
Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks


Abstract
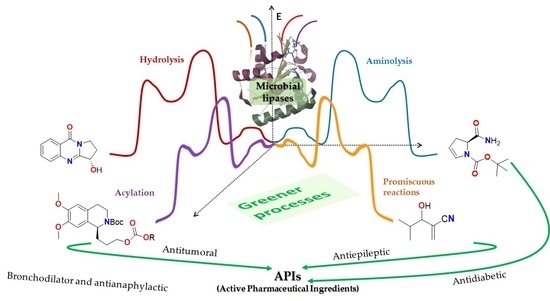
1. Introduction
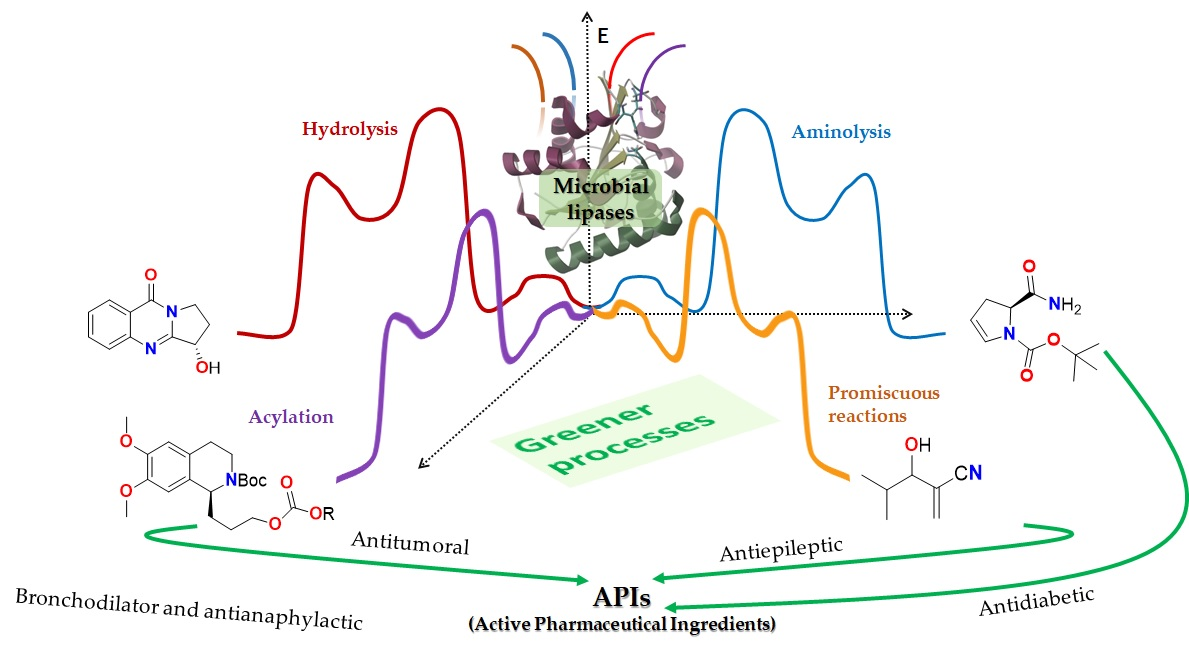
2. Microbial Lipases: Structure and Function
2.1. Structure
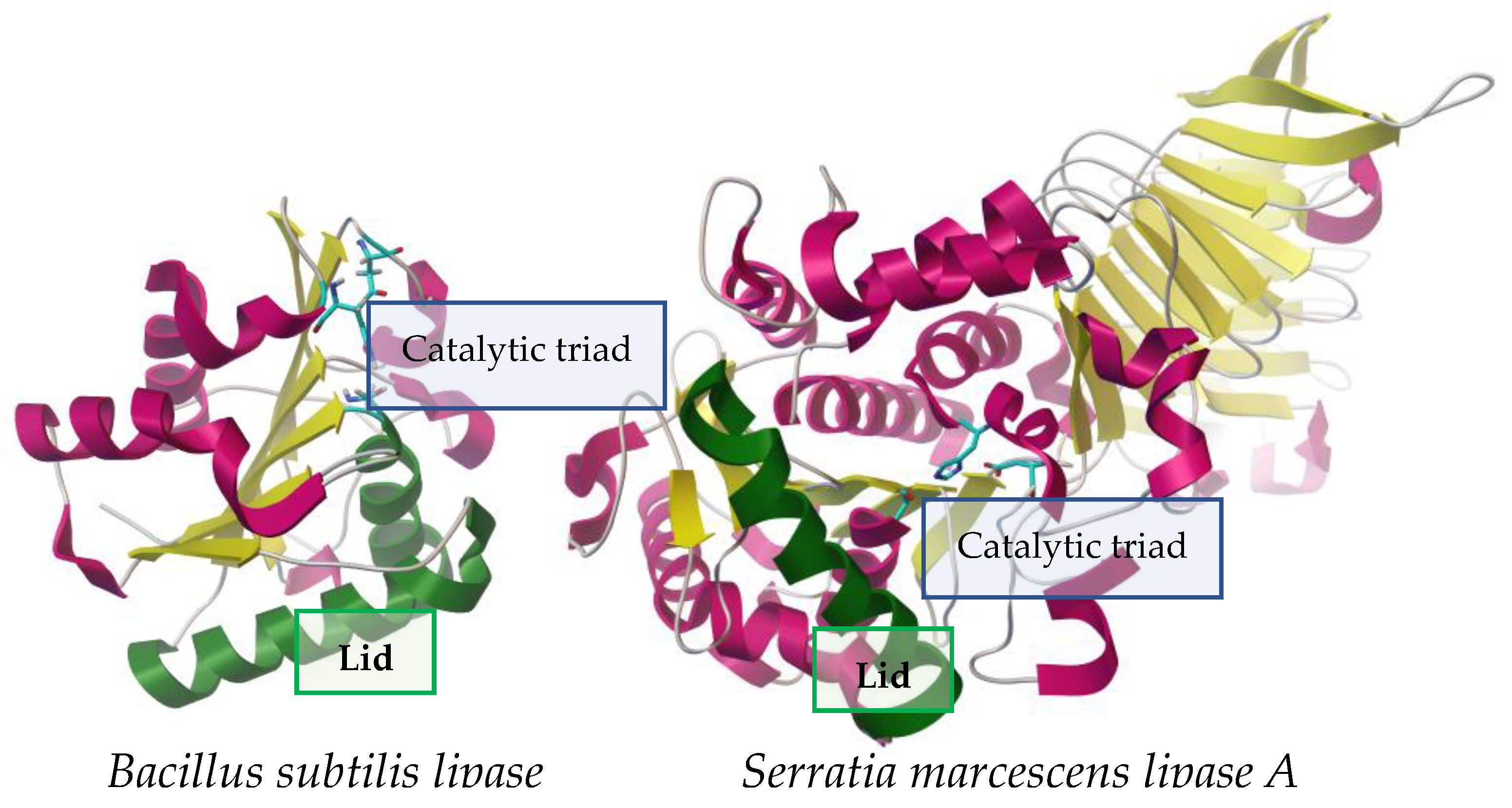
