1. Introduction
Approximately 6.2 million Americans aged 65 and more have Alzheimer’s disease (AD)-related dementia. The number is increasing at an alarming pace and so is the mortality caused by AD. Between 2000 and 2019, the number of deaths due to AD increased by 145% [
1]. In this context, AD is the neuronal degeneration associated with β-amyloid (Aβ) plaques. Such plaques are composed of the amyloid peptide, and the 42-residue form β amyloid (Aβ1–42) is the predominant peptide causing AD [
2]. Until now, amyloid-targeting therapies remain a prominent avenue for research as well [
3]. The n-butylidenephthalide (BP) was initially extracted from the Chinese herbal medicine
Angelica sinensis and can also be produced by chemical synthesis. BP can reduce Aβ deposits and the hyperphosphorylated status of Tau protein (p-tau) according to in vitro studies [
4]. These results suggest that BP has the potential to treat AD. However, evidence of the BP effect on AD animal models is lacking. Thus, the efficacy of BP in reducing Aβ in AD is still worth exploring through in vivo studies.
Multiple genes play crucial roles in regulating Aβ produced in the brain. They include
amyloid precursor protein (
APP) [
5],
beta-site APP-cleaving enzyme-1 (
BACE1) [
6],
presenilin-1 (
PSEN1), and
presenilin-2 (
PSEN2) [
7]. Previous studies in the literature report that small molecule drugs can regulate γ-secretase by binding to
PSEN1; which becomes a viable treatment strategy for treating AD [
8]. In this study, we will discuss the effect of BP on genes associated with AD, and we consider
PSEN1 as the primary target. In recent years, several experiments demonstrated that aberrant miRNA expression is associated with Aβ production in AD [
9,
10]. For instance, loss of miR-29a/b-1 in sporadic AD correlates with increased
BACE1/β-secretase expression and contributes to the increase of Aβ production [
9]. Likewise, overexpression of miR-16 can cause decreased APP expression [
10]. In this study, we analyze the PSEN1 binding affinity of BP through molecular docking calculation. In addition, we also explore other genes and miRNA that might be regulated by BP. In specific, the cell models of trisomy 21 (Ts21) induced pluripotent stem cells (iPSCs)-derived neurons [
4] and human β-CTF/C6 cells [
11] were used to investigate the mechanism of action of BP in the treatment of AD.
Furthermore, we used positron emission tomography-computed tomography (PET/CT) and Morris water maze to discuss the effect of BP on the behaviors of 3xTg-AD mice [
12]. The Aβ deposition in the brain of 3xTg mice was detected by [18F]-Florbetaben PET/CT, and the spatial learning ability was tested by Morris water maze [
13]. Moreover, we evaluated the association of miRNA-mRNA by establishing the miRNA mutant mice generated by CRISP/Cas9 technology and by analyzing the data collected using brain samples obtained from patients with AD [
14]. Through these investigations, we aim to understand the potential of BP in treating AD in an in vivo setting and further explore the cellular and molecular mechanisms underlying BP’s effect in reducing Aβ levels.
3. Discussion
This study aimed to determine the molecular mechanism underlying the action of BP in AD. In vitro results indicated that the interaction between lnc-CYP3A43-2 and miR-29b-2-5p is implicated directly in Aβ formation. We also identified PSEN1, which is involved in Aβ production, as a target of miR-29b-2-5p, both in vitro and in vivo. We further validated the negative correlation between miR-29b-2-5p and PSEN1 expression in clinical human AD samples. We realized that a further, more extensive sample analysis is helpful in solidifying our hypothesis. Nevertheless, these results concisely emphasized the importance of the lncRNA (LncCYP3A43-2)-miRNA(miR29-2-5p)-mRNA (PSEN1) network in AD pathogenesis.
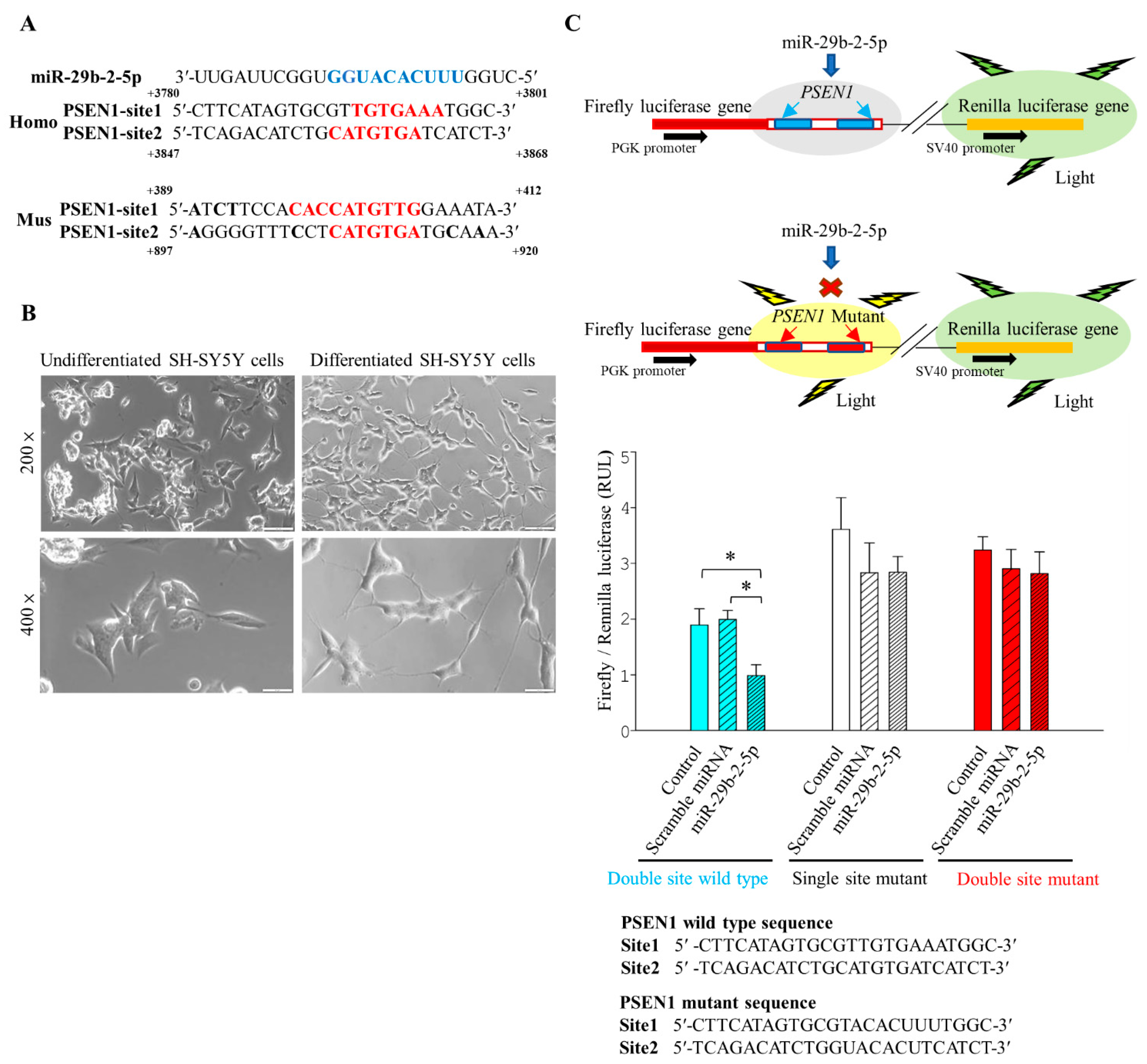
An early intervention strategy for AD treatment was to inhibit the cleavage of γ-secretase and reduce the production of Aβ [
15]. Recent research focused on investigating the use of γ-secretase modulators (GSMs) as AD treatments [
30]. In fact, γ-Secretase contains PSEN1, Nicastrin, Anterior pharynx-defective 1, and Presenilin enhancer 2.
