
Effects of Chronic Kidney Disease on Nanomechanics of the Endothelial Glycocalyx Are Mediated by the Mineralocorticoid Receptor

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
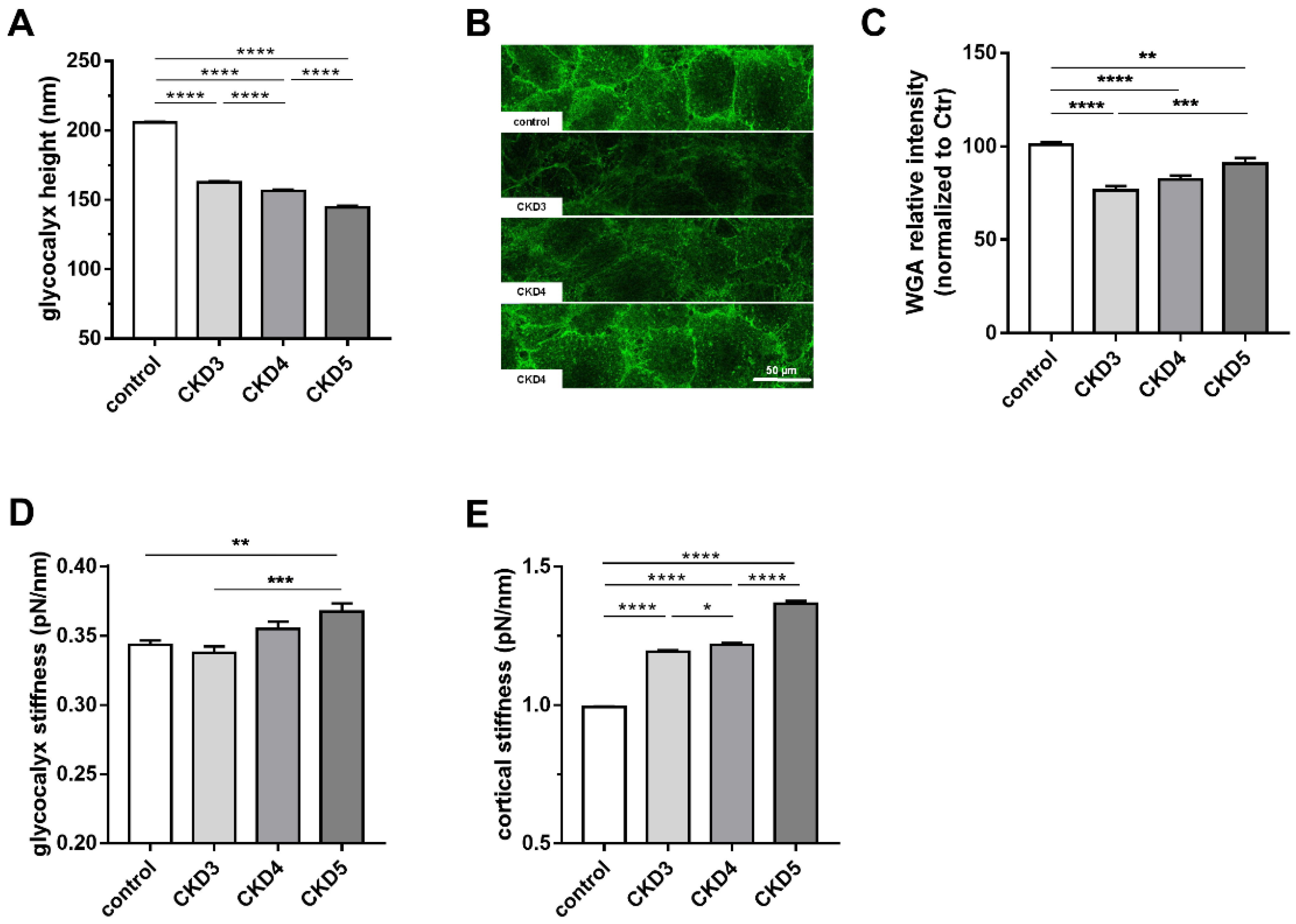
2.1. The eGC Height Decreases Stage-Dependently in Children with CKD
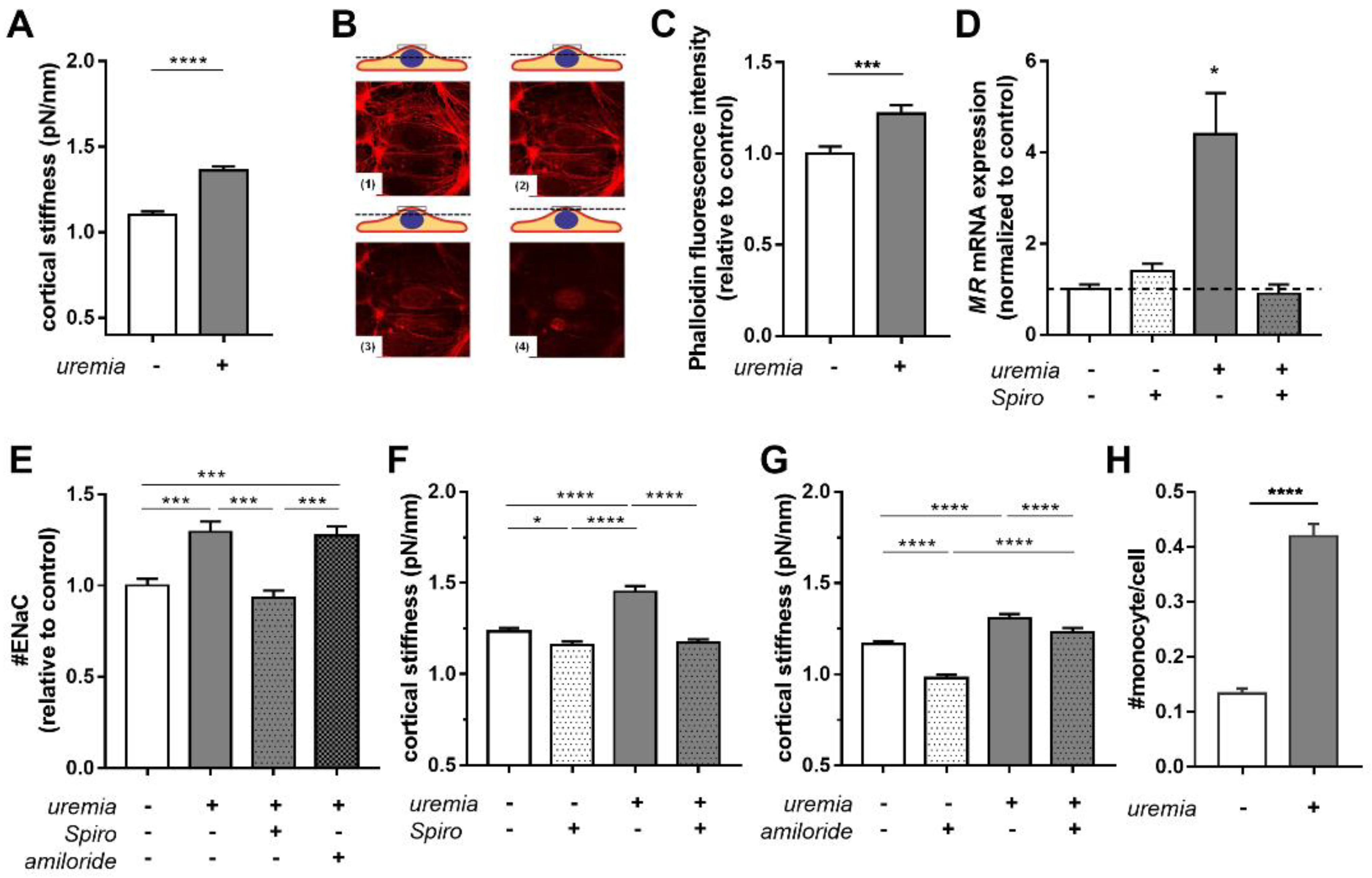
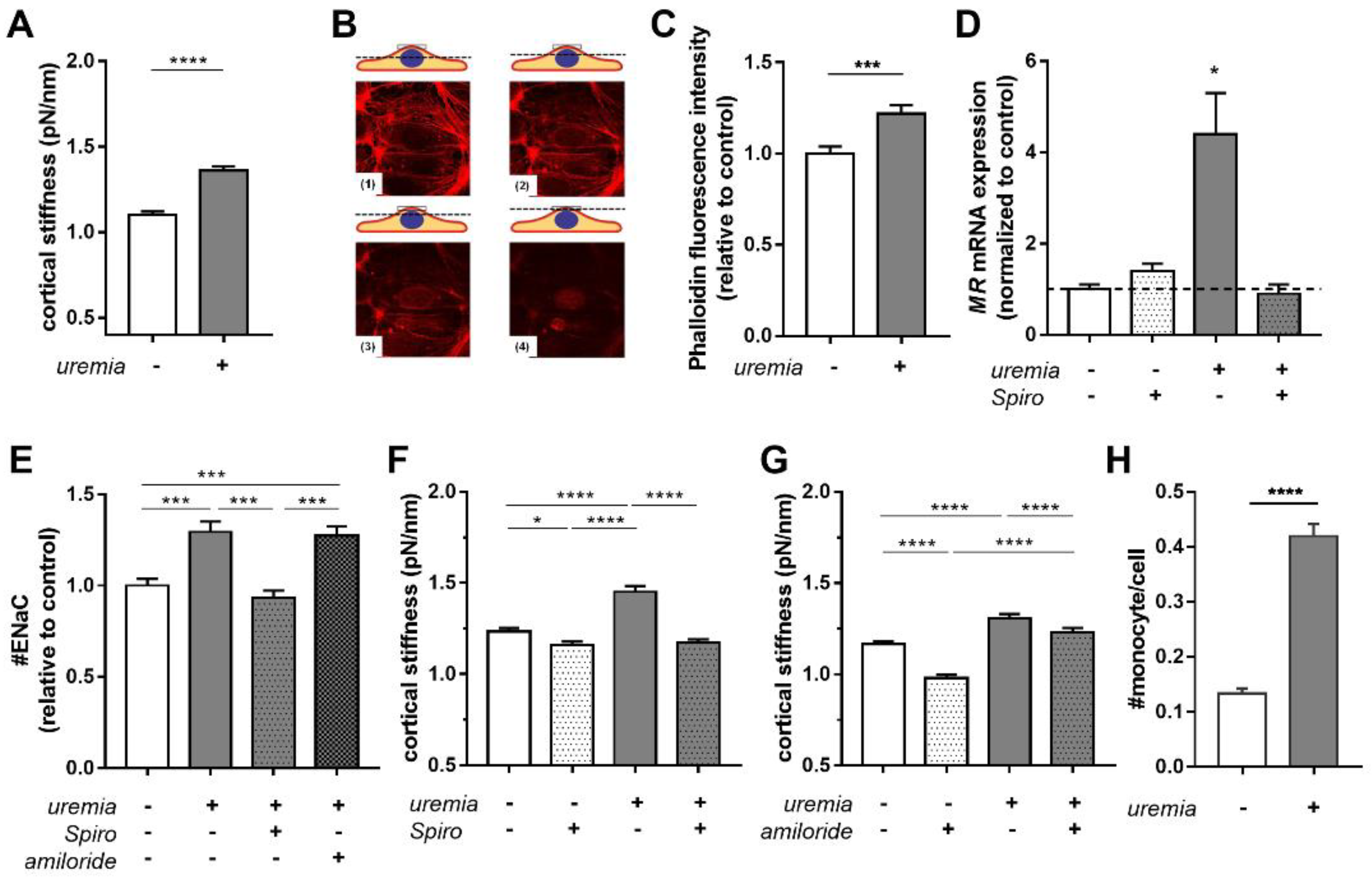
2.2. Uremic Serum Induces Cortical Stiffness and Increases Actin Content in Endothelial Cells
2.3. Uremic Serum Increases Cortical Stiffness in an MR-Dependent Fashion
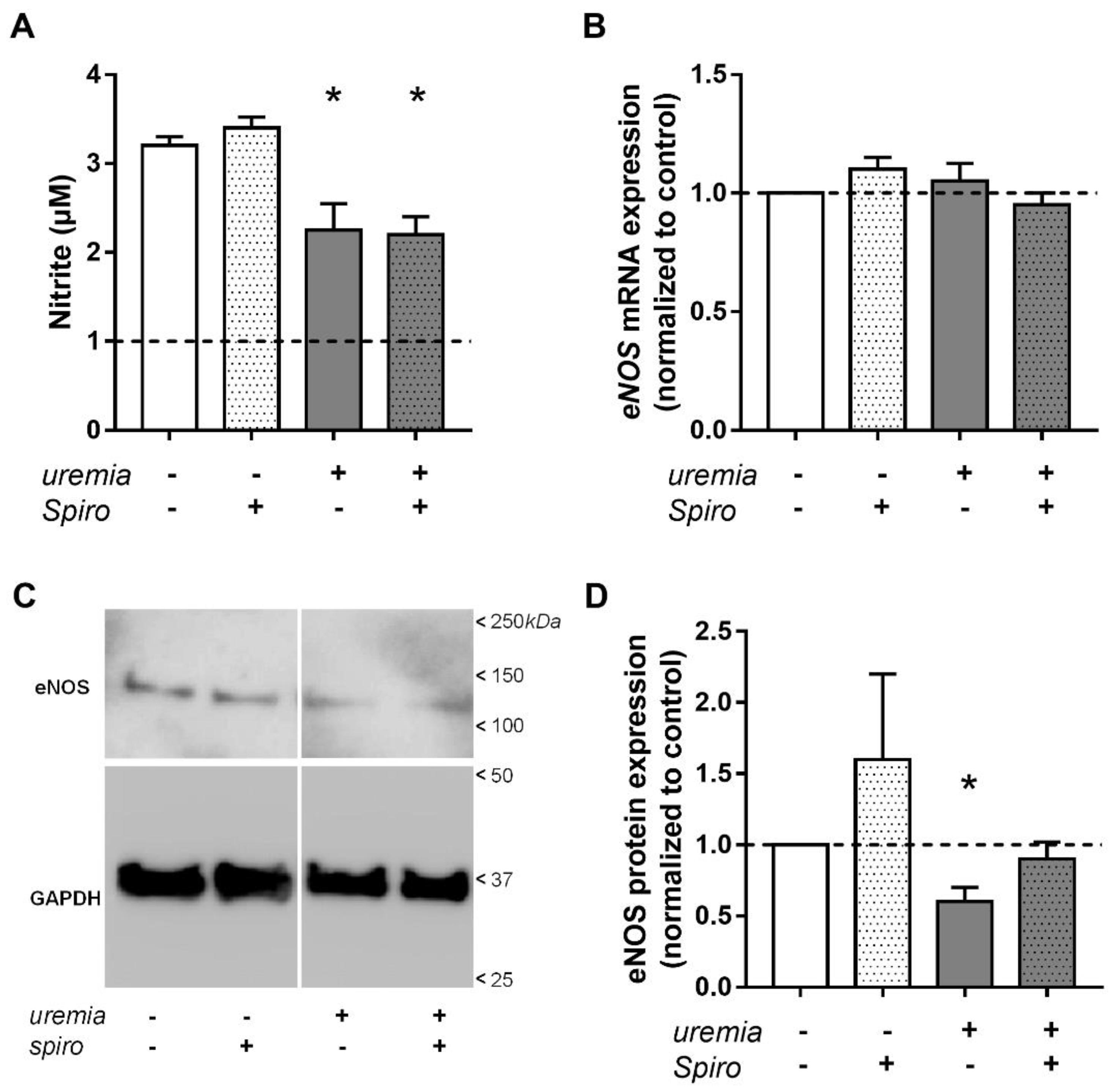
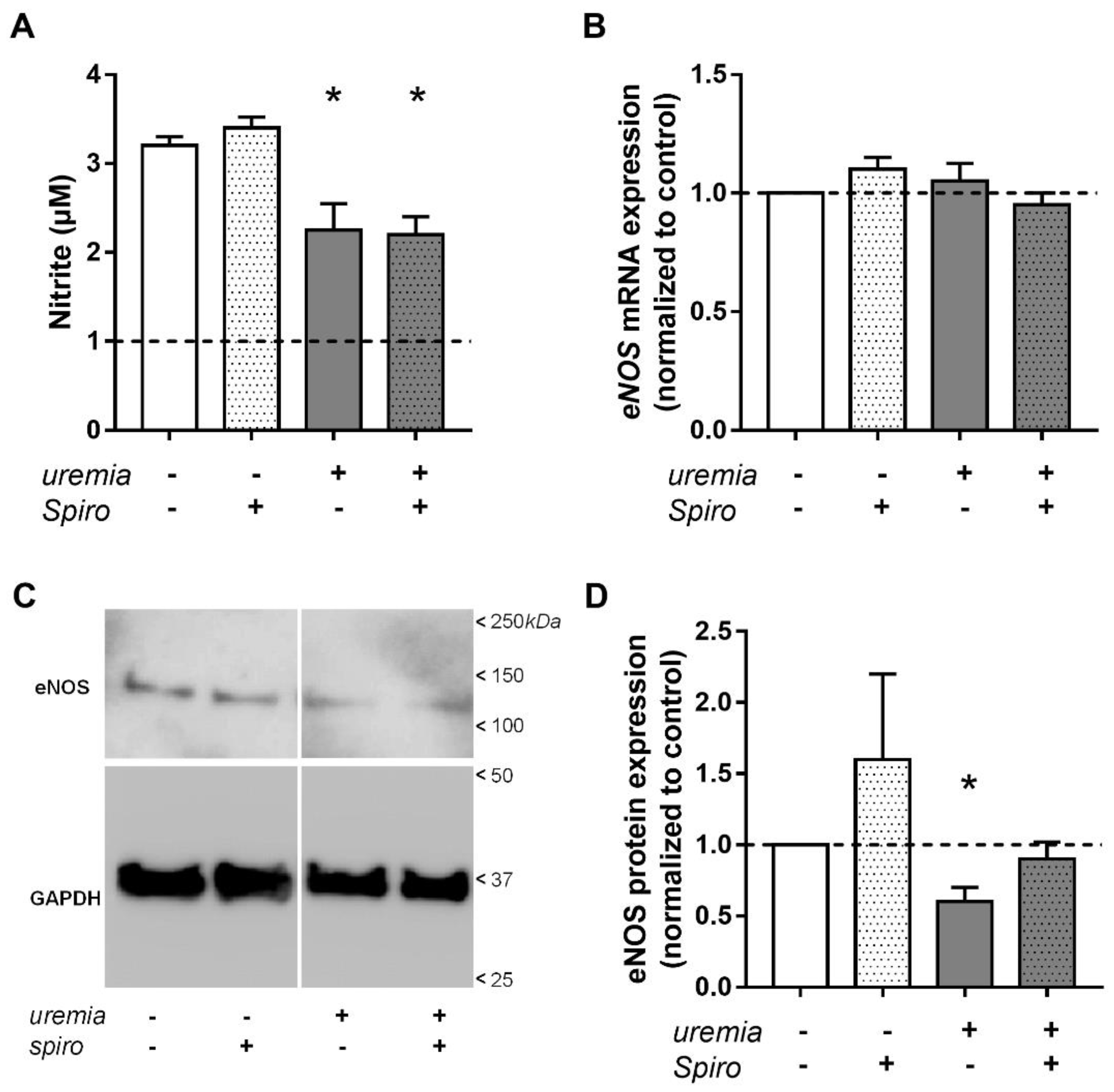
2.4. Uremic Serum Decreases eNOS Protein Abundance and NO Bioavailability
2.5. Correlation with Clinical Data of Children with CKD
3. Discussion
4. Materials and Methods
4.1. Cell Isolation and Culture
4.2. Human Uremic Serum
4.3. Immunofluorescence Staining
4.4. Quantification of Gene Expression
4.5. Western Blot
4.6. Quantification of NO Production
4.7. Monocyte Adhesion Assay
4.8. Atomic Force Microscopy Measurements
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Collaborators The following principal investigators contributed to the 4C Study: | |
|---|---|
| Austria: | Gerard Cortina, Children’s Hospital, Innsbruck; Klaus Arbeiter, University Children’s Hospital, Vienna. |
| Czech Republic: | Jiri Dusek, University Hospital Motol, Prague |
| France: | Jerome Harambat, Hôpital des Enfants, Bordeaux; Bruno Ranchin, Hôpital Femme Mère Enfant et Université de Lyon; Michel Fischbach, Ariane Zaloszyc, Hôpital de Hautepierre, Strasbourg. |
| Germany: | Uwe Querfeld, Jutta Gellermann, Charité Children’s Hospital, Berlin; Sandra Habbig, Max Liebau, University Children’s Hospital, Cologne; Matthias Galiano, University Children’s Hospital, Erlangen; Rainer Büscher, University Children’s Hospital, Essen; Charlotte Gimpel, Center for Pediatrics and Adolescent Medicine, Freiburg; Matthias Kemper, Jun