2.2. Mechanism of Action: Interfacial Activation and Catalysis
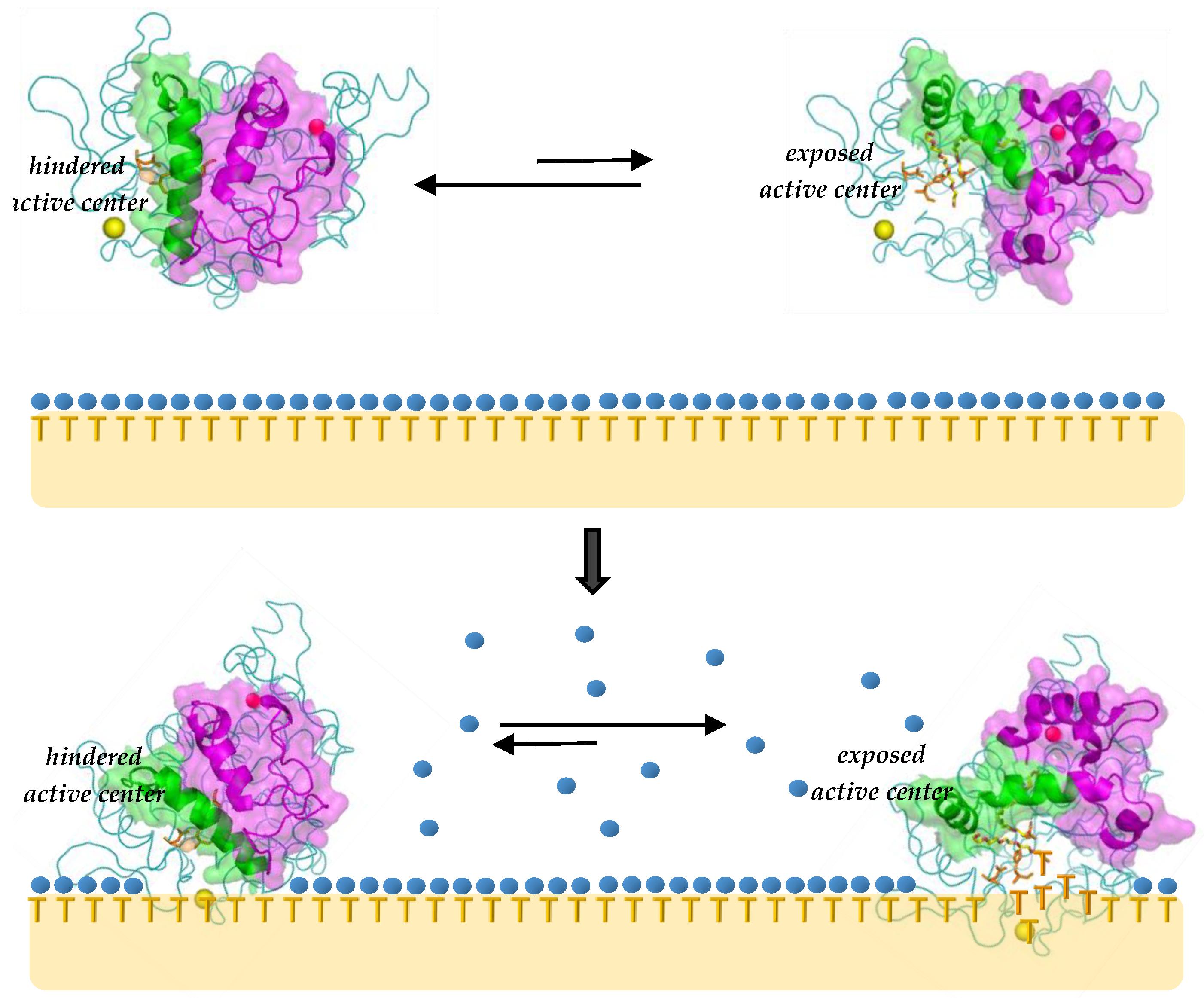
2.2.1. Interfacial Activation
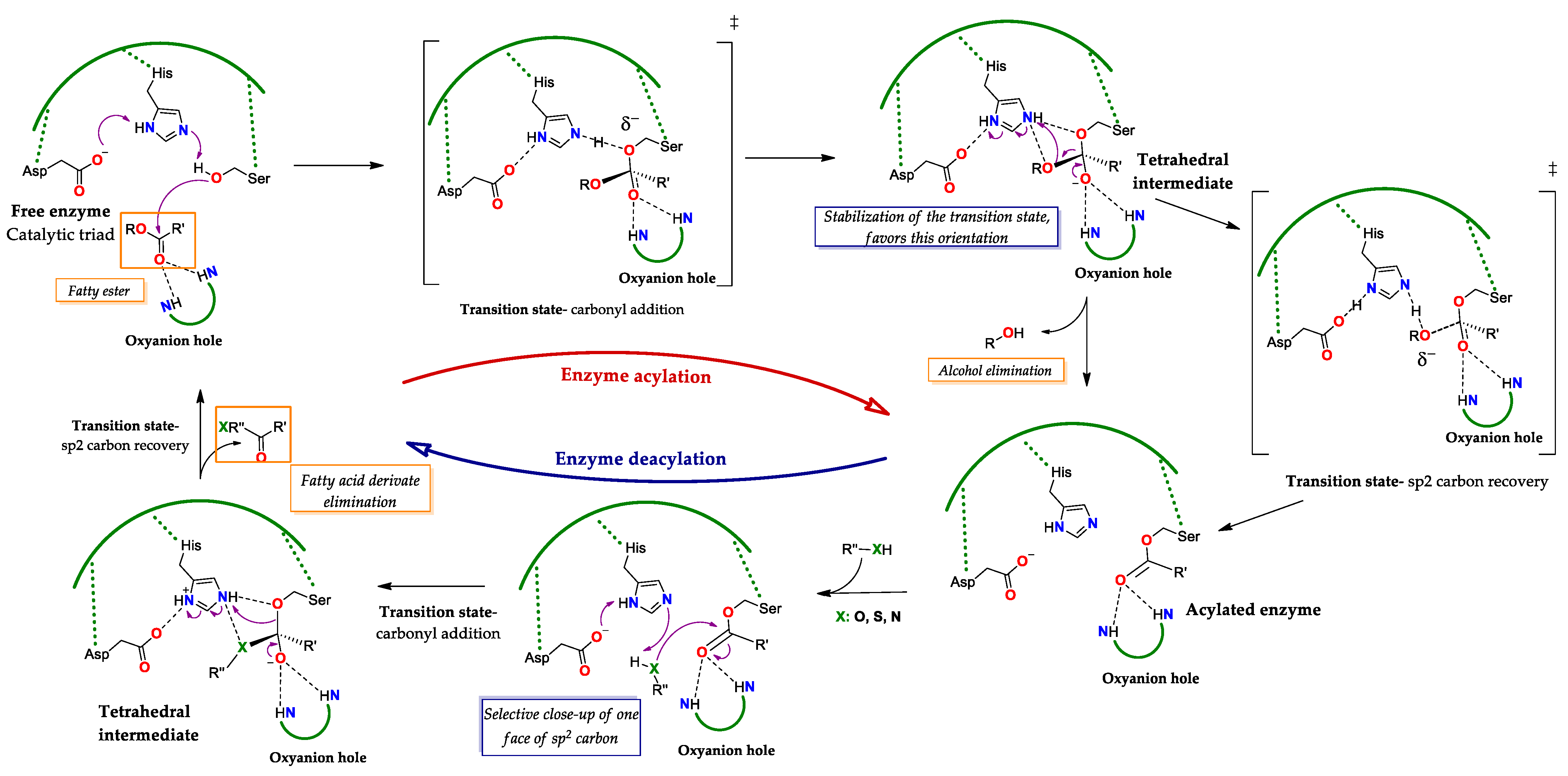
2.2.2. Catalysis of Lipases Functioning as Serine Hydrolases
2.2.3. Selectivity of Lipases