Increasing evidence indicates that targeting
PSEN1 regulation reduced the production of β-amyloid, the main component for Aβ plaque, without affecting the Notch signaling pathway. Besides, it does not affect NICD formation [
18,
31]. Our data demonstrated that BP treatment did not affect NICD production. We speculated that the action of BP is through regulating LncCYP3A43-2/miR-29b-2-5p and in turn downregulating
PSEN1 expression rather than inhibiting all four components of γ-secretase. This explained why Notch signaling was not affected by BP treatment. Further in-depth studies are needed before forming a complete picture of BP effects on improving AD.
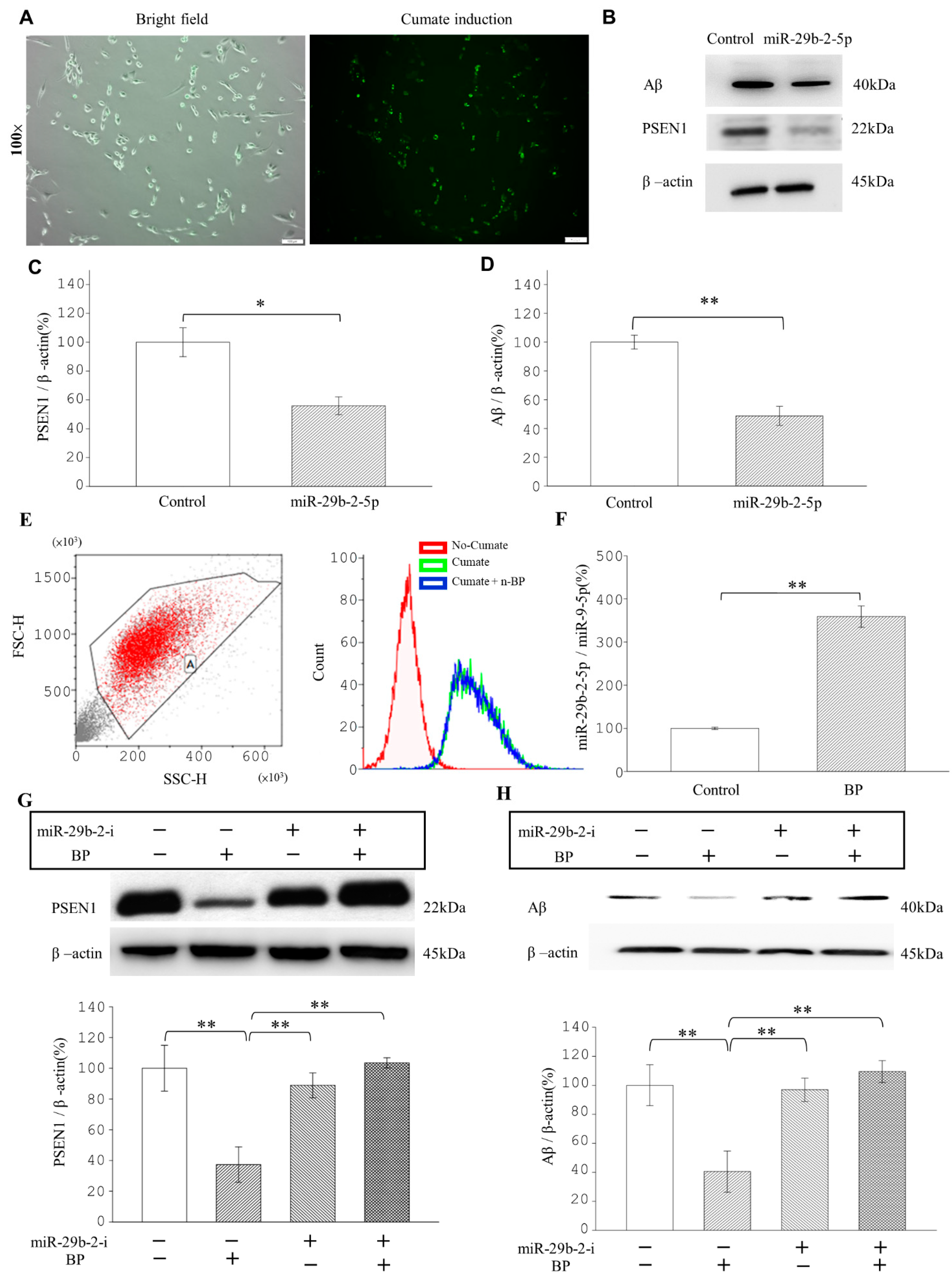
The Ts21-neuron has
APP and
SOD1 gene alterations; the model displayed Aβ production from APP source and intermediate processes in neurons. Our study disclosed that BP did not inhibit APP directly but rather worked via reducing
PSEN1 expression in Ts-21 neurons. A previous study showed that the LncRNAs localized in the cytoplasm were involved in the modulation of mRNA stability [
32], and these lncRNAs played a crucial role in fine-tuning and regulating gene expression via miRNAs and acting as competing endogenous RNAs (ceRNAs) [
33].
PSEN1 regulates the enzymatic cleavage of γ-secretase of APP protein and Notch [
34,
35]. In this study, we found that BP can regulate
PSEN1. BP overlapped with γ-secretase inhibitor DAPT in its binding to the active site of the PSEN1 through molecular docking calculation. However, the PSEN1 binding affinity of BP was not as strong as that of γ-secretase inhibitor DAPT by molecular docking simulations (
Figure 1D).