Oh, UKE University Children’s Hospital, Hamburg; Anette Melk, Daniela Thurn-Valassina, Hannover Medical School, Hannover; Anke Doyon, Elke Wühl, Franz Schaefer, Center for Pediatrics and Adolescent Medicine, Heidelberg; Ulrike John, Center for Pediatrics and Adolescent Medicine, Jena; Simone Wygoda, City Hospital St. Georg, Leipzig; Nicola Jeck, KfH Kidney Center for Children, Marburg; Birgitta Kranz, University Children’s Hospital, Muenster; Marianne Wigger, Children’s Hospital, Rostock. |
| Italy: | Francesca Mencarelli, S. Orsola-Malpighi Hospital, Bologna; Francesca Lugani, Istituto Giannina Gaslini, Genova; Sara Testa, Giovanni Montini, William Morello, Fondazione Ospedale Maggiore Policlinico, Milano; Enrico Vidal, Elisa Benetti, Luisa Murer, Pediatric Nephrology, Dialysis & Transplant Unit, Padova; Ciara Matteucci, Stefano Picca, Ospedale Bambino Gesù, Rome. |
| Lithuania | Augustina Jankauskiene, Karolis Azukaitis, University Children’s Hospital, Vilnius |
| Poland | Aleksandra Zurowska, Ilona Zagozozon, Pediatric and Adolescent Nephrology, Gdansk; Dorota Drodz, University Children’s Hospital, Krakow; Tomasz Urasinski, Clinic of Pediatrics, Szczecin; Mieczyslaw Litwin, Anna Niemirska, Lukasz Obycki, Children’s Memorial Health Institute, Warsaw; Maria Szczepanska, Zabrze. |
| Portugal | Ana Texeira, Hospital Sao Joao, Porto |
| Serbia | Amira Peco-Antic, Dusan Paripovic, University Children’s Hospital, Belgrade. |
| Switzerland | Giacomo Simonetti, Inselspital, Bern; Guido Laube, University Children’s Hospital, Zurich. |
| Turkey | Ali Anarat, Aysun K. Bayazit, Cukurova University, Adana; Fatos Yalcinkaya, University Faculty of Medicine, Ankara; Esra Baskin, Baskent University Faculty of Medicine, Ankara; Nilgun Cakar, Diskapi Children’s Hospital, Ankara; Oguz Soylemezoglu, Gazi University Hospital, Ankara; Ali Duzova, Yelda Bilginer, Hacettepe Medical Faculty, Ankara; Hakan Erdogan, Dortcelik Children’s