Keyword] |
| #9 | Spinocerebellar ataxia with epilepsy [Title:Abstract:Keyword] |
| #10 | ataxia neuropathy spectrum [Title:Abstract:Keyword] |
| #11 | MIRAS [Title:Abstract:Keyword] |
| #12 | SANDO [Title:Abstract:Keyword] |
| #13 | #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 [Hits 486]. |
| #14 | Diet: Ketogenic [MESH] |
| #15 | Diet theraphy [MESH] |
| #16 | Glycemic Index [MESH] |
| #17 | “ketogenic diet” [Title:Abstract:Keyword] |
| #18 | “High fat diet” [Title:Abstract:Keyword] |
| #19 | “Atkins diet” [Title:Abstract:Keyword] |
| #20 | #14 OR #15 OR #16 OR #17 OR #18 OR #19 [Hits: 8234] #13 AND #20 [Hits 6] |
| Search | Query |
|---|---|
| #1 | DNA directed DNA polymerase gamma [Map term] |
| #2 | Alpers disease [Map term] |
| #3 | Complex 1 deficiency [Map term] |
| #4 | Mitochondrial encephalopathy [Map term] |
| #5 | Multiple acyl coa dehydrogenase deficiency [Map term] |
| #6 | POLG [Keyword] |
| #7 | Alpers huttenlocher [Keyword] |
| #8 | MEMSA [Keyword] |
| #9 | SCAE [Keyword] |
| #10 | Spinocerebellar ataxia with epilepsy [Keyword] |
| #11 | MIRAS [Keyword] |
| #12 | SANDO [Keyword] |
| #13 | #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #11 OR #12 [Hits: 5334]. |
| #14 | Low glycemic index diet [Map term] |
| #15 | Low fat diet [Map term] |
| #16 | Methionine/choline deficient diet [Map term] |
| #17 | Ketogenic diet [Map term] |
| #18 | High methionine diet [Map term] |
| #19 | Low carbohydrate diet [Map term] |
| #20 | High-protein low-carbohydrate diet [Map term] |
| #21 | Lipid diet [ Map term] |
| #22 | Atkins diet [Map term] |
| #23 | Protein diet [Map term] |
| #24 | Mediterranean diet [Map term] |
| #25 | Ketogenic diet [Title] |
| #26 | Low glycemic-index diet [Title] |
| #27 | Low carbohydrate diet [Title] |
| #28 | Modified Atkins diet [Title] |
| #29 | #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR#26 OR #27 OR #28 [Hits:89936]. #13 AND #29 [Hits:136]. |
Appendix B
| Study/Year | Reason for Exclusion |
|---|---|
| Hattori et al., 2001 [50] | MD not caused by POLG variant |
| Bachmann-Gagescu et al., 2009 [51] | MD not caused by POLG variant |
| Kumagi, 1999 [52] | MD not caused by POLG variant |
| Barzegar & Hashemilar, 2007 [53] | Missing information about diet intervention |
| Malojcic et al., 2004 [54] | Not clinical proven POLG mutation (LEIGH) |
| Seessle et al., 2012 [55] | Not clinical proven POLG mutation (MELAS) |
| Roe & Brunengraber, 2015 [56] | Not clinical proven POLG mutation |
| Daniel et al., 2015 [57] | No diet intervention |
| Di pisa et al., 2012 [58] | MD not caused by POLG variant |
| Pons et al., 2000 [59] | MD not caused by POLG variant |
| Gong et al., 2021 [60] | Not clinical proven POLG mutation (LEIGH) |
| Teitelbaum et al., 2002 [61] | Missing information about diet intervention |
| Morava et al., 2006 [62] | Not clinical proven POLG mutation |
| Beghin et al., 2016 [63] | Not clinical proven POLG mutation |
| Laugel et al., 2007 [64] | Not clinical proven POLG mutation (LEIGH NDUFV1 mutation) |
| Llingworth et al., 2012 [65] | Authors contacted for access |
| Zweers et al., 2020 [66] | Not clinical proven POLG mutation |
| Nangia et al., 2012 [67] | Not clinical proven POLG mutation |
| Kang et al., 2006 [68] | Not clinical proven POLG mutation |