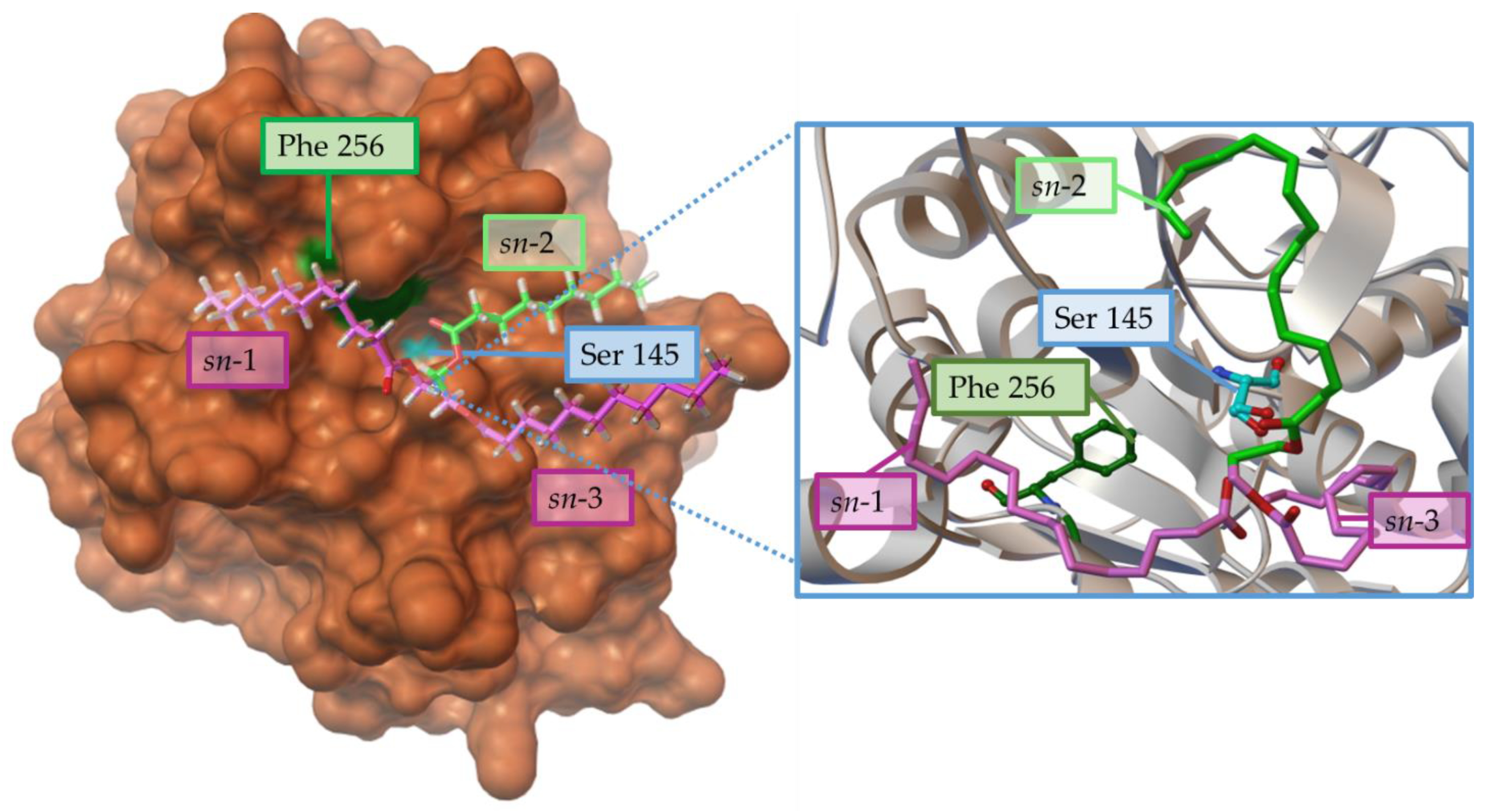
Chain Length
Cis/Trans Selectivity
Preferences between tri, di- and Mono Glycerides
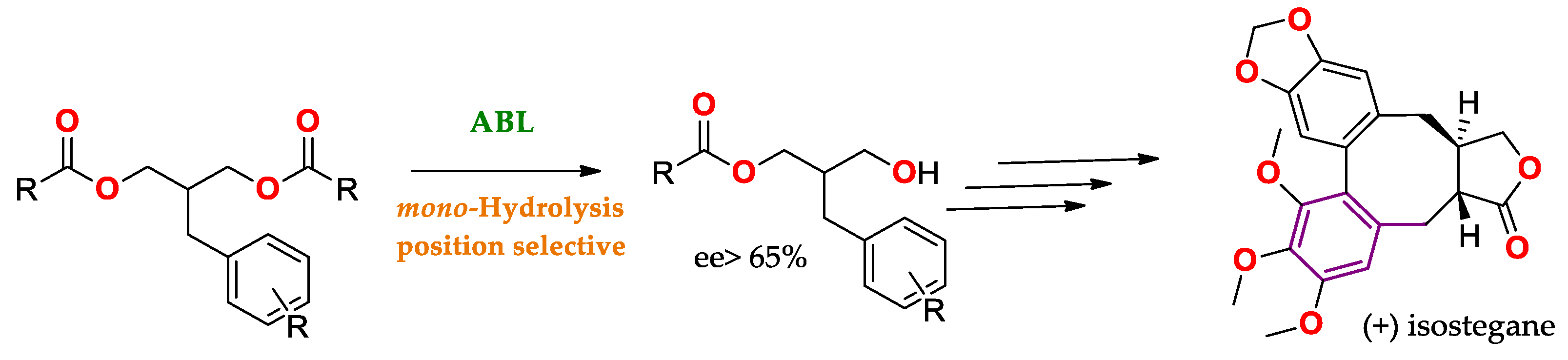
Regioselectivity
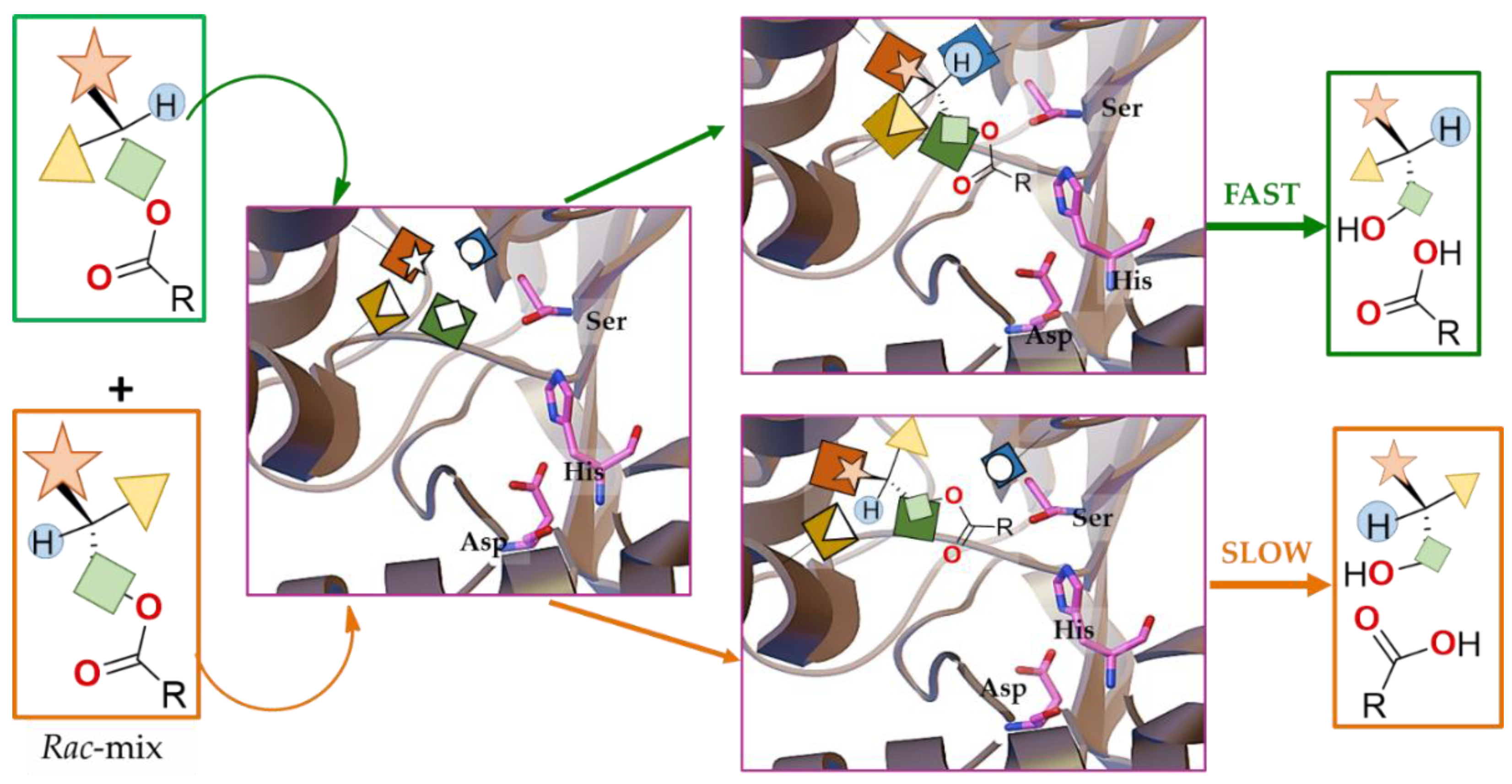
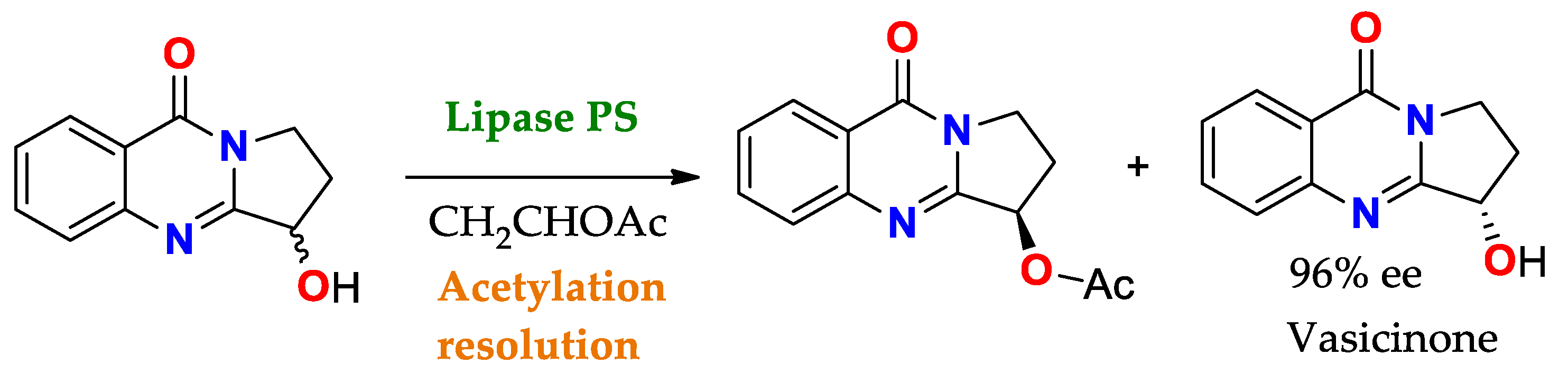