Hospital, Bursa; Osman Donmez, Uludag University, Bursa; Ayse Balat, University of Gaziantep; Aysel Kiyak, Bakirkoy Children’s Hospital, Istanbul; Salim Caliskan, Nur Canpolat, Mahmut Civilibal, Istanbul University Cerrahpasa Faculty of Medicine, Istanbul; Cengiz Candan, Goztepe Educational and Research Hospital, Istanbul; Sevinc Emre, Alev Yilmaz, Istanbul Medical Faculty, Istanbul; Harika Alpay, Marmara University Medical Faculty, Istanbul; Gul Ozcelik, Sisli Educational and Research Hospital, Istanbul; Sevgi Mir, Betul Sözeri, Ipek K. Bulut, Ege University Medical Faculty; Izmir; Nejat Aksu, Onder Yavascan, Tepecik Training and Research Hospital, Izmir; Yilmaz Tabel, Inonu University, Malatya; Pelin Ertan, Celal Bayar University, Manisa; Ebru Yilmaz, Children’s Hospital, Sanliurfa. |
| United Kingdom | Rukshana Shroff, Great Ormond Street Hospital, London. |
References
- Stam, F.; van Guldener, C.; Becker, A.; Dekker, J.M.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. Endothelial Dysfunction Contributes to Renal Function-Associated Cardiovascular Mortality in a Population with Mild Renal Insufficiency: The Hoorn Study. J. Am. Soc. Nephrol. 2006, 17, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Vila Cuenca, M.; Hordijk, P.L.; Vervloet, M.G. Most Exposed: The Endothelium in Chronic Kidney Disease. Nephrol. Dial. Transplant. 2020, 35, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Cosgun, Z.C.; Fels, B.; Kusche-Vihrog, K. Nanomechanics of the Endothelial Glycocalyx: From Structure to Function. Am. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2021, 12, 37–71. [Google Scholar] [CrossRef]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef]