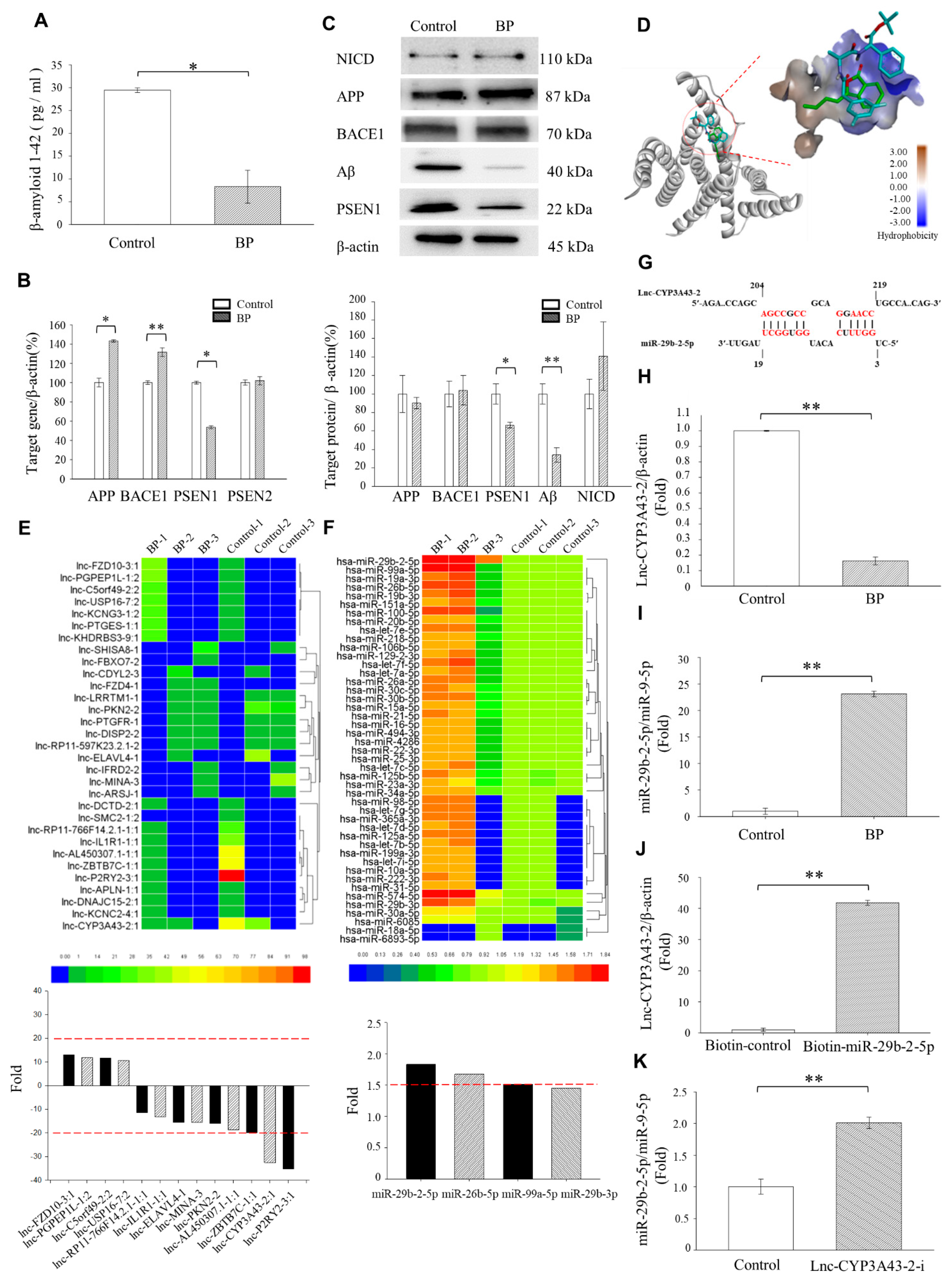
We found that the LncCYP3A43-2 levels were significantly reduced by BP treatment in Ts-21 neurons, and this resulted in a subsequent upregulation of miR-29b-2-5p, confirming the hypothesis that lncRNA acts as an miRNA sponge (
Figure 1H,K). Likewise, our data demonstrated that the therapeutic potential of BP in treating AD was through regulating miRNA-29b-2-5p and consequently reducing
PSEN1 expression (
Figure 2 and
Figure 3). The β-CTF/C6 cells could overexpress
PSEN1 and Aβ that induce neurotoxicity and cause neuronal death. Therefore, this cell line is suitable for observing the action of drugs on
PSEN1 [
11]. When miR-29b-2-5p was suppressed, BP no longer exerted its effect on PSEN1 (
Figure 3G,H). These data provide strong evidence that BP acted via regulating lncRNA-miRNA-mRNA (lnc-CYP3A43-2 -miR-29b-2-5p-
PSEN1) network and in turn reduced β-amyloid accumulation.
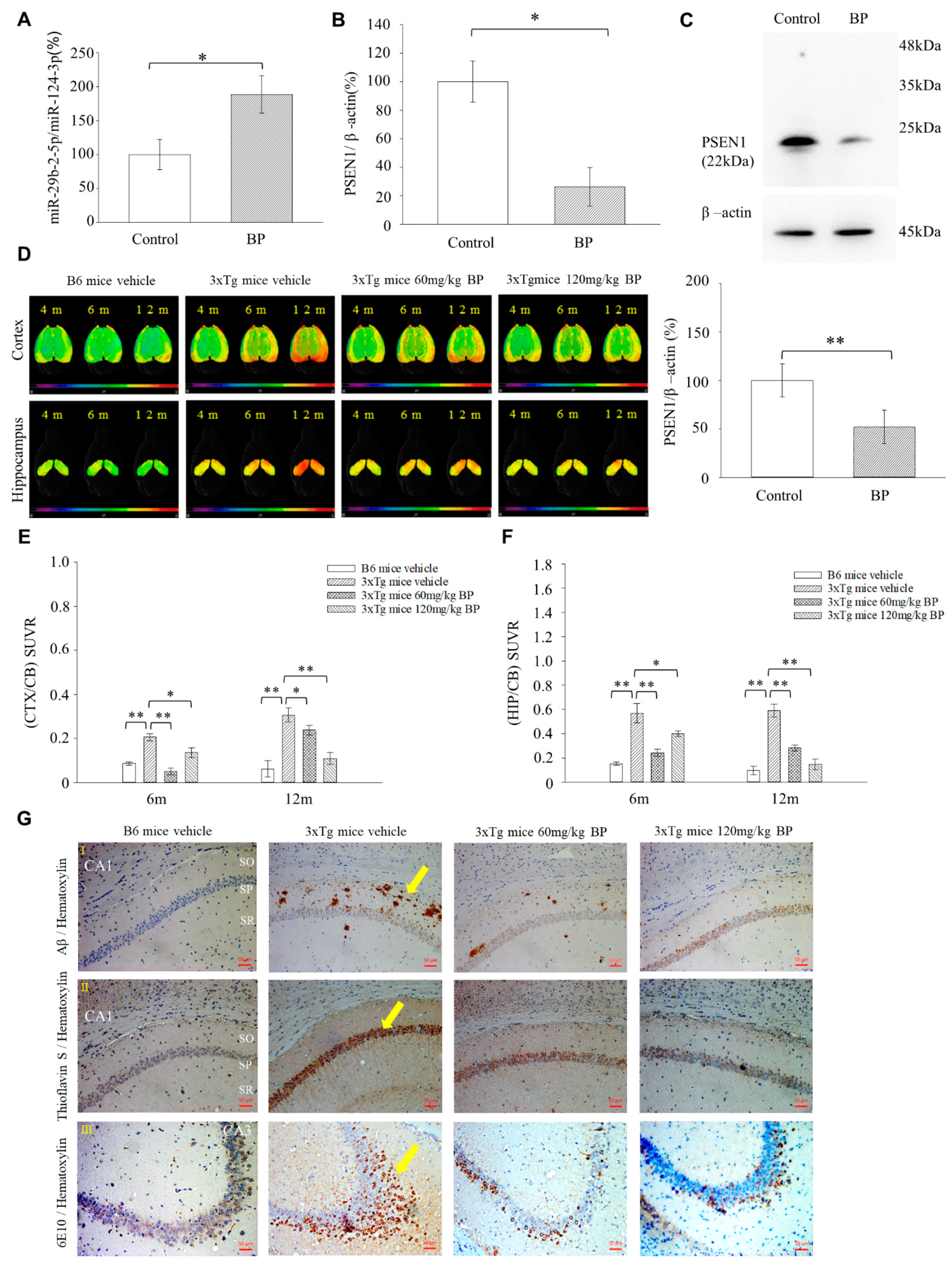
Herein, we are the first to present the therapeutic potential of BP in treating AD using a mouse model (3xTg-AD mice). Our results show that 3xTg AD mice treated with BP had significantly reduced Aβ accumulation in the hippocampus and cortex regions as compared to untreated controls, and this finding is further supported by our [18F]-FBB PET study (
Figure 4D,F). The level of Aβ reduction in the cortex and hippocampus of 3xTg AD mice was related to the concentration of BP used in treatment. In specific, in the cortex, 60 mg/kg BP treatment could not inhibit Aβ accumulation, whereas 120 mg/kg BP treatment could. In the hippocampus, we