Enantiomeric/Prochiral Preference (Regioselectivity in Chiral Triglycerides)
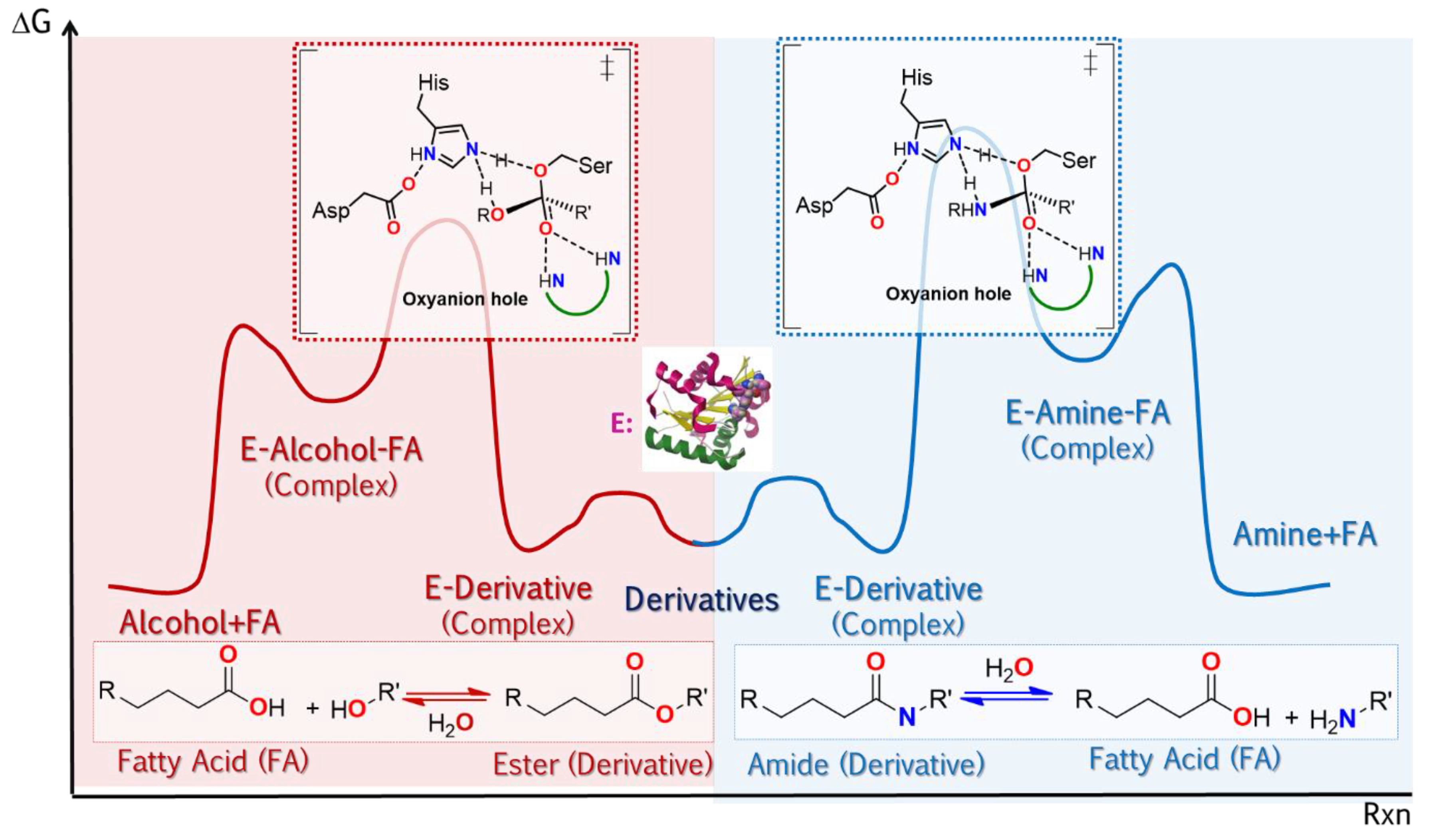
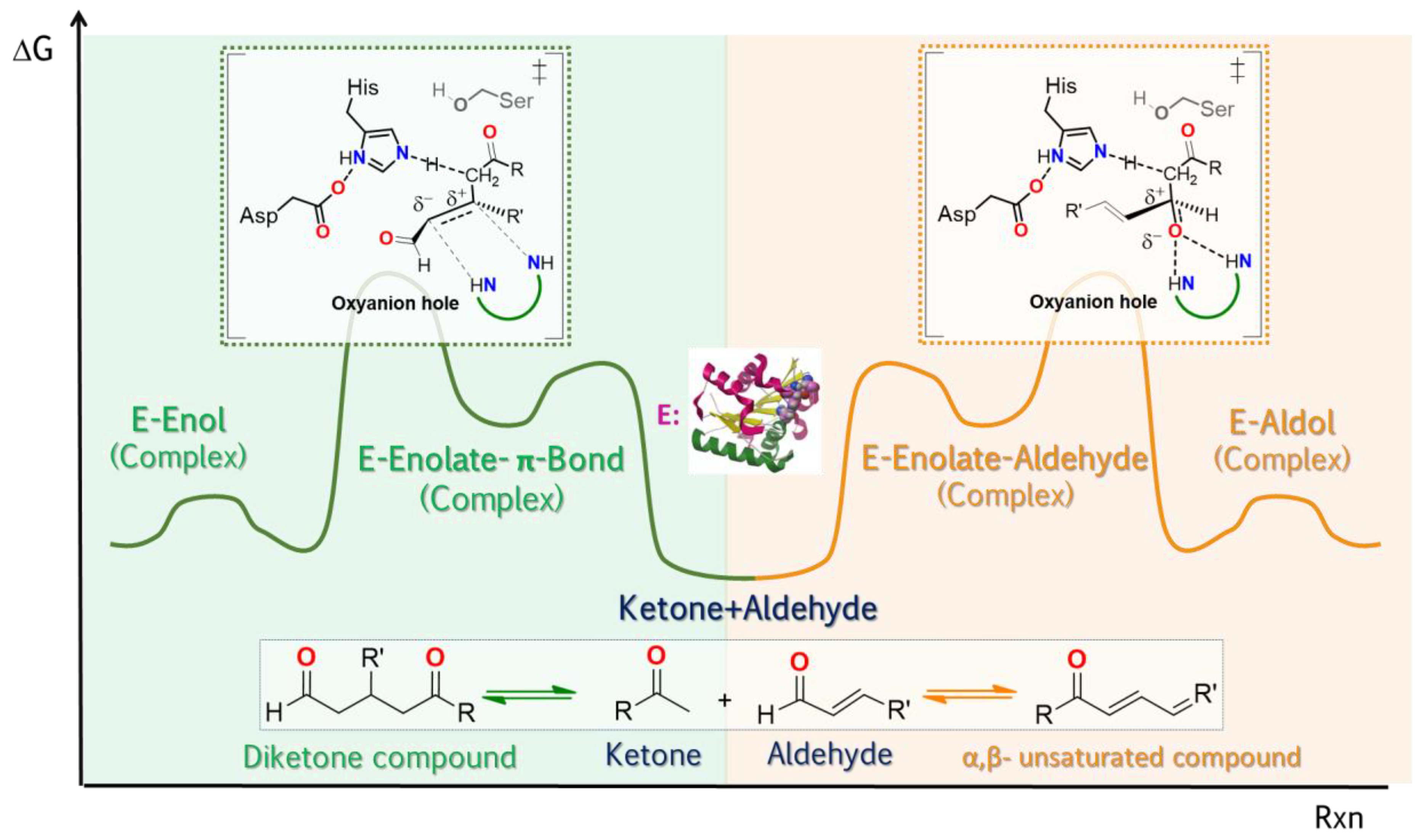
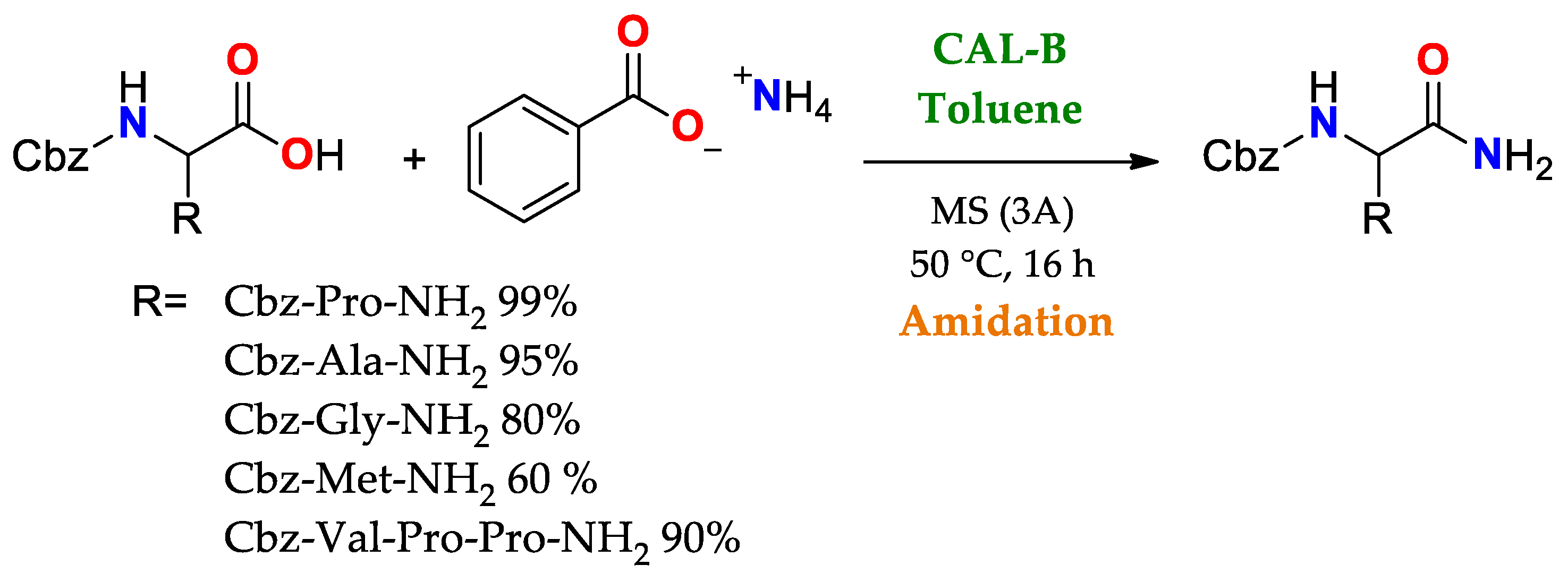
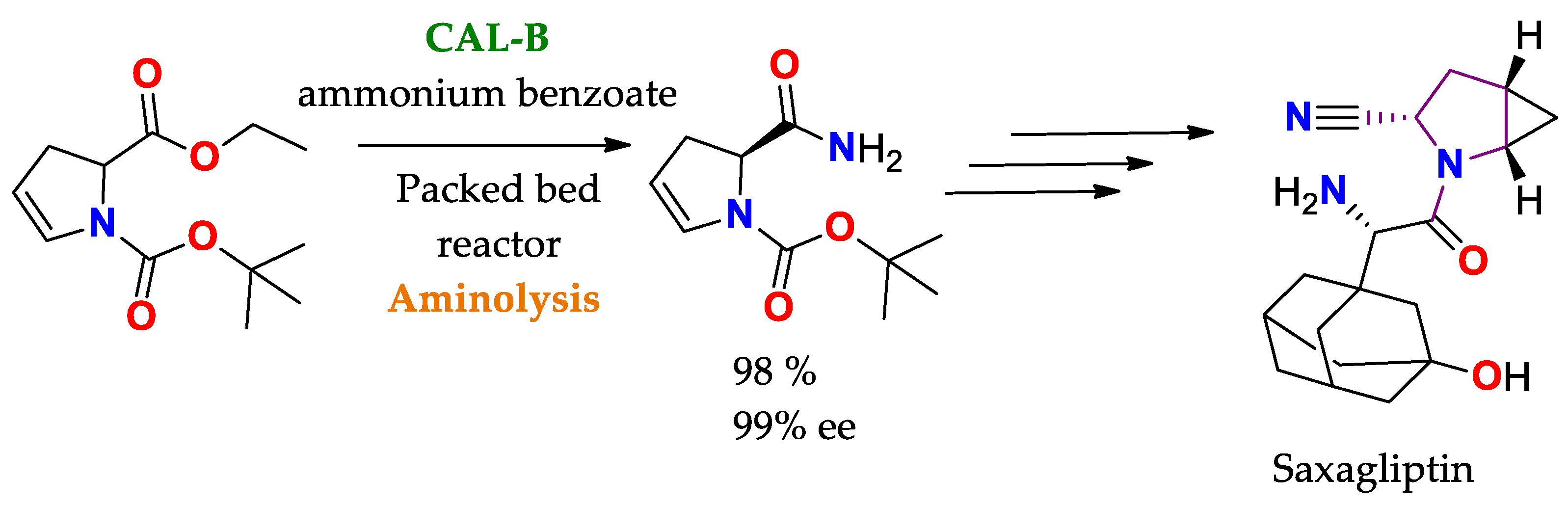