- Lang, F. Stiff Endothelial Cell Syndrome in Vascular Inflammation and Mineralocorticoid Excess. Hypertension 2011, 57, 146–147. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Jeggle, P.; Oberleithner, H. The Role of ENaC in Vascular Endothelium. Pflugers Arch. 2013. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Tarjus, A.; Fels, J.; Jaisser, F. The Epithelial Na+ Channel: A New Player in the Vasculature. Curr. Opin. Nephrol. Hypertens. 2014, 23, 143–148. [Google Scholar] [CrossRef]
- Jeggle, P.; Callies, C.; Tarjus, A.; Fassot, C.; Fels, J.; Oberleithner, H.; Jaisser, F.; Kusche-Vihrog, K. Epithelial Sodium Channel Stiffens the Vascular Endothelium in Vitro and in Liddle Mice. Hypertension 2013, 61, 1053–1059. [Google Scholar] [CrossRef]
- Perez, F.R.; Venegas, F.; Gonzalez, M.; Andres, S.; Vallejos, C.; Riquelme, G.; Sierralta, J.; Michea, L. Endothelial Epithelial Sodium Channel Inhibition Activates Endothelial Nitric Oxide Synthase via Phosphoinositide 3-Kinase/Akt in Small-Diameter Mesenteric Arteries. Hypertension 2009, 53, 1000–1007. [Google Scholar] [CrossRef]
- Tarjus, A.; Maase, M.; Jeggle, P.; Martinez-Martinez, E.; Fassot, C.; Loufrani, L.; Henrion, D.; Hansen, P.B.L.; Kusche-Vihrog, K.; Jaisser, F. The Endothelial ΑENaC Contributes to Vascular Endothelial Function in Vivo. PLoS ONE 2017, 12, e0185319. [Google Scholar] [CrossRef] [PubMed]
- Padberg, J.-S.; Wiesinger, A.; di Marco, G.S.; Reuter, S.; Grabner, A.; Kentrup, D.; Lukasz, A.; Oberleithner, H.; Pavenstädt, H.; Brand, M.; et al. Damage of the Endothelial Glycocalyx in Chronic Kidney Disease. Atherosclerosis 2014, 234, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Dane, M.J.; Khairoun, M.; Lee, D.H.; van den Berg, B.M.; Eskens, B.J.; Boels, M.G.; van Teeffelen, J.W.; Rops, A.L.; van der Vlag, J.; van Zonneveld, A.J.; et al. Association of Kidney Function with Changes in the Endothelial Surface Layer. Clin. J. Am. Soc. Nephrol. 2014, 9, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Liew, H.; Roberts, M.A.; Pope, A.; McMahon, L.P. Endothelial Glycocalyx Damage in Kidney Disease Correlates with Uraemic Toxins and Endothelial Dysfunction. BMC Nephrol. 2021, 22, 21. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.S.D.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How Do Uremic Toxins Affect the Endothelium? Toxins 2020, 12, 412. [Google Scholar] [CrossRef]