| Craigen et al., 1996 [69] | Not clinical proven POLG mutation LEIGH (E3 deficiency Maple syrup urine disease) |
| Wijburg et al., 1992 [70] | Not clinical proven POLG mutation LEIGH (PDHC deficiency) |
| Ahola et al., 2016 [71] | MD not caused by POLG variant |
| Wexler et al., 1997 [72] | MD not caused by POLG variant (PDHC deficiency) |
| Barnerias et al., 2010 [73] | MD not caused by POLG variant |
| Suntum et al., 2017 [74] | MD not caused by POLG variant |
| Deberles et al., 2020 [75] | MD not caused by POLG variant |
| Kang et al., 2007 [76] | MD not caused by POLG variant |
| Theunissen et al., 2017 [77] | MD not caused by POLG variant |
| Berio, A 1994 [78] | Authors contacted for access |
| Kanabus et al., 2016 [79] | Other MD disease (LEIGH) Supplement intervention |
| Peterson, P. L, 1995 [80] | Not clinical proven POLG |
| Villarroya et al., 2019 [81] | Authors contacted for access |
| Foschi et al., 2015 [82] | MD not caused by POLG variant |
| Sato-shirai et al., 2021 [83] | MD not caused by POLG variant (LEIGH ECHS1D gene) |
Appendix C
| General Recommendation (Nordic Nutrition Recommendations) | 4:1/CKD | MAD/Atkins | LGIT | MCT-KD | |
|---|---|---|---|---|---|
| Fedt | 25–40 E% | 90 E% | 70 E% | 45 E% | 70 E% |
| Protein | 10–20 E% | 7 E% | 25 E% | 28 E% | 10 E% |
| Kulhydrater | 45–60 E% | 3 E% | 5 E% | 27 E% | 20 E% |
References
- Popov, L.-D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Kennedy, W.D.; Yin, Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009, 139, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Saneto, R.P.; Cohen, B.H.; Copeland, W.C.; Naviaux, R.K. Alpers-Huttenlocher Syndrome. Pediatr. Neurol. 2013, 48, 167–178. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef]
- Tzoulis, C.; Tran, G.T.; Coxhead, J.; Bertelsen, B.; Lilleng, P.K.; Balafkan, N.; Payne, B.; Miletic, H.; Chinnery, P.F.; Bindoff, L.A. Molecular Pathogenesis of Polymerase Gamma–Related Neurodegeneration. Ann. Neurol. 2014, 76, 66–81. [Google Scholar] [CrossRef]
- Saneto, R.P.; Naviaux, R.K. Polymerase gamma disease through the ages. Dev. Disabil. Res. Rev. 2010, 16, 163–174. [Google Scholar] [CrossRef]
- Wiltshire, E.; Davidzon, G.; DiMauro, S.; Akman, H.O.; Sadleir, L.; Haas, L.; Zuccollo, J.; McEwen, A.; Thorburn, D.R. Juvenile Alpers Disease. Arch. Neurol. 2008, 65, 121–124. [Google Scholar] [CrossRef]
- Harding, B.N. Progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher syndrome): A personal review. J. Child Neurol. 1990, 5, 273–287. [Google Scholar] [CrossRef]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; de Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Naviaux, R.K.; Nguyen, K.V. POLG mutations associated with Alpers syndrome and mitochondrial DNA depletion. Ann. Neurol. 2005, 58, 491. [Google Scholar] [CrossRef]
- Van Goethem, G.; Martin, J.J.; Dermaut, B.; Löfgren, A.; Wibail, A.; Ververken, D.; Tack, P.; Dehaene, I.; Van Zandijcke, M.; Moonen, M.; et al. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul. Disord. 2003, 13, 133–142. [Google Scholar] [CrossRef]
- Mutations of Mitochondrial DNA Polymerase γA Are a Frequent Cause of Autosomal Dominant or Recessive Progressive External Ophthalmoplegia-Lamantea-2002-Annals of Neurology-Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/full/10.1002/ana.10278?sid=nlm%3Apubmed (accessed on 8 May 2022).