2.2.4. Reaction Specificity of Lipases
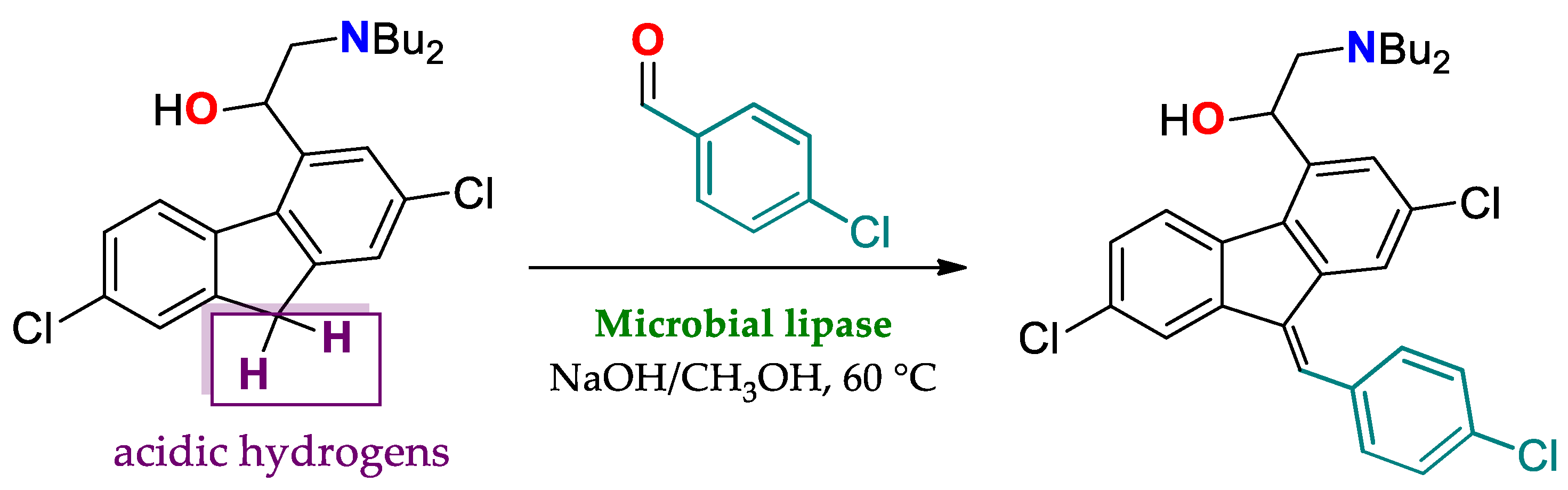
Molecular Origins of the Catalytic Promiscuity of Lipases
3. Immobilization of Lipases
3.1. Immobilization of Lipases and Effects on Their Properties in Reactions of Interest