- Epstein, M. Aldosterone and Mineralocorticoid Receptor Signaling as Determinants of Cardiovascular and Renal Injury: From Hans Selye to the Present. Am. J. Nephrol. 2021, 52, 209–216. [Google Scholar] [CrossRef]
- Agarwal, R.; Anker, S.D.; Bakris, G.; Filippatos, G.; Pitt, B.; Rossing, P.; Ruilope, L.; Gebel, M.; Kolkhof, P.; Nowack, C.; et al. Investigating New Treatment Opportunities for Patients with Chronic Kidney Disease in Type 2 Diabetes: The Role of Finerenone. Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Urbanova, K.; Blanqué, A.; Wilhelmi, M.; Schillers, H.; Kliche, K.; Pavenstädt, H.; Brand, E.; Oberleithner, H. C-Reactive Protein Makes Human Endothelium Stiff and Tight. Hypertension 2011, 57, 231–237. [Google Scholar] [CrossRef]
- McIntyre, C.W.; Harrison, L.E.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating Endotoxemia: A Novel Factor in Systemic Inflammation and Cardiovascular Disease in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Meuwese, M.C.; Mooij, H.L.; van Lieshout, M.H.; Hayden, A.; Levi, M.; Meijers, J.C.; Ince, C.; Kastelein, J.J.; Vink, H.; et al. Tumor Necrosis Factor-Alpha Inhibition Protects against Endotoxin-Induced Endothelial Glycocalyx Perturbation. Atherosclerosis 2009, 202, 296–303. [Google Scholar] [CrossRef]
- Holle, J.; Querfeld, U.; Kirchner, M.; Anninos, A.; Okun, J.; Thurn-Valsassina, D.; Bayazit, A.; Niemirska, A.; Canpolat, N.; Bulut, I.K.; et al. Indoxyl Sulfate Associates with Cardiovascular Phenotype in Children with Chronic Kidney Disease. Pediatr. Nephrol. 