- Tzoulis, C.; Engelsen, B.A.; Telstad, W.; Aasly, J.; Zeviani, M.; Winterthun, S.; Ferrari, G.; Aarseth, J.H.; Bindoff, L.A. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: A study of 26 cases. Brain 2006, 129, 1685–1692. [Google Scholar] [CrossRef]
- Engelsen, B.A.; Tzoulis, C.; Karlsen, B.; Lillebø, A.; Laegreid, L.M.; Aasly, J.; Zeviani, M.; Bindoff, L.A. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain 2008, 131, 818–828. [Google Scholar] [CrossRef]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- Van der Louw, E.; van den Hurk, D.; Neal, E.; Leiendecker, B.; Fitzsimmon, G.; Dority, L.; Thompson, L.; Marchió, M.; Dudzińska, M.; Dressler, A.; et al. Ketogenic diet guidelines for infants with refractory epilepsy. Eur. J. Paediatr. Neurol. 2016, 20, 798–809. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Jaramillo, J.G.; Clarke, K.; Soto, A. Ketotherapeutics for neurodegenerative diseases. Int. Rev. Neurobiol. 2020, 155, 141–168. [Google Scholar] [CrossRef]
- Lutas, A.; Yellen, G. The ketogenic diet: Metabolic influences on brain excitability and epilepsy. Trends Neurosci. 2013, 36, 32–40. [Google Scholar] [CrossRef]
- Danial, N.N.; Hartman, A.L.; Stafstrom, C.E.; Thio, L.L. How does the ketogenic diet work? Four potential mechanisms. J. Child Neurol. 2013, 28, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Koessler, M.; Haberlandt, E.; Karall, D.; Baumann, M.; Höller, A.; Scholl-Bürgi, S. Ketogenic diet in a patient with refractory status epilepticus due to POLG mutation. JIMD Rep. 2021, 57, 3–8. [Google Scholar] [CrossRef]
- Scalais, E.; Francois, B.; Schlesser, P.; Stevens, R.; Nuttin, C.; Martin, J.-J.; Van Coster, R.; Seneca, S.; Roels, F.; Van Goethem, G.; et al. Polymerase gamma deficiency (POLG): Clinical course in a child with a two stage evolution from infantile myocerebrohepatopathy spectrum to an Alpers syndrome and neuropathological findings of Leigh’s encephalopathy. Eur. J. Paediatr. Neurol. 2012, 16, 542–548. [Google Scholar] [CrossRef]
- Khan, A.; Trevenen, C.; Wei, X.-C.; Sarnat, H.B.; Payne, E.; Kirton, A. Alpers syndrome: The natural history of a case highlighting neuroimaging, neuropathology, and fat metabolism. J. Child Neurol. 2012, 27, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Joshi, C.N.; Greenberg, C.R.; Mhanni, A.A.; Salman, M.S. Ketogenic diet in Alpers-Huttenlocher syndrome. Pediatr. Neurol. 2009, 40, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Spiegler, J.; Stefanova, I.; Hellenbroich, Y.; Sperner, J. Bowel obstruction in patients with Alpers-Huttenlocher syndrome. Neuropediatrics 2011, 42, 194–196. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, S.E.; Ream, M.A.; Richardson, C.; Mikati, M.A.; Trescher, W.H.; Byler, D.L.; Sather, J.D.; Michael, E.H.; Urbanik, K.B.; Richards, J.L.; et al. The ketogenic diet for the treatment of pediatric status epilepticus. Pediatr. Neurol. 2014, 50, 101–103. [Google Scholar] [CrossRef]
- Martikainen, M.H.; Päivärinta, M.; Jääskeläinen, S.; Majamaa, K. Successful treatment of POLG-related mitochondrial epilepsy with antiepileptic drugs and low glycaemic index diet. Epileptic Disord. 2012, 14, 438–441. [Google Scholar] [CrossRef]
- Cardenas, J.F.; Amato, R.S. Compound heterozygous polymerase gamma gene mutation in a patient with Alpers disease. Semin. Pediatr. Neurol. 2010, 17, 62–64. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. BMJ 2009, 339, b2700. [Google Scholar] [CrossRef] [Green Version]