observed a similar situation: 60 mg/kg BP treatment could maintain the Aβ level in the hippocampus, whereas 120 mg/kg BP treatment could decrease Aβ accumulation. Recent literature showed that in an animal model of Spinocerebellar Ataxia Type 3, BP induced autophagy and the elimination of aggregates [
36]. Thus, we speculate that BP at high concentrations, such as 120 mg/Kg, can play a role in promoting autophagy for the elimination of Aβ in the hippocampus of 3xTg AD mice in addition to its primary function of reducing
PSEN1. The 3xTg mice also produced p-Tau in the hippocampus and cortex regions detected by [18F]-THK5351 [
37]. In this context, our data suggests that BP affects the levels of p-Tau in the cortex but not in the hippocampus, although this needs further studies to derive definite conclusions (
Figure S9). The discrepancies in its effect on Aβ and p-Tau might imply that BP elicits its effects through distinct mechanisms in the cortex and hippocampus for treating AD. However, further investigations are needed.
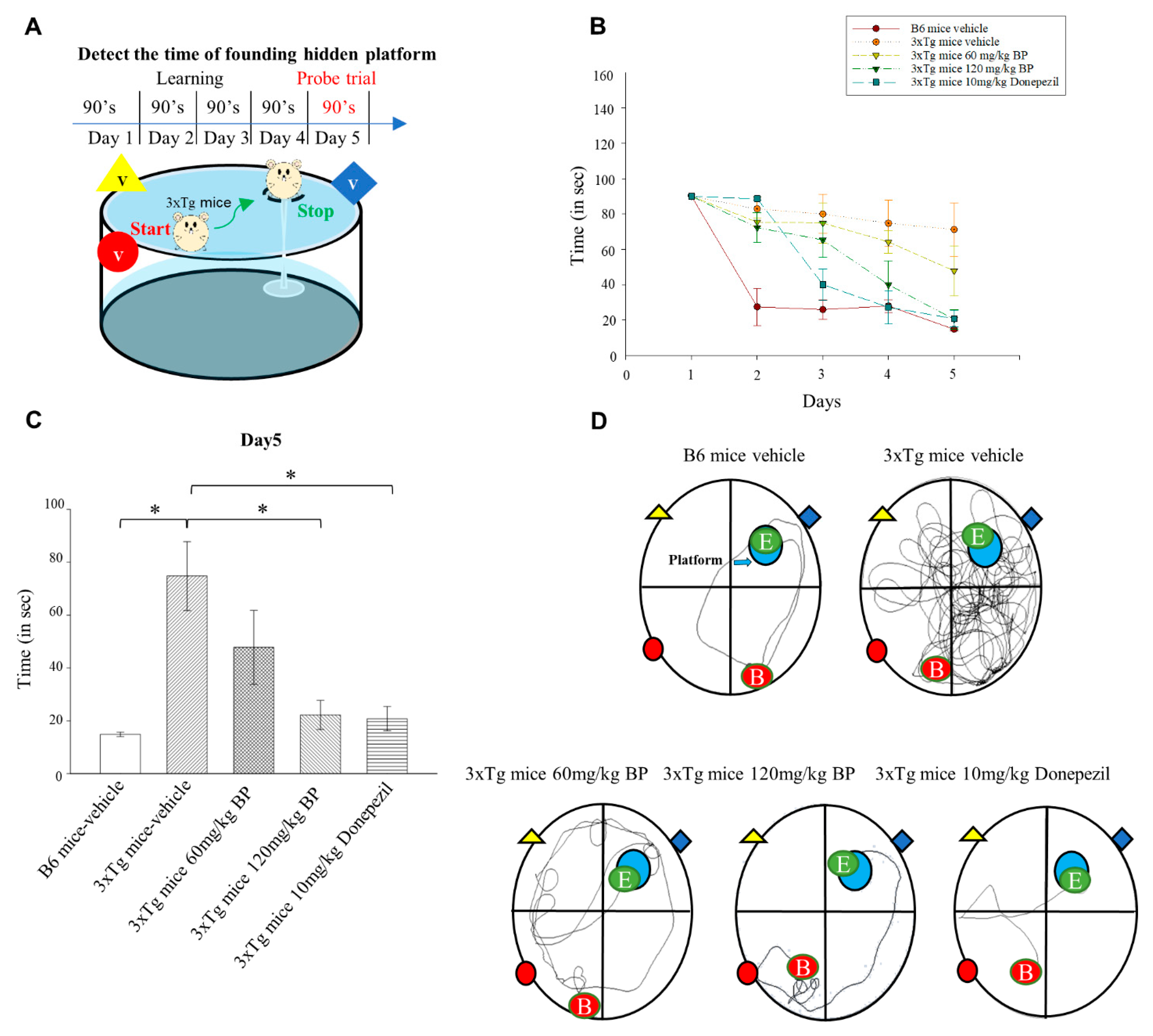
We tested the effect of BP on the performance of 3xTg AD mice in the Morris water maze. In addition, we compared the efficacy of BP with the clinically used drug Donepezil. Donepezil is an FDA-approved cholinesterase inhibitor for treating AD. However, Donepezil’s mechanism does not directly involve PSEN1 and Aβ [
38]. We found that the efficacy of BP is comparable to that of Donepezil when it comes to Aβ reduction. Our study suggested that BP facilitates spatial learning and memory in 3xTg AD mice (
Figure 5).