2019, 34, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-Bound Uremic Toxins Stimulate Crosstalk between Leukocytes and Vessel Wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Briet, M.; Boutouyrie, P.; Laurent, S.; London, G.M. Arterial Stiffness and Pulse Pressure in CKD and ESRD. Kidney Int. 2012, 82, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.L.; Palmisano, J.N.; Benjamin, E.J.; Larson, M.G.; Vasan, R.S.; Mitchell, G.F.; Hamburg, N.M. Microvascular Function Contributes to the Relation Between Aortic Stiffness and Cardiovascular Events: The Framingham Heart Study. Circ. Cardiovasc. Imaging 2016, 9, e004979. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Lee, C.S.; Chiu, Y.W.; Kuo, H.T.; Lee, S.C.; Hwang, S.J.; Kuo, M.C.; Chen, H.C. Angiopoietin-2, Angiopoietin-1 and Subclinical Cardiovascular Disease in Chronic Kidney Disease. Sci. Rep. 2016, 6, 39400. [Google Scholar] [CrossRef]
- Baylis, C. Nitric Oxide Deficiency in Chronic Kidney Disease. Am. J. Physiol. Renal. Physiol. 2008, 294, F1–F9. [Google Scholar] [CrossRef]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef]
- Lukasz, A.; Hillgruber, C.; Oberleithner, H.; Kusche-Vihrog, K.; Pavenstädt, H.; Rovas, A.; Hesse, B.; Goerge, T.; Kümpers, P. Endothelial Glycocalyx Breakdown Is Mediated by Angiopoietin-2. Cardiovasc. Res. 2017, 113, 671–680. [Google Scholar] [CrossRef]
- Querfeld, U.; Anarat, A.; Bayazit, A.K.; Bakkaloglu, A.S.; Bilginer, Y.; Caliskan, S.; Civilibal, M.; Doyon, A.; Duzova, A.; Kracht, D.; et al. The Cardiovascular Comorbidity in Children with Chronic Kidney Disease (4C) Study: Objectives, Design, and Methodology. Clin. J. Am. Soc. Nephrol. 2010, 5, 1642–1648. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Muñoz, A.; Schneider, M.F.; Mak, R.H.; Kaskel, F.; Warady, B.A.; Furth, S.L. New Equations to Estimate GFR in Children with CKD. J. Am. Soc. Nephrol. 2009, 20, 629–637. [Google Scholar] [CrossRef]