- Gagnier, J.J.; Kienle, G.; Altman, D.G.; Moher, D.; Sox, H.; Riley, D. The CARE guidelines: Consensus-based clinical case report guideline development. J. Clin. Epidemiol. 2014, 67, 46–51. [Google Scholar] [CrossRef]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.E.; Samsonsen, C.; Brodtkorb, E.; Ostergaard, E.; de Coo, R.; et al. The impact of gender, puberty, and pregnancy in patients with POLG disease. Ann. Clin. Transl. Neurol. 2020, 7, 2019–2025. [Google Scholar] [CrossRef]
- Wong, M.; Moss, R.L. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J. Neurosci. 1992, 12, 3217–3225. [Google Scholar] [CrossRef]
- Cramer, J.A.; Gordon, J.; Schachter, S.; Devinsky, O. Women with epilepsy: Hormonal issues from menarche through menopause. Epilepsy Behav. 2007, 11, 160–178. [Google Scholar] [CrossRef]
- Schiff, M.; Bénit, P.; El-Khoury, R.; Schlemmer, D.; Benoist, J.-F.; Rustin, P. Mouse studies to shape clinical trials for mitochondrial diseases: High fat diet in Harlequin mice. PLoS ONE 2011, 6, e28823. [Google Scholar] [CrossRef]
- Harun-Or-Rashid, M.; Inman, D.M. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. J. Neuroinflamm. 2018, 15, 313. [Google Scholar] [CrossRef]
- Castro-Gago, M.; González-Conde, V.; Fernández-Seara, M.J.; Rodrigo-Sáez, E.; Fernández-Cebrián, S.; Alonso-Martín, A.; Campos, Y.; Arenas, J.; Eirís-Puñal, J. Early mitochondrial encephalomyopathy due to complex IV deficiency consistent with Alpers-Huttenlocher syndrome: Report of two cases. Rev. Neurol. 1999, 29, 912–917. [Google Scholar]
- Greene, A.E.; Todorova, M.T.; McGowan, R.; Seyfried, T.N. Caloric Restriction Inhibits Seizure Susceptibility in Epileptic EL Mice by Reducing Blood Glucose. Epilepsia 2001, 42, 1371–1378. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, H.-C.; Lee, E.J.; Lee, J.S.; Kim, H.D. Low glycemic index treatment in patients with drug-resistant epilepsy. Brain Dev. 2017, 39, 687–692. [Google Scholar] [CrossRef]
- Muzykewicz, D.A.; Lyczkowski, D.A.; Memon, N.; Conant, K.D.; Pfeifer, H.H.; Thiele, E.A. Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia 2009, 50, 1118–1126. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Li, X.-J.; Jiang, W.-L.; Sun, H.-B.; Liu, J. Efficacy of and patient compliance with a ketogenic diet in adults with intractable epilepsy: A meta-analysis. J. Clin. Neurol. 2015, 11, 26–31. [Google Scholar] [CrossRef]
- Cabrera, A.M.; Fain, H.; Fain, B.; Muniategui, J.; Buiras, V.M.; Galicchio, S.; Cacchia, P.A.; Retamero, M.; Ocampo, R.P.; Porto, M.B. Treatment of refractory epilepsy. A comparison between classic ketogenic diet and modified Atkins diet in terms of efficacy, adherence, and undesirable effects. Nutr. Hosp. 2021, 38, 1144–1148. [Google Scholar] [CrossRef]
- Zarnowska, I.; Luszczki, J.J.; Zarnowski, T.; Buszewicz, G.; Madro, R.; Czuczwar, S.J.; Gasior, M. Pharmacodynamic and pharmacokinetic interactions between common antiepileptic drugs and acetone, the chief anticonvulsant ketone body elevated in the ketogenic diet in mice. Epilepsia 2009, 50, 1132–1140. [Google Scholar] [CrossRef]
- Bronsky, J.; Campoy, C.; Braegger, C.; Braegger, C.; Bronsky, J.; Cai, W.; Campoy, C.; Carnielli, V.; Darmaun, D.; Decsi, T.; et al. ESPGHAN/ESPEN/ESPR/CSPEN guidelines on pediatric parenteral nutrition: Vitamins. Clin. Nutr. 2018, 37, 2366–2378. [Google Scholar] [CrossRef]
- Riley, D.S.; Barber, M.S.; Kienle, G.S.; Aronson, J.K.; von Schoen-Angerer, T.; Tugwell, P.; Kiene, H.; Helfand, M.; Altman, D.G.; Sox, H.; et al. CARE guidelines for case reports: Explanation and elaboration document. J. Clin. Epidemiol. 2017, 89, 218–235. [Google Scholar] [CrossRef]
- Howick, J.; Phillips, B.; Ball, C.; Sackett, D.; Badenoch, D. Oxford Centre for Evidence-Based Medicine Levels of Evidence; University of Oxford, Centre for Evidence-Based Medicine: Oxford, UK, 2009; Volume 5. [Google Scholar]