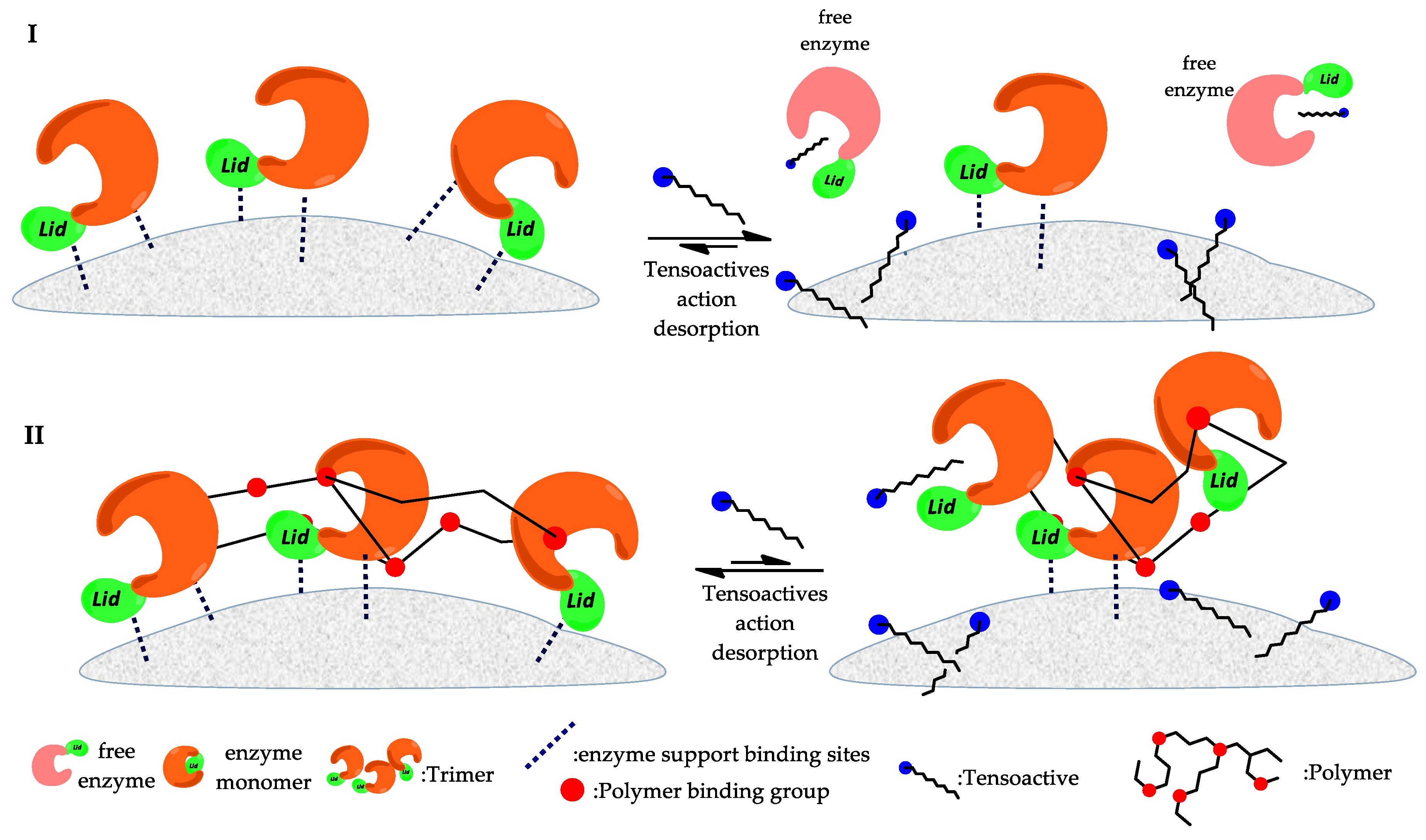
3.1.1. Immobilization by Hydrophobic Interaction
3.1.2. Ion Exchange Immobilization
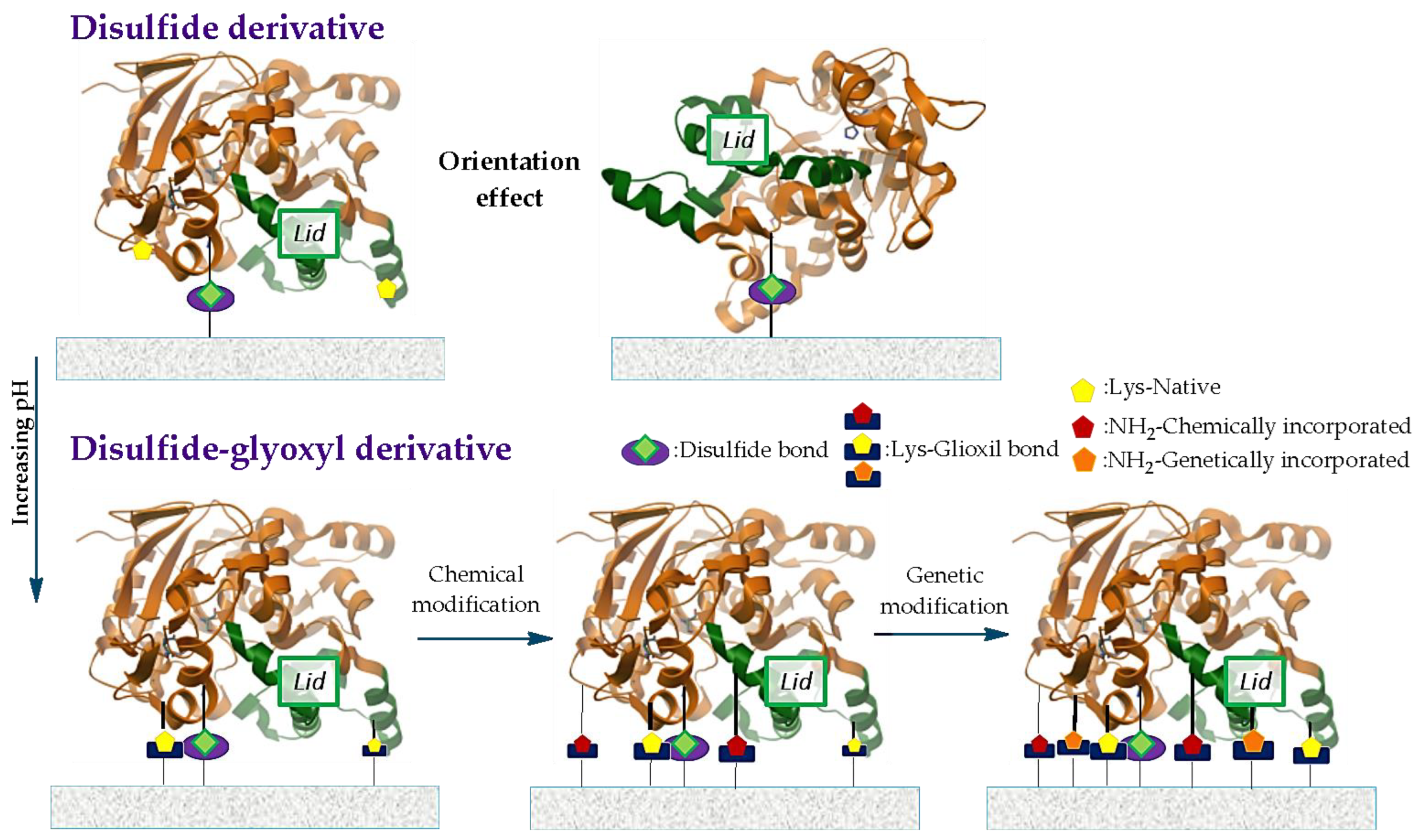
3.1.3. Covalent Immobilization
3.1.4. Comparisons between Types of Immobilizations