- Cazana-Perez, V.; Cidad, P.; Donate-Correa, J.; Martin-Nunez, E.; Lopez-Lopez, J.R.; Perez-Garcia, M.T.; Giraldez, T.; Navarro-Gonzalez, J.F.; Alvarez de la Rosa, D. Phenotypic Modulation of Cultured Primary Human Aortic Vascular Smooth Muscle Cells by Uremic Serum. Front. Physiol. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Klucka, J.; Stourac, P.; Krikava, I.; Stoudek, R.; Toukalkova, M.; Michalek, P.; Cerny, V. The Czech RSI 2016 study group: Rapid sequence induction in the Czech Republic 2016: Survey. Anesteziol. Intenziv. 2017, 28, 232–239. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative C(T) Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Snell, F.D.; Snell, C.T. Colorimetric Methods of Analysis, Including Some Turbidimetric and Nephelometric Methods, 3rd ed.; Van Nostrand: New York, NY, USA, 1949; pp. 804–805. [Google Scholar]
- Privat, C.; Lantoine, F.; Bedioui, F.; Millanvoye van Brussel, E.; Devynck, J.; Devynck, M.A. Nitric Oxide Production by Endothelial Cells: Comparison of Three Methods of Quantification. Life Sci. 1997, 61, 1193–1202. [Google Scholar] [CrossRef]
- Schierke, F.; Wyrwoll, M.J.; Wisdorf, M.; Niedzielski, L.; Maase, M.; Ruck, T.; Meuth, S.G.; Kusche-Vihrog, K. Nanomechanics of the Endothelial Glycocalyx Contribute to Na+-Induced Vascular Inflammation. Sci. Rep. 2017, 7, 46476. [Google Scholar] [CrossRef]
- Peters, W.; Drueppel, V.; Kusche-Vihrog, K.; Schubert, C.; Oberleithner, H. Nanomechanics and Sodium Permeability of Endothelial Surface Layer Modulated by Hawthorn Extract WS 1442. PLoS ONE 2012, 7, e29972. [Google Scholar] [CrossRef]
- Wiesinger, A.; Peters, W.; Chappell, D.; Kentrup, D.; Reuter, S.; Pavenstädt, H.; Oberleithner, H.; Kümpers, P. Nanomechanics of the Endothelial Glycocalyx in Experimental Sepsis. PLoS ONE 2013, 8, e80905. [Google Scholar] [CrossRef]

| CKD Stage | Log IS | Log PCS | Angpt1 | Angpt2 | cIMT-SDS | PWV-SDS | |
|---|---|---|---|---|---|---|---|
| eGC Height | |||||||
| r = | −0.67 | −0.37 | −0.50 | −0.13 | −0.06 | −0.07 | −0.23 |
| p = | <0.01 | 0.04 | 0.01 | 0.28 | 0.40 | 0.37 | 0.13 |
| eGC Stiffness | |||||||