- McHugh, M.L. Interrater reliability: The kappa statistic. Biochem. Med. 2012, 22, 276–282. [Google Scholar] [CrossRef]
- Hattori, Y.; Matsuda, M.; Eizawa, T.; Nakajima, K. A case of mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), showing temporary improvement during the treatment with eicosapentaenoic acid ethyl ester. Rinsho Shinkeigaku 2001, 41, 668–672. [Google Scholar]
- Bachmann-Gagescu, R.; Merritt, J.L., 2nd; Hahn, S.H. A cognitively normal PDH-deficient 18-year-old man carrying the R263G mutation in the PDHA1 gene. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), 123–126. [Google Scholar] [CrossRef]
- Kumagai, R.; Ichikawa, K.; Yasui, T.; Kageyama, Y.; Miyabayashi, S. Adult leigh syndrome: Treatment with intravenous soybean oil for acute central respiratory failure. Eur. J. Neurol. 1999, 6, 613–615. [Google Scholar] [CrossRef]
- Barzegar, M.; Hashemilar, M. Alpers Disease: Report of two Familial Cases. Pak. J. Med. Sci. 2007, 23, 643. [Google Scholar]
- Malojcic, B.; Brinar, V.; Poser, C.; Djakovic, V. An adult case of Leigh disease. Clin. Neurol. Neurosurg. 2004, 106, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Seessle, J.; Stremmel, W.; Ebinger, F.; Merle, U. An unusual case of paralytic ileus. Z. Gastroenterol. 2012, 50, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.R.; Brunengraber, H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: Review of 15 years Experience. Mol. Genet. Metab. 2015, 116, 260–268. [Google Scholar] [CrossRef]
- Daniel, R.; Singh, M.; O’Rourke, K. Another “Complex” Case: Complex I Deficiency Secondary to Acyl-CoA Dehydrogenase 9 Mutation. Am. J. Med. Sci. 2015, 350, 423–424. [Google Scholar] [CrossRef]
- Di Pisa, V.; Cecconi, I.; Gentile, V.; Di Pietro, E.; Marchiani, V.; Verrotti, A.; Franzoni, E. Case report of pyruvate dehydrogenase deficiency with unusual increase of fats during ketogenic diet treatment. J. Child Neurol. 2012, 27, 1593–1596. [Google Scholar] [CrossRef]
- Pons, R.; Cavadini, P.; Baratta, S.; Invernizzi, F.; Lamantea, E.; Garavaglia, B.; Taroni, F. Clinical and molecular heterogeneity in very-long-chain acyl-coenzyme A dehydrogenase deficiency. Pediatr. Neurol. 2000, 22, 98–105. [Google Scholar] [CrossRef]
- Gong, K.; Xie, L.; Wu, Z.-S.; Xie, X.; Zhang, X.-X.; Chen, J.-L. Clinical exome sequencing reveals a mutation in PDHA1 in Leigh syndrome: A case of a Chinese boy with lethal neuropathy. Mol. Genet. Genom. Med. 2021, 9, e1651. [Google Scholar] [CrossRef]
- Teitelbaum, J.E.; Berde, C.B.; Nurko, S.; Buonomo, C.; Perez-Atayde, A.R.; Fox, V.L. Diagnosis and management of MNGIE syndrome in children: Case report and review of the literature. J. Pediatr. Gastroenterol. Nutr. 2002, 35, 377–383. [Google Scholar] [CrossRef]
- Morava, E.; Rodenburg, R.; van Essen, H.Z.; De Vries, M.; Smeitink, J. Dietary intervention and oxidative phosphorylation capacity. J. Inherit. Metab. Dis. 2006, 29, 589. [Google Scholar] [CrossRef]
- Béghin, L.; Coopman, S.; Schiff, M.; Vamecq, J.; Mention-Mulliez, K.; Hankard, R.; Cuisset, J.M.; Ogier, H.; Gottrand, F.; Dobbelaere, D. Doubling diet fat on sugar ratio in children with mitochondrial OXPHOS disorders: Effects of a randomized trial on resting energy expenditure, diet induced thermogenesis and body composition. Clin. Nutr. 2016, 35, 1414–1422. [Google Scholar] [CrossRef]
- Laugel, V.; This-Bernd, V.; Cormier-Daire, V.; Speeg-Schatz, C.; de Saint-Martin, A.; Fischbach, M. Early-onset ophthalmoplegia in Leigh-like syndrome due to NDUFV1 mutations. Pediatr. Neurol. 2007, 36, 54–57. [Google Scholar] [CrossRef]