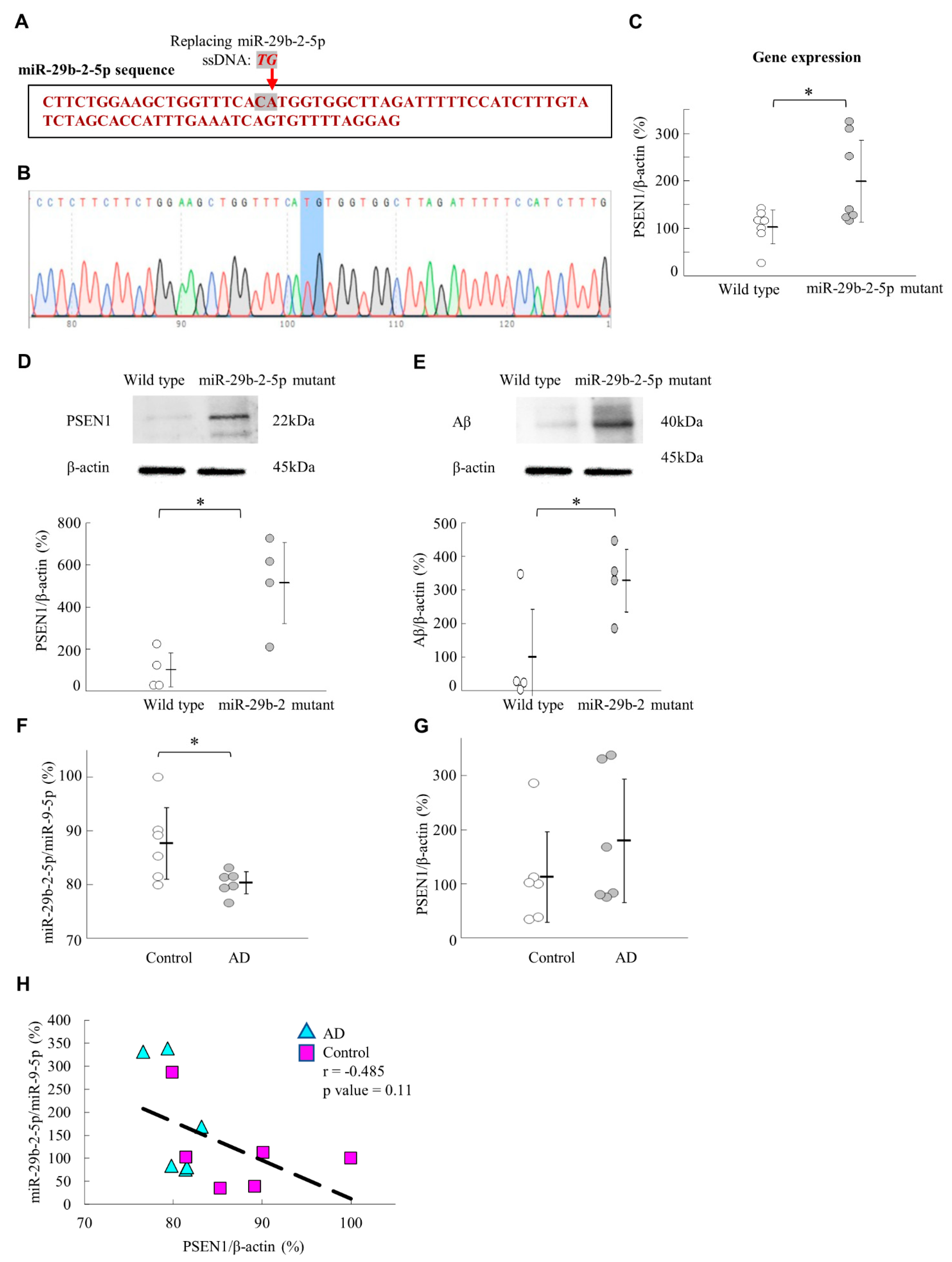
We generated miR-29b-2-5p mutant mice using CRISPR/Cas9 technique. The results showed that the expression levels of
PSEN1 and Aβ were upregulated in the hippocampus of these mice (
Figure 6A–E). This result supported the evidence regarding the role of miR-29b-2-5p in AD and its regulation by
PSEN1. On the other hand, we showed that miR-29b-2-5p levels were downregulated in sample brain tissue of patients with AD (
Figure 6F). This result suggested the critical role that miR-29b-2-5p plays in the pathogenesis of, and it deserves further investigation to see if cerebrospinal fluid samples from patients with AD have a similar trend in the future. In our study, miRNA-29b-2-5p was significantly decreased in the BA9 tissue samples of patients with Alzheimer’s.
In conclusion, we proposed that BP can effectively reduce amyloid production and promote cognitive function in the 3xTg AD mouse model. Furthermore, miR-29b-2-5p might be a good therapeutic target for treating early AD. This is attributed to its ability to regulate PSEN1 expression via lnc-CYP3A43-2/miRNA-29b-2-5p. However, some obstacles remain to be tackled when considering miRNA therapy for AD, including inappropriate biodistribution, disruption, and saturation of endogenous RNA machinery, and untoward side effects [
39]. We propose modifying the structure of miR-29b-2-5p through wrapping it with lipophilic nanoparticles, exosomes, or combining tissue-specific targets in the hippocampus. These methods are expected to solve the problems of non-specificity to the brain and instability.
4. Methods and Materials
4.1. Culture of Ts21-iPSCs
The Ts21-iPSC cells were reprogrammed from allantoic fluid-derived trisomy 21 iPS cells as previously described [
4]. These cells displayed the characteristics of AD phenotype with accumulated amyloid deposits and Tau protein hyperphosphorylation. The iPSCs were cultured in Essential 8 Medium (Thermo Fisher, A1517001, Waltham, MA, USA) containing 1% Essential 8 Flex Supplement (Thermo Fisher, A28584-01) and 1% penicillin-streptomycin (Thermo Fisher, 15140-122) [
4]. When the cells reached complete confluence, they were washed once with 1× PBS before adding Accutase (Thermo Fisher, A1110501) to dissociate into small clumps and seeded on 1% Matrigel matrix (Corning, 354230, Corning city, NY, USA) for neural maturation as previously described [
4]. The iPSCs were maintained at 37 °C in a 5% CO
2 incubator, cell culture medium was replaced once daily. To promote the differentiation of Ts21 multifunctional stem cells into neuronal cells, they were treated with Accutase (Thermo Fisher, A1110501) and cultured in DMEM/F12 HEPES (Thermo Fisher, 11330032) medium containing KnockOut Serum Replacement (Thermo Fisher, 10828028). These cells displayed as suspension of embryoid bodies [
4]. After one day, the medium was changed to high-glucose DMEM (Thermo Fisher, 11965092) and KnockOut DMEM/F-12 (Thermo Fisher, 12660012) containing nerve-inducing components and cultured for another two days. The medium was changed every two days until Day 30.