4. Applications of Free or Immobilized Lipases Related to APIs
4.1. Additions on Carbonyl Compounds: Lipases Substituting Lyases
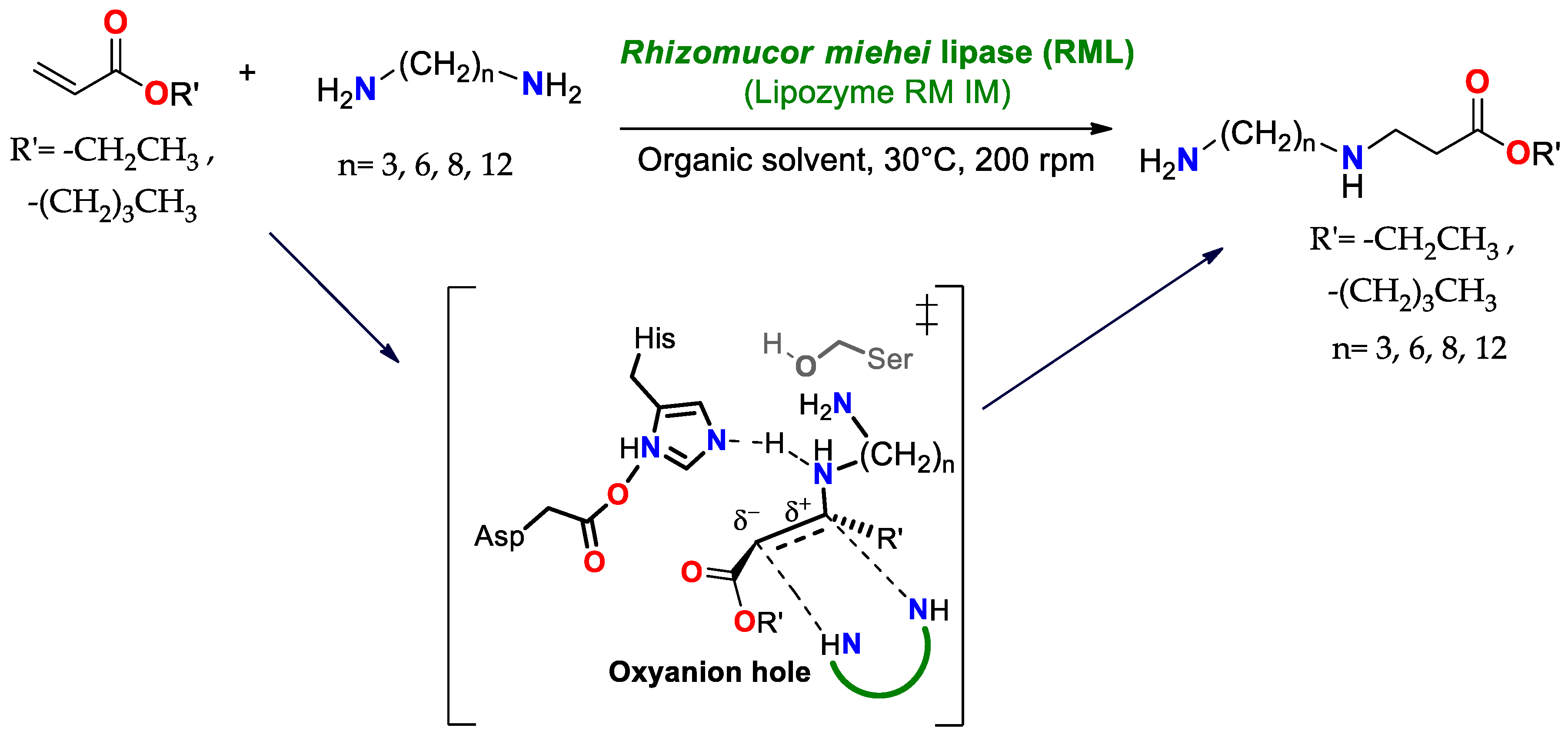
4.1.1. Aza-Michael Addition Reaction
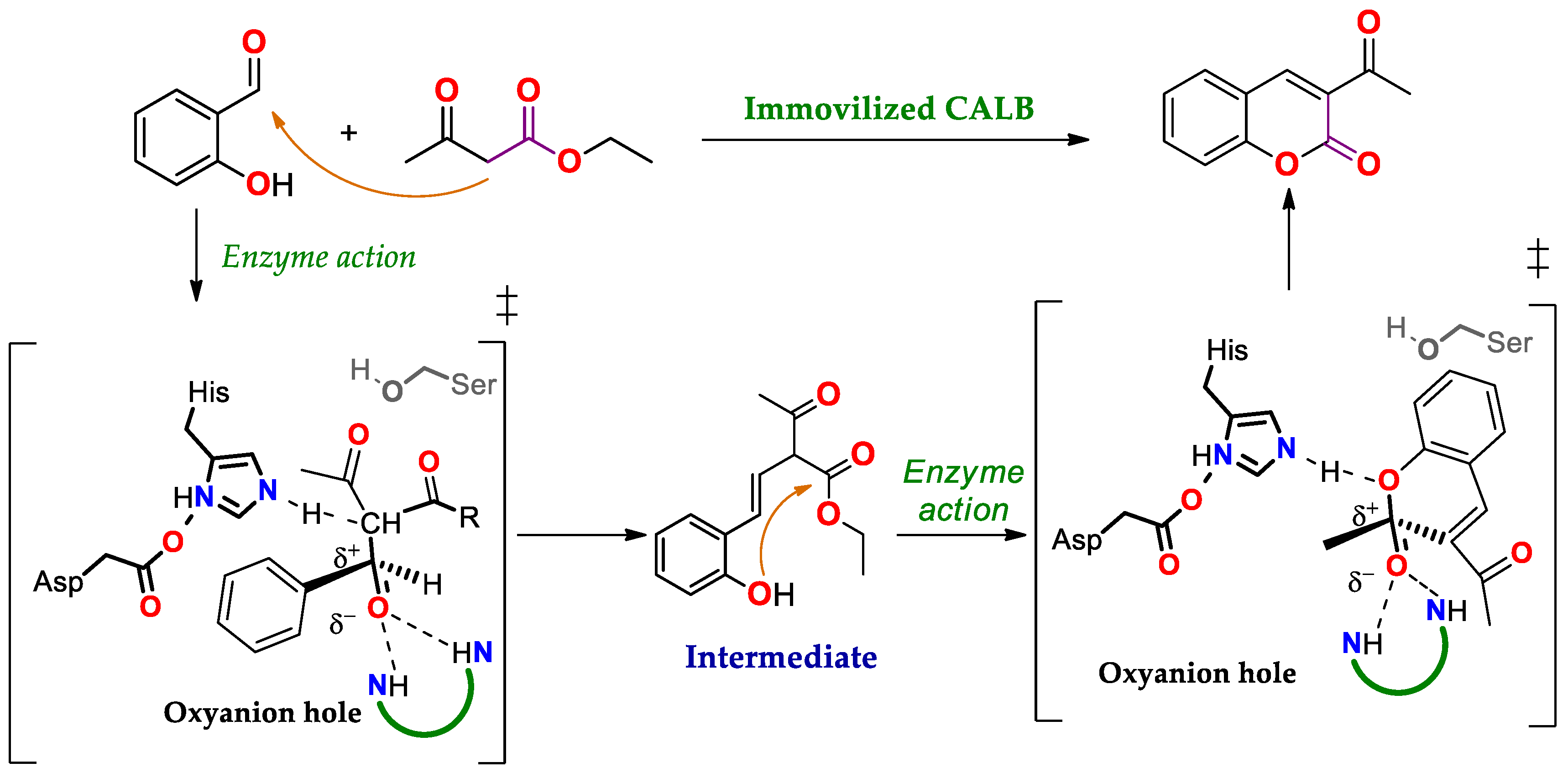
4.1.2. Knoevenagel Condensation