- Illingworth, M.A.; Boyd, S.G.; Varadkar, S.; Rahman, S. Epileptic phenotypes in children with proven mitochondrial disease and epilepsy. J. Inherit. Metab. Dis. 2012, 35, S120. [Google Scholar]
- Zweers, H.; Smit, D.; Leij, S.; Wanten, G.; Janssen, M.C.H. Individual dietary intervention in adult patients with mitochondrial disease due to the m.3243 A>G mutation. Nutrition 2020, 69, 110544. [Google Scholar] [CrossRef]
- Nangia, S.; Caraballo, R.H.; Kang, H.-C.; Nordli, D.R.; Scheffer, I.E. Is the ketogenic diet effective in specific epilepsy syndromes? Epilepsy Res. 2012, 100, 252–257. [Google Scholar] [CrossRef]
- Kang, H.-C.; Kim, H.D.; Lee, Y.M.; Han, S.H. Landau-Kleffner syndrome with mitochondrial respiratory chain-complex I deficiency. Pediatr. Neurol. 2006, 35, 158–161. [Google Scholar] [CrossRef]
- Craigen, W.J. Leigh disease with deficiency of lipoamide dehydrogenase: Treatment failure with dichloroacetate. Pediatr. Neurol. 1996, 14, 69–71. [Google Scholar] [CrossRef]
- Wijburg, F.A.; Barth, P.G.; Bindoff, L.A.; Birch-Machin, M.A.; van der Blij, J.F.; Ruitenbeek, W.; Turnbull, D.M.; Schutgens, R.B. Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: Results of treatment with a ketogenic diet. Neuropediatrics 1992, 23, 147–152. [Google Scholar] [CrossRef]
- Ahola, S.; Auranen, M.; Isohanni, P.; Niemisalo, S.; Urho, N.; Buzkova, J.; Velagapudi, V.; Lundbom, N.; Hakkarainen, A.; Muurinen, T.; et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 2016, 8, 1234–1247. [Google Scholar] [CrossRef]
- Wexler, I.D.; Hemalatha, S.G.; McConnell, J.; Buist, N.R.; Dahl, H.H.; Berry, S.A.; Cederbaum, S.D.; Patel, M.S.; Kerr, D.S. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology 1997, 49, 1655–1661. [Google Scholar] [CrossRef]
- Barnerias, C.; Saudubray, J.-M.; Touati, G.; De Lonlay, P.; Dulac, O.; Ponsot, G.; Marsac, C.; Brivet, M.; Desguerre, I. Pyruvate dehydrogenase complex deficiency: Four neurological phenotypes with differing pathogenesis. Dev. Med. Child Neurol. 2010, 52, e1–e9. [Google Scholar] [CrossRef]
- Suntum, T.; Allen, N.; Pagano, S.; Jaworski, M.L.; Duncan, L.; Lee, C.C. Remembering MUDPILES: A Case of Unexplained Metabolic Acidosis. Hosp. Pediatr. 2017, 7, 357–360. [Google Scholar] [CrossRef]
- Deberles, E.; Maragnes, P.; Penniello-Valette, M.-J.; Allouche, S.; Joubert, M. Reversal of Cardiac Hypertrophy with a Ketogenic Diet in a Child With Mitochondrial Disease and Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2020, 36, 1690.e1–1690.e3. [Google Scholar] [CrossRef]
- Kang, H.-C.; Lee, Y.-M.; Kim, H.D.; Lee, J.S.; Slama, A. Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects. Epilepsia 2007, 48, 82–88. [Google Scholar] [CrossRef]
- Theunissen, T.E.; Gerards, M.; Hellebrekers, D.M.; Van Tienen, F.H.; Kamps, R.; Sallevelt, S.C.; Hartog, E.N.; Scholte, H.R.; Verdijk, R.M.; Schoonderwoerd, K.; et al. Selection and characterization of palmitic acid responsive patients with an OXPHOS complex i defect. Front. Mol. Neurosci. 2017, 10, 336. [Google Scholar] [CrossRef]
- Berio, A. The Kearns-Sayre syndrome. Pediatr. Med. Chir. 1994, 16, 167–171. [Google Scholar]
- Kanabus, M.; Fassone, E.; Hughes, S.D.; Bilooei, S.F.; Rutherford, T.; Donnell, M.O.; Heales, S.J.R.; Rahman, S. The pleiotropic effects of decanoic acid treatment on mitochondrial function in fibroblasts from patients with complex I deficient Leigh syndrome. J. Inherit. Metab. Dis. 2016, 39, 415–426. [Google Scholar] [CrossRef]
- Peterson, P.L. The treatment of mitochondrial myopathies and encephalomyopathies. Biochim. Biophys. Acta 1995, 1271, 275–280. [Google Scholar] [CrossRef]
- ESPGHAN 52nd Annual Meeting Abstracts. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 1–1243. [CrossRef]