4.2. β-CTF/C6 Cells
The human C-terminal 99-residue fragment (β-CTF) of amyloid precursor protein was introduced into the C6 rat glioma cell line using a lentivector (System Biosciences LV500A-1, Palo Alto, USA) as previously described [
11]. The production of Aβ40 and Aβ42 peptides was induced by a Cumate-inducible gene expression system (System Biosciences, PBQM100A-1) [
11]. The culture medium was Ham’s F12K (Thermo Fisher, 21127022) containing 2.5% FBS, 15% horse serum (Thermo Fisher, 26050088), and 1% penicillin-streptomycin (Thermo Fisher, 15140122). The cell line was kindly provided by Professor Yi-Cheng Chen from the Department of Medicine, Mackay Medical College, Taipei, Taiwan.
4.3. SH-SY5Y Cell Model
The human SH-SY5Y cell line was isolated from SK-N-SH, a metastatic bone tumor biopsy cell line, and cultured for 5–7 days in DMEM (Thermo Fisher, 11965092) supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher, 16000044). When the cells were grown to 80% confluence in the flask, the serum-containing medium was replaced with Neurobasal Medium (Thermo Fisher, 21103049), containing B27 supplement (Thermo Fisher, 17504044), GlutaMAX (Thermo Fisher, 35050061), and 10 µM retinoic acid (Merck, R2625, Darmstadt, Germany) to promote the differentiation and growth of neuronal phenotypes.
4.4. n-Butylidenephthalide
n-Butylidenephthalide (3-Butylidenephthalide, BP) was purchased from Sigma-Aldrich (purity ≥96%, #W333301, St. Louis, MO, USA) as a mixture of cis and trans isomers. The stock solution of BP was prepared in a mixture of 99% alcohol, chloroform, and pure water in the ratio of 1:1.5:1.5:6, respectively. Olive oil (Oro del, Desierto, Spain) was used to dilute the stock BP solution to the desired concentration for animal use. The reported effective concentrations of BP for neuronal degeneration in animal models were around 100~250 mg/kg, and did not shorten mice survival [
40]. We chose the first effective concentration (120 mg/kg/day) to treat 3xTg AD mice, and the low concentration group was 60 mg/kg/day, they were administered orally. It was well documented that BP efficiently crossed the blood–brain barrier [
41]. The vehicle group was treated with olive oil. Mice of the same age were randomly assigned to each group and subjected to the same training. These treatments on 3xTg-AD mice were started from the age of 4 to 14 months.
4.5. Cell Viability under BP Treatment
To examine the protective ability of BP against Aβ-induced toxicity, CTF/C6 cells were treated with a designed concentration of BP. For viability assay, 1 × 10
4 CTF/C6 cells based on the result shown in
Figure S6 in supplemental information, were incubated with 0, 50, 100, 150, and 200 μM of BP in a 96-well microtiter plate for 72 h at 37 °C incubator containing 5% CO
2. The incubated cells treated with or without BP were then assayed for cell viability. Cell viability was assayed by adding 10 μL MTT solution to each well, and wells were incubated for another 2–4 h at room temperature. The optical density was determined at 450 nm by using a microplate reader (Thermo Fisher, Multiskan GO).
4.6. Molecular Docking Calculation
The binding energy or the molecular docking calculation between BP and PSEN1 protein was performed with the Lamarckian Genetic Algorithm (LGA) by Autodock 4.2 software [
20]. The structures of Presenillin1 protein (PDB ID: 5FN2) [
19] were obtained from the RCSB Protein Data Bank [
42]. The co-crystallized ligands of protein were all removed. Besides, the protein structure was modified by adding polar hydrogens and Kallman united atom charges for docking calculation by AutoDock Tool 1.5.6 interfaces (ADT) [
43]. BP was optimized with MMFF94 force field by ChemBio3D version 11.0 software (Cambridge Soft Corp, Cambridge, UK). Hydrogens and Gasteiger charges were also added to the ligand for the docking study by ADT. The Grid box calculated by AutoGrid program was centered at the x = 116.95, y = 123.86 and z = 125.29, with dimensions 52 × 76 × 58 Å grid points at a spacing of 0.375Å. All docking parameters were set to default except for the following parameter: maximum number of energy evaluation increased to 25,000,000 per run. The docking results were analyzed with cluster analysis, and the simulation model was shown by BIOVIA Discovery Studio 4.5 Visualizer software (BIOVIA Corp., San Diego, CA, USA). AutoDock analysis was conducted by Dr. Yang-Chang Wu.
4.7. Predicting miRNA Interaction That Could Interfere with Gene Expression
miRNA interaction at the 3′-end of the gene which interfere with the gene expression (Target detection) was performed using the software RNA22 v2 miRNA.
4.8. Predicting lncRNA Interaction That Could Interfere with miRNA Expression
Target detection was performed using the software of Freiburg RNA Tools to predict the lncRNA interaction that could interfere with miRNA expression.
4.9. Microarray
The cells were treated with Trizol reagent (Thermo Fisher, 15596-018), and the miRNA of Ts21 neurons was detected using the human miRNA microarray chip (Agilent Technologies, G4872A-070156, Santa Clara, CA, USA). Each group was subjected to three replicates, and miRNAs exceeding two-fold expression levels were considered significant. Long noncoding RNA microarray were purchased from Agilent (Agilent Technologies, G4851C). Microarray analysis was conducted to evaluate expression levels of these lnRNA between the BP-treated and untreated (control) groups.
4.10. Extracellular Protein Measurement by Sandwich ELISA
After neuronal differentiation of Ts21 iPSCs (5 × 105 cells), the conditioned mediums were collected and centrifuged at 1200 rpm for 5 min. These supernatants were stored at −80 °C. They were analyzed by sandwich ELISA (IBL-international, 27711, Hamburg, Germany), and Aβ42 quantitation were carried out within 30 min (Thermo Scientific, Multiskan GO).
4.11. Real-Time PCR
Primers were prepared and diluted to 20 μM/μL, and cDNA samples were diluted to 50 ng/μL. A 96-well plate was ready for RT-PCR. and then, 10 μL SYBR Green PCR Master Mix (Thermo Fisher, A25741), 2 μL cDNA, 2 μL primer, and 6 μL RNase-free water were added to each well and gently mixed. Gene expression was detected using the QuantStudio™ 3 Real-Time PCR System. The primers for mRNA are listed in
Table 3.