| r = | 0.30 | 0.33 | 0.35 | 0.14 | −0.11 | 0.04 | 0.37 |
| p = | 0.07 | 0.06 | 0.05 | 0.27 | 0.30 | 0.42 | 0.03 |
| Cortex Stiffness | |||||||
| r = | 0.43 | −0.01 | 0.10 | 0.29 | 0.82 | −0.22 | 0.37 |
| p = | 0.02 | 0.48 | 0.34 | 0.10 | <0.01 | 0.15 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fels, B.; Beyer, A.; Cazaña-Pérez, V.; Giraldez, T.; Navarro-González, J.F.; Alvarez de la Rosa, D.; Schaefer, F.; Bayazit, A.K.; Obrycki, Ł.; Ranchin, B.; et al. Effects of Chronic Kidney Disease on Nanomechanics of the Endothelial Glycocalyx Are Mediated by the Mineralocorticoid Receptor. Int. J. Mol. Sci. 2022, 23, 10659. https://doi.org/10.3390/ijms231810659
Fels B, Beyer A, Cazaña-Pérez V, Giraldez T, Navarro-González JF, Alvarez de la Rosa D, Schaefer F, Bayazit AK, Obrycki Ł, Ranchin B, et al. Effects of Chronic Kidney Disease on Nanomechanics of the Endothelial Glycocalyx Are Mediated by the Mineralocorticoid Receptor. International Journal of Molecular Sciences. 2022; 23(18):10659. https://doi.org/10.3390/ijms231810659
Chicago/Turabian StyleFels, Benedikt, Arne Beyer, Violeta Cazaña-Pérez, Teresa Giraldez, Juan F. Navarro-González, Diego Alvarez de la Rosa, Franz Schaefer, Aysun K. Bayazit, Łukasz Obrycki, Bruno Ranchin, and et al. 2022. "Effects of Chronic Kidney Disease on Nanomechanics of the Endothelial Glycocalyx Are Mediated by the Mineralocorticoid Receptor" International Journal of Molecular Sciences 23, no. 18: 10659. https://doi.org/10.3390/ijms231810659
APA StyleFels, B., Beyer, A., Cazaña-Pérez, V., Giraldez, T., Navarro-González, J. F., Alvarez de la Rosa, D., Schaefer, F., Bayazit, A. K., Obrycki, Ł., Ranchin, B., Holle, J., Querfeld, U., & Kusche-Vihrog, K. (2022). Effects of Chronic Kidney Disease on Nanomechanics of the Endothelial Glycocalyx Are Mediated by the Mineralocorticoid Receptor. International Journal of Molecular Sciences, 23(18), 10659. https://doi.org/10.3390/ijms231810659