- Foschi, F.G.; Morelli, M.C.; Savini, S.; Dall’Aglio, A.C.; Lanzi, A.; Cescon, M.; Ercolani, G.; Cucchetti, A.; Pinna, A.D.; Stefanini, G.F. Urea cycle disorders: A case report of a successful treatment with liver transplant and a literature review. World J. Gastroenterol. 2015, 21, 4063–4068. [Google Scholar] [CrossRef] [PubMed]
- Sato-Shirai, I.; Ogawa, E.; Arisaka, A.; Osaka, H.; Murayama, K.; Kuwajima, M.; Watanabe, M.; Ichimoto, K.; Ohtake, A.; Kumada, S. Valine-restricted diet for patients with ECHS1 deficiency: Divergent clinical outcomes in two Japanese siblings. Brain Dev. 2021, 43, 308–313. [Google Scholar] [CrossRef]
| Authors | POLG Mutation | Complex Deficiencies | Gender | Symptoms at Onset | Age at Onset of Symptoms | Diagnosis | Age at Diagnosis | Age at Death | Reason for Death | Co-Medication | Valproate |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Scalais et al., 2011 [24] | C.1399G>A + C.2542G>A | Complex IV | F | Hypoglycemia, hyperlactatemia | 3–5 mo. | AS | 3–5 y | 5 y | Respiratory failure | AED | Avoided |
| Koessler et al., 2021 [23] | C.1399G>A | N/A | F | Refractory status epilepticus | 16 y | AHS | 16 y | 16 y | Apnea | LEV+PB+LCM | 2 days |
| Khan et al., 2011 [25] | C.1399>A + C.3562T>C | Complex IV+V | M | Epileptic seizures | 9 mo. | AHS | 9 mo. | 14 mo. | Heart failure, Respiratory failure | N/A | N/A |
| Joshi et al., 2009 [26] | C.2243G>C + C.2480+1g>A | Normal | F | Status epilepticus | 31 mo. | AHS | 55 mo. | 66 mo. | Respiratory failure | LEV+NP+ESM | Avoided |
| Spiegler et al., 2011 [27] | C.911 T>G + C3434insGAGG | N/A | M | Developmental delay | 15 mo. | AHS | 33 mo. | 35 mo. | N/A | N/A | Short period |
| Spiegler et al., 2011 [27] | C.1399G>A + C.844T>G | N/A | F | Developmental delay | 18 mo. | AHS | 45 mo. | 46 mo. | N/A * | LCM+TPM | Short period |
| O’Connor et al., 2014 [28] | N/A ** | N/A | M | Status epilepticus | 10 mo. | AHS | N/A | N/A **** | Liver failure | N/A *** | N/A |
| Martikainen et al., 2012 [29] | C.2243G>C | N/A | M | Seizures, headaches, visual and speech disturbances | 26 y | N/A | N/A | Alive in the process | Alive in the process | PHT+OXC+LEV | N/A |
| Cardenas and Amato, 2010 [30] | C.911T>G + C.1174C>G + 3240–3242 duplication | N/A | F | Epilepsia partialias continua | 14 mo. | AHS | 14 mo. | 19 mo. | Liver failure, status epilepticus | Multidrug | Avoided |
| Authors | Diet Treatment | Beta-Hydroxybutyrate | Diet Registration | Age at Initiating Diet Treatment | Duration of Diet Treatment | Nutritional Supplements |
|---|---|---|---|---|---|---|
| Scalais et al., 2011 [24] | LCT-restrictions * | N/A | N/A | 5.5 mo. (63 days after onset) | N/A | N/A |
| Koessler et al., 2021 [23] | 4:1 KD | >2 mmol/L day 5 on KD | N/A | 16 y (9 days after onset) | 3 mo. | riboflavin, Q10, thiamine |
| Khan et al., 2011 [25] | KD | N/A | N/A | 13 mo. | N/A | N/A |
| Joshi et al., 2009 [26] | CKD | N/A | N/A | 55 mo. | Until death (66 mo.) | Carnitine |
| Spiegler et al., 2011 [27] | KD (PEG) | N/A | N/A | 33 mo. | Discontinued after 2 weeks | N/A |
| Spiegler et al., 2011 [27] | KD (PEG: Ketocal advanced) | N/A | N/A | 44 mo. | Until death (46 mo.) | N/A |
| O´Connor et al., 2014 [28] | 4:1 KD | 5.1 mmol/L | N/A | 10.5 mo. (15 days after onset) | Until death | N/A |
| Martikainen et al., 2012 [29] | LGIT | N/A | N/A | 26 y (7 days after onset) | Continued | N/A |
| Cardenas and Amato, 2010 [30] | KD (NG) | Measured ** | N/A | 14 mo. | N/A | N/A |
| Scalais et al., 2011 [24] | Koessler et al., 2021 [23] | Khan et al., 2011 [25] | Joshi et al., 2009 [26] | Spiegler et al., 2011 [27] | Spiegler et al., 2011 [27] | O´Connor et al., 2014 [28] | Martikainen et al., 2012 [29] | Cardenas and Amato, 2010 [30] | |