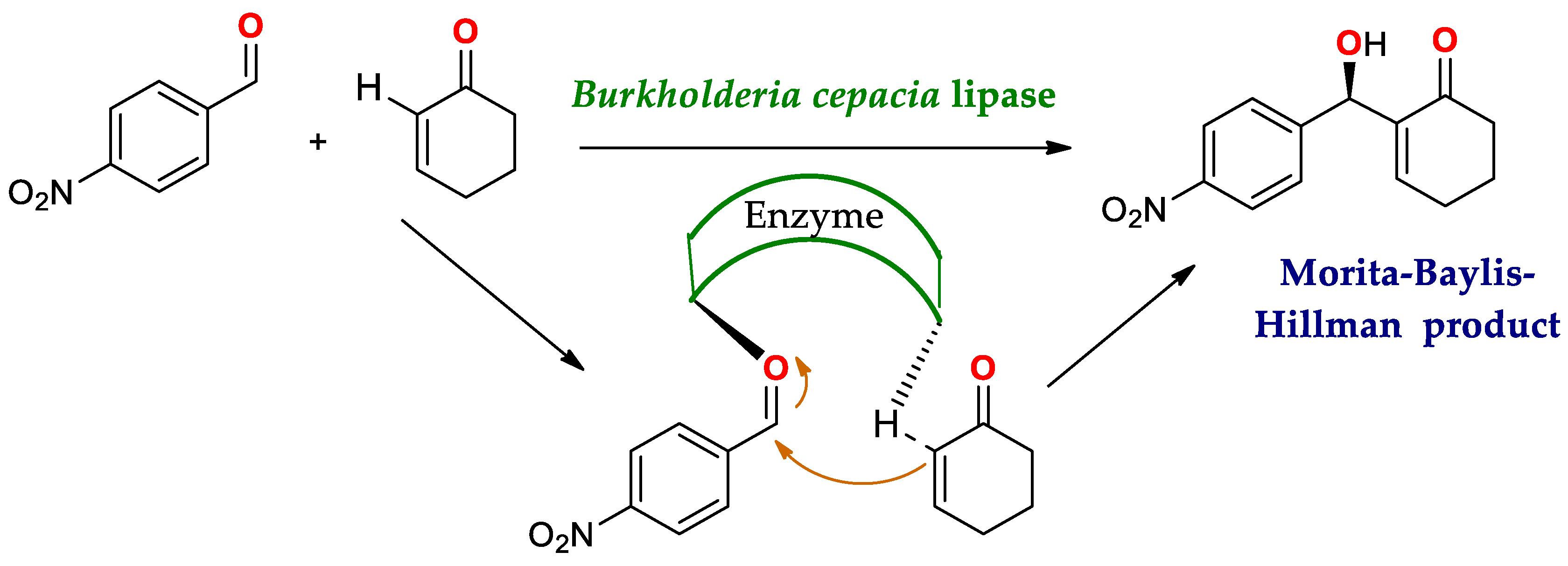
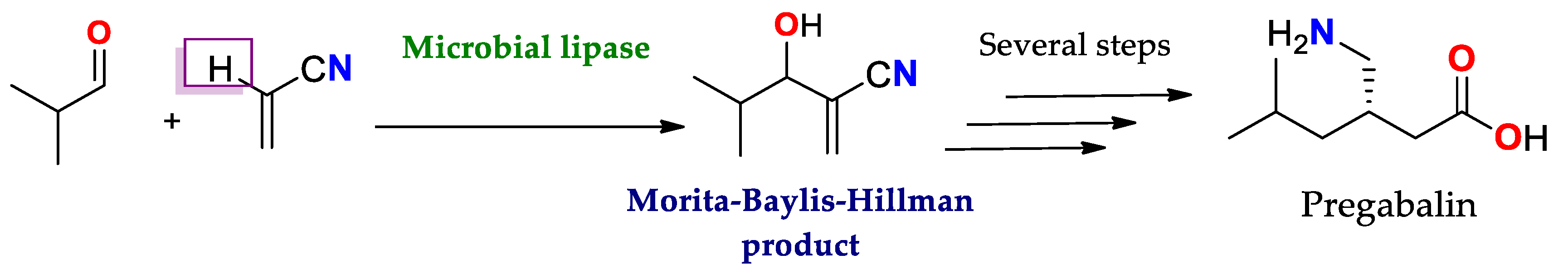
4.1.3. Reaction of Morita–Baylis–Hillman (MBH)
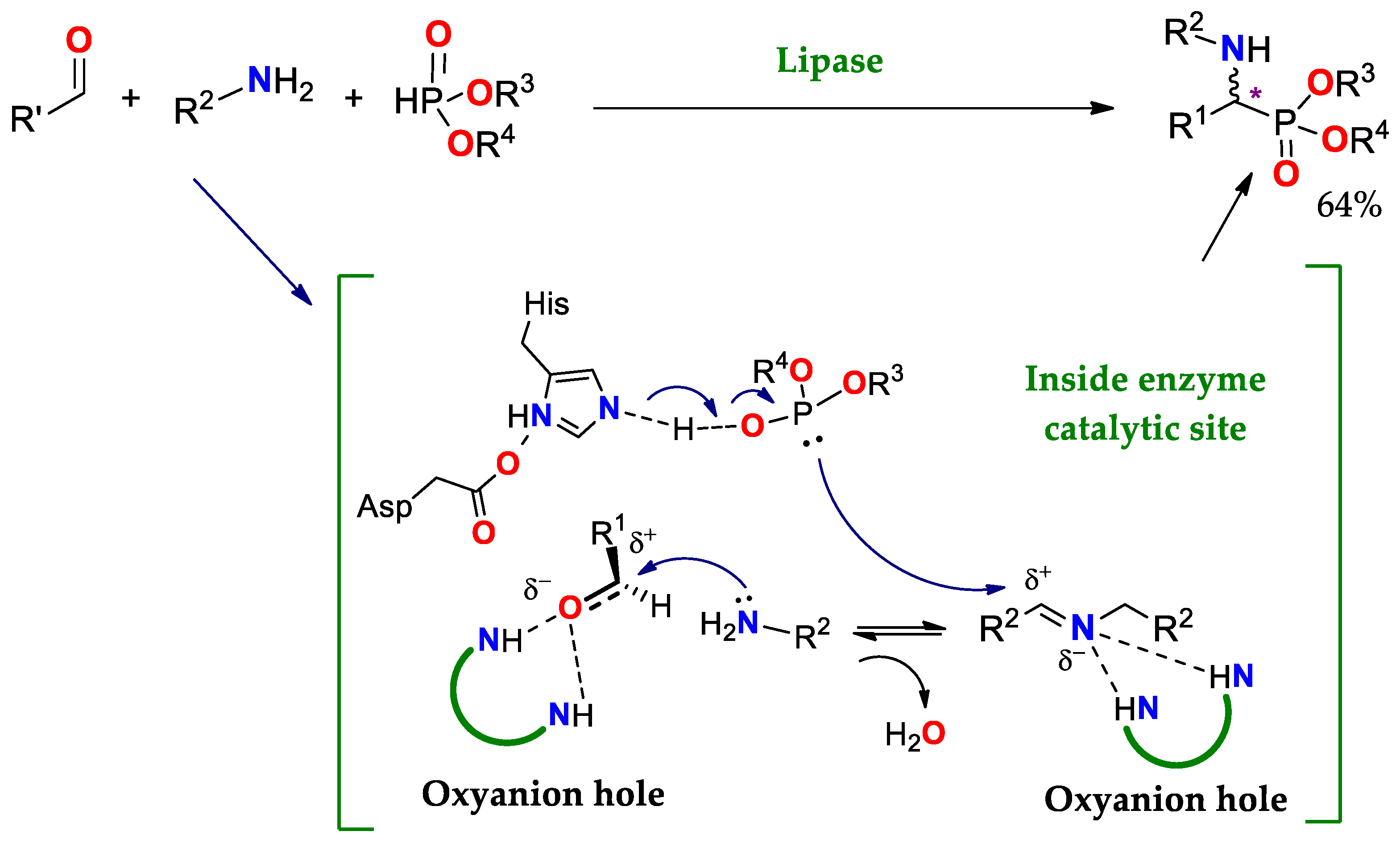
4.1.4. Kabachnik–Fields Reaction (KF)
4.1.5. Lipase Acting in an Addition Reaction Type Markovnikov