4.12. Western Blot Analysis
Cells were analyzed by western blotting using 4–12% SDS gradient gels (NP0322PK2; Invitrogen) and the following antibodies were used: APP (Abcam, ab32136, Cambridge, UK), BACE1 (Abcam, ab183612), PSEN1 (Cell Signaling Technology, D39D1, Danvers, MA, USA), cleaved Notch1 (Cell Signaling Technology, 4147), Aβ1-42 (Abcam, ab201060), and anti-β-actin (Abcam, ab8227). Electrophoreses were performed using 15% Tris-Glycine SDS gels for Aβ and 12% Tris-Glycine SDS gradient gels for PSEN1 in 12-well gel, and the resulting bands were transferred by western blotting. The protein concentration of the loading sample was 30 μg. Immunoreactivity was analyzed by chemiluminescence (GE Healthcare, Piscataway, NJ, USA). The protein marker ranges from 10–180 kDa were used (Tris-glycine-SDS running buffer) (GeneDireX, PM006-0500, Taoyuan City, Taiwan). The transfer buffer was 25% Methanol and 10% TG-SDS buffer (Avantor, 0783-5L, Radnor, PA, USA). The setup time and voltage are 90 min and 400 mA by gel transfer to PVDF member. The chemiluminescence signal was observed using a digital image analyzer (Fuji Film Inc, LAS-3000, Tokyo, Japan).
4.13. Transfection
One hundred microliter transfection solutions were prepared and supplemented with 10 nM miRNA-29b-2-5p mimic or miRNA-29b-2-5p -siRNA in a 4.5:1 mixture. They were then incubated for 10 min at 23 °C before cell transfection (Lonza, V4XP-3024, Basel, Switzerland). Cells were trypsinized and counted, 1.0 × 106 cells were then seeded in six-well culture plate. After centrifugation at 335× g for 5 min, the respective siRNA solution was mixed with cells and placed in a tube shocker. The cells were transfected by electroporation (Lonza, V4XP-3024) and transferred to a new six-well culture plate to be cultured overnight in an incubator at 37 °C with 5% CO2. Before transfection, 75 pmol of biotinylated hsa-29b-2-5p miRNA was diluted in 200 μL complete neuron basal (NB) medium (Thermo Fisher, 21103049). This mixture was transferred to another 1.7 mL microfuge tube containing Lipofectamine 3000 reagent (Thermo Fisher, L3000001) diluted in 200 μL of Opti-MEM media (Thermo Fisher, 31985088). These cells were again cultured at 37 °C with 5% CO2 for at least 36 h. The preparation of lnc-CYP3A43-2 siRNA and miR-29b-2-5p-i experiments were the same as stated above. The miR-29b-2-5p-i sequence: 5′-CUAAGCCACCAUGUGAAACCAG-3′.
4.14. Flow Cytometry
β-CTF/C6 cells emit endogenous fluorescence after activation by Cumate (System Biosciences, QM100A-1). For analysis, 5 × 105 cells were washed in 500 μL PBS and centrifuged at 1500 rpm for 5 min. The supernatant was discarded, and the centrifugation was repeated once more. The centrifuged cells were resuspended in 500 μL PBS and evaluated using a flow cytometer (Beckman, Coulter CytoFLEX S Flow Cytometry, Brea, CA, USA).
4.15. Dual-Luciferase Assay System
Primers were annealed and digested with Sacl and Xbal restriction enzymes. The digested fragment was purified and cloned into pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, E1330, Madison, WI, USA). The cloned fragment included the miR-29b-2-5p targeted PSEN1 sites (
Figure 2). The reporter gene was transfected using the following Dual-Luciferase Reporter Assay System (Promega, E1910): (1) firefly luciferase (
Photinus pyralis) with a molecular weight of 61 kDa and OD measurement at approximately 560 nm and (2)
Renilla luciferase (
Renilla reniformis) with a molecular weight of 31 kDa and OD measurement at approximately 480 nm. After transfection into cells, fluorescence was emitted without stimulation by drugs. The comparison of luminescence intensity revealed the expression of the reporter gene.
4.16. Biotin-Based Pulldown Assay
For this assay, 1 × 10
6 Ts21-neuron cells were seeded and cultured overnight at 37 °C in a 5% CO
2 incubator. Thereafter, a solution containing 75 picomoles of biotinylated miRNA was prepared in a total 200 µL MEM. This mixture was transferred to another microfuge tube containing Liposome-based transfection reagent (8 µL/well) diluted in 200 µL MEM. They were mixed gently and incubated at room temperature to allow for the formation of the transfection complexes. After that, the transfection complexes were added in a drop-wise manner to the cells, and they were cultured for at least 36 h. Streptavidin Coated Magnetic Beads (Merck Millipore, LSKMAGT10, Burlington, MA, USA) were prepared, and 100 µL of bead wash buffer (10 mM Tris-Cl pH 7.5, 0.5 mM EDTA, 1 M NaCl) was added to the beads and washed by vortexing at room temperature. The tube containing beads was then placed on the magnet for 2 min. The resulting supernatant was carefully removed and discarded, and the previous step was repeated three times. Next, 100 µL of RNase freeing solution (0.1 M NaOH, 0.05 M NaCl) was added to the beads and mixed, and the supernatant was discarded. This step was repeated twice more. Bead resuspension solution (0.5 M NaCl) was added, and the mixture was vortexed and incubated at room temperature for 5 min. The resuspended beads were placed on the magnet for 2 min, and the supernatant was removed. After that, 200 µL of bead blocking solution (1 µg/µL BSA) was added, and they were incubated at 4 °C for over-night on a multi-tube rotator. Transfected Ts21-neuron cells were harvested by gentle scraping using a sterile cell scraper within a laminar hood, each sample was placed into a new microfuge tube. Complete pull-down wash buffer (10 mM KCl, 1.5 mM MgCl2, 10 mM Tris-Cl pH 7.5, 5 mM DTT, 1 M NaCl, 0.5% IGEPAL, 60 U/mL Superase, and 1× Protease Inhibitor cocktail) were prepared. 150 µL of ice-cold CPDW buffer was added to the beads, vortexed, and incubated at room temperature for 30–60 s. The supernatant was removed and discarded, and this step was repeated two more times. The beads were resuspended in 100 µL nuclease-free water and incubated on ice. Total RNAs were extracted, cDNA synthesis was performed and subjected to qPCR as previously described [
44].
4.17. AD Animal Model
Experiments were conducted using transgenic mice (3xTg) harboring the following three transgenes: presenilin PS1M146V knock-in mutation (PS1M146V), human transgenes and the APP Swedish mutation (APPSwe), and microtubule-associated protein tau (tauP30IL) (The Jackson Laboratory, Sacramento, CA, USA). As these mice age, they gradually exhibit synaptic dysfunction neuropathy; Aβ protein accumulates in the later stage, and plaques and neurofibrillary tangles are formed. At the age of 3–6 months, Aβ deposits are detected in some regions of the brain. At the age of 12 months, the pathological features include highly phosphorylated tau protein and neurofibrillary tangles in the hippocampus. In this study, only female mice were used.