|---|---|---|---|---|---|---|---|---|---|
| Title | I | I | I | N | N | N | N | N | I |
| Keywords | I | Y | Y | N | I | I | I | I | N |
| Abstract | Y | Y | Y | Y | I | I | Y | Y | Y |
| Introduction | Y | Y | Y | Y | Y | Y | Y | Y | N |
| Patient information | Y | Y | Y | Y | I | I | I | I | Y |
| Clinical findings | Y | I | Y | Y | Y | Y | I | I | I |
| Timeline | Y | Y | Y | Y | I | I | I | I | I |
| Diagnostic assessment | Y | Y | Y | Y | Y | Y | I | Y | Y |
| Therapeutic intervention | I | Y | I | Y | I | I | I | I | I |
| Follow-up and outcomes | I | Y | I | I | I | I | I | I | I |
| Discussion | Y | Y | Y | Y | Y | Y | I | Y | I |
| Patient perspective | N | N | N | Y | N | N | N | I | N |
| Informed consent | I | I | Y | I | I | I | I | I | I |
| Authors | Reduction in Epileptic Seizures | EEG Improvements | Clinical Symptom Improvements | Effect on AEDs |
|---|---|---|---|---|
| Scalais et al., 2011 [24] | % | % | X ****** | N/A |
| Koessler et al., 2021 [23] | % | X | X ***** | X ************ |
| Khan et al., 2011 [25] | X | % | X******** | N/A |
| Joshi et al., 2009 [26] | X ** | X | X * | N/A |
| Spiegler et al., 2011 [27] | % *** | % | % | N/A |
| Spiegler et al., 2011 [27] | X | % | X ********* | N/A |
| O´Connor et al., 2014 [28] | X | X | X ********** | N/A |
| Martikainen et al., 2012 [29] | X | % | X **** | N/A |
| Cardenas and Amato, 2010 [30] | X ******* | % | X *********** | X *********** |
| Authors | ASAT Pre Diet Treatment | ASAT Post Diet Treatment | ALAT Pre Diet Treatment | ALAT Post Diet Treatment | GGT Pre Diet Treatment | GGT Post Diet Treatment |
|---|---|---|---|---|---|---|
| Scalais et al., 2011 [24] | 244 Ul/l | 100 Ul/L **** | 273 Ul/l | 47 Ul/l **** | N/A | N/A |
| Koessler et al., 2021 [23] | N/A | 704 U/l * | N/A | 782 U/l * | N/A | 1700 U/l |
| Khan et al., 2011 [25] | Moderately elevated ** | N/A *** | Moderately elevated ** | N/A | Moderately elevated | N/A *** |
| Joshi et al., 2009 [26] | 51–73 U/L | 94–112 U/L | 30–47 U/L | 60–101 U/L | N/A | N/A |
| Spiegler et al., 2011 [27] | Moderately elevated ** | 81 U/L ***** | Moderately elevated ** | 61 U/L ***** | N/A | N/A |
| Spiegler et al., 2011 [27] | Moderately elevated ** | 163 U/L ***** | Moderately elevated ** | 130 U/L ***** | N/A | N/A |
| O´Connor et al., 2014 [28] | N/A | N/A | N/A | N/A | N/A | N/A |
| Martikainen et al., 2012 [29] | N/A | N/A | N/A | N/A | N/A | N/A |
| Cardenas and Amato, 2010 [30] | N/A | N/A | N/A | N/A | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, Z.O.; Holm-Yildiz, S.; Dysgaard, T. Nutritional Interventions for Patients with Mitochondrial POLG-Related Diseases: A Systematic Review on Efficacy and Safety. Int. J. Mol. Sci. 2022, 23, 10658. https://doi.org/10.3390/ijms231810658
Pedersen ZO, Holm-Yildiz S, Dysgaard T. Nutritional Interventions for Patients with Mitochondrial POLG-Related Diseases: A Systematic Review on Efficacy and Safety. International Journal of Molecular Sciences. 2022; 23(18):10658. https://doi.org/10.3390/ijms231810658
Chicago/Turabian StylePedersen, Zandra Overgaard, Sonja Holm-Yildiz, and Tina Dysgaard. 2022. "Nutritional Interventions for Patients with Mitochondrial POLG-Related Diseases: A Systematic Review on Efficacy and Safety" International Journal of Molecular Sciences 23, no. 18: 10658. https://doi.org/10.3390/ijms231810658
APA StylePedersen, Z. O., Holm-Yildiz, S., & Dysgaard, T. (2022). Nutritional Interventions for Patients with Mitochondrial POLG-Related Diseases: A Systematic Review on Efficacy and Safety. International Journal of Molecular Sciences, 23(18), 10658. https://doi.org/10.3390/ijms231810658