4.18. Gene-Edited for miR29b-2-5p in C57BL/6 Mouse
Mouse zygotes were obtained by mating super-ovulated B6 females and males (National Laboratory Animal Center, Taiwan). Thus, sgRNA, Cas9 nuclease, and ssDNA were mixed just before the microinjection into the pronuclei of zygotes. The injected embryos were incubated at 37 °C until they were transferred into the pseudopregnant females on the same day. Mouse genomic DNA was extracted from the tail tips of pups, and the genomic sequences around the mutant site were PCR-amplified using the following primers: miR29b-2-5p F 5′-ACAGTTTCTTGTGCAGACATCGG-3′, Chchd23B 5′-GGGACCACTTCTCATTGCCATAG-3′. The obtained PCR products were directly sequenced. The animals were kindly provided by Professor Si-Tse Jiang of the National Laboratory Animal Center, Taiwan. miR-29b-2-5p-mutant mice were obtained from the National Laboratory Animal Center, National Applied Research Laboratories, Taiwan (109-NLAC-AA-017).
4.19. PET-CT Imaging
The accumulation of Aβ deposits in 3xTg mice occurs over a long period of time. Using [18F]-Florbetaben (FBB) or [18F]-THK5351 PET scan, the accumulated amyloid-like protein in the brain can be observed without sacrificing the animals. Mice were anesthetized by inhalation of 2–3% isoflurane and then injected with [18F]-FBB (3 mCi) through tail vein. We waited for 30 min to allow for [18F]-Florbetaben or [18F]-THK5351 uptake, and then static PET images were recorded for a total of 30 min using Sedecal SuperArgus PET 2r (Sedecal, Madrid, Spain). List mode data were acquired for 30 min per scan using an energy window of 250–700 keV. To improve resolution and sensitivity, the acquired images were reconstructed using the 2D OSEM algorithm. The image voxel size was 0.387 × 0.397 × 0.775 mm. Quantitative image analysis of the mouse brain was performed using PMOD (PMOD Technologies, v3.8, Zürich, Switzerland). Mouse brain images were aligned to a standard mouse brain template using PMOD’s PFUS module rigid matching tool. The regions of interest of each mouse brain, including the frontal cortex, hippocampus, and cerebellum, were then manually defined and expressed as standard uptake values. The SUVR of each brain region was expressed as cortex/cerebellum and hippocampus/cerebellum. All SUVRs were calculated using the cerebellum as a reference region.
4.20. Immunohistochemistry Staining
Brain slices obtained from four mice in each group were simultaneously stained with purified anti-β-Amyloid antibody (Cell Signaling, #9888) by immunohistochemistry. Briefly, after 20 min of antigen retrieval using 88% formic acid at room temperature, the slices were mixed with 500× diluted Aβ antibody and incubated overnight at 4 °C. This was followed by hematoxylin (Leica, 3801522, Wetzlar, Germany) counterstaining, and DAB (TA-060-QHSX and TA-002-QHCX, Thermo Fisher) was used as a chromogen for detection. The photographs of Aβ staining were captured using a microscope (Olympus, VS120-S6, Tokyo, Japan). Amyloid-beta 1-42 plaque load was analyzed only in the CA1 to CA3 of the hippocampus region. The quantification of Aβ deposits was performed using the ImageJ software (National Institutes of Health, Bethesda, ML, USA). Thioflavin S (Th-S) staining was performed in which Th-S (Sigma-Aldrich, #230456) was prepared with distilled water. The tissue slides were first deparaffinized and hydrated with distilled water and then stained with hematoxylin for 5 min. They were washed with running water for 5 s and rinsed with distilled water. Finally, they were soaked in 1% Th-S for 5 s at room temperature, differentiated in 70% alcohol for 5 s, and rinsed in distilled water. In addition, 6E10 staining was also performed, during which slides were incubated with 3% H2O2 solution for 10 min to quench endogenous peroxidase activity at room temperature. They were rinsed later with 1× TBS and blocked using a blocking buffer. Finally, the slides were incubated with 6E10 (Biolegend, SIG-39320, San Diego, CA, USA) at room temperature, rinsed with 1× TBS, and labeled with the secondary antibody.
4.21. Quantification of Hippocampus miRNA Expressions in 3xTg and miR-29b-2-5p Mutant Mice
The analysis of hippocampus miRNAs was conducted on 6-month-old female miR-29b-2-5p mutant mice (n = 4 for each group) and 14-month-old female 3xTg mice (n = 7 for each group). All animal procedures have been approved by the animal committee of Hualien Tzu Chi Hospital and Tzu Chi University (#108-04, Hualien Tzu Chi hospital). Total RNA was extracted from hippocampus using RNeasy Mini kit (Qiagen, 74106, Hilden, Germany), and miRNAs were purified using the miRNeasy Mini Kit (Qiagen, 217004). cDNAs were synthesized using the TaqMan Advanced miRNA cDNA Synthesis Kit (Thermo Fisher, A28007). The primer pairs purchased were the mmu-miR-29b-2-5p (Thermo Fisher, mmu481675_mir) and mmu-miR-124-3p (Thermo Fisher, mmu480901_mir,). The quantification of these miRNAs was conducted by real-time PCR (Thermo Fisher, A28567).
4.22. Morris Water Maze Evaluation of Learning and Memory
Mice were trained in a swimming pool with a rest platform hidden under opaque water for 5 days. On Day 1, 120 s were allowed for mice to find the platform, and if the mice could not find the platform on time, we led them to the platform. On Days 2–5, the time allowed to reach the rest platform was decreased to 90 s; this training was also conducted twice. On Day 5, the time required to reach the rest of the platform was recorded. The water maze analysis system used in this study was Smart V3.0 Basic Pack.
4.23. Quantification of miRNA Expression in Human or Mouse Brain
Frozen brain specimens isolated from the BA9 of the frontal cortex were obtained from the NIH NeuroBioBanK. Control participants (
n = 6) and patients with AD (
n = 6) were referred by Dr. Catherine Cahill (NBTR NeuroBioBank Req, #1311, Bronx, NY, USA). Specimens were stored at −80 °C. Human brain BA9 tissue specimens were examined at Harvard Medical School, Massachusetts General Hospital. The information on the human brain samples of controls and patients with AD are listed in
Table 4. RNA was purified using the miRNeasy MiniKit (Qiagen, 217004), and cDNA was synthesized using the TaqMan Advanced miRNA cDNA Synthesis Kit (Thermo Fisher, A28007). The product was detected using real-time PCR and quantified. The targets included hsa-miR-29b-3p (Thermo Fisher, 478369_mir), hsa-miR-29b-2-5p (Thermo Fisher, 478003_mir), hsa-miR-9-5p (Thermo Fisher, 000583_mir). The expression was quantified in absolute terms as miRNA copy counts/5 ng of total RNA, and hsa-miR-9-5p was used as an internal control. Regular cDNA synthesis from mRNA was performed using either RevertAid RT Kit or SYBR Green Master Mix (Thermo Fisher, 4368577).
4.24. Statistical Analysis
ANOVA was performed for the comparison between multiple groups. LSD methods were used as post hoc tests for pairwise comparisons, and Dunnett’s tests compared a fixed group and other groups. Repeated measures ANOVA was performed for the comparison of subjects with multiple measurements. The student’s t-test was performed to compare two groups. All tests were two-tailed, and data sets were statistically significant when p < 0.05. Statistical analyses were performed using IBM SPSS 20.0.0 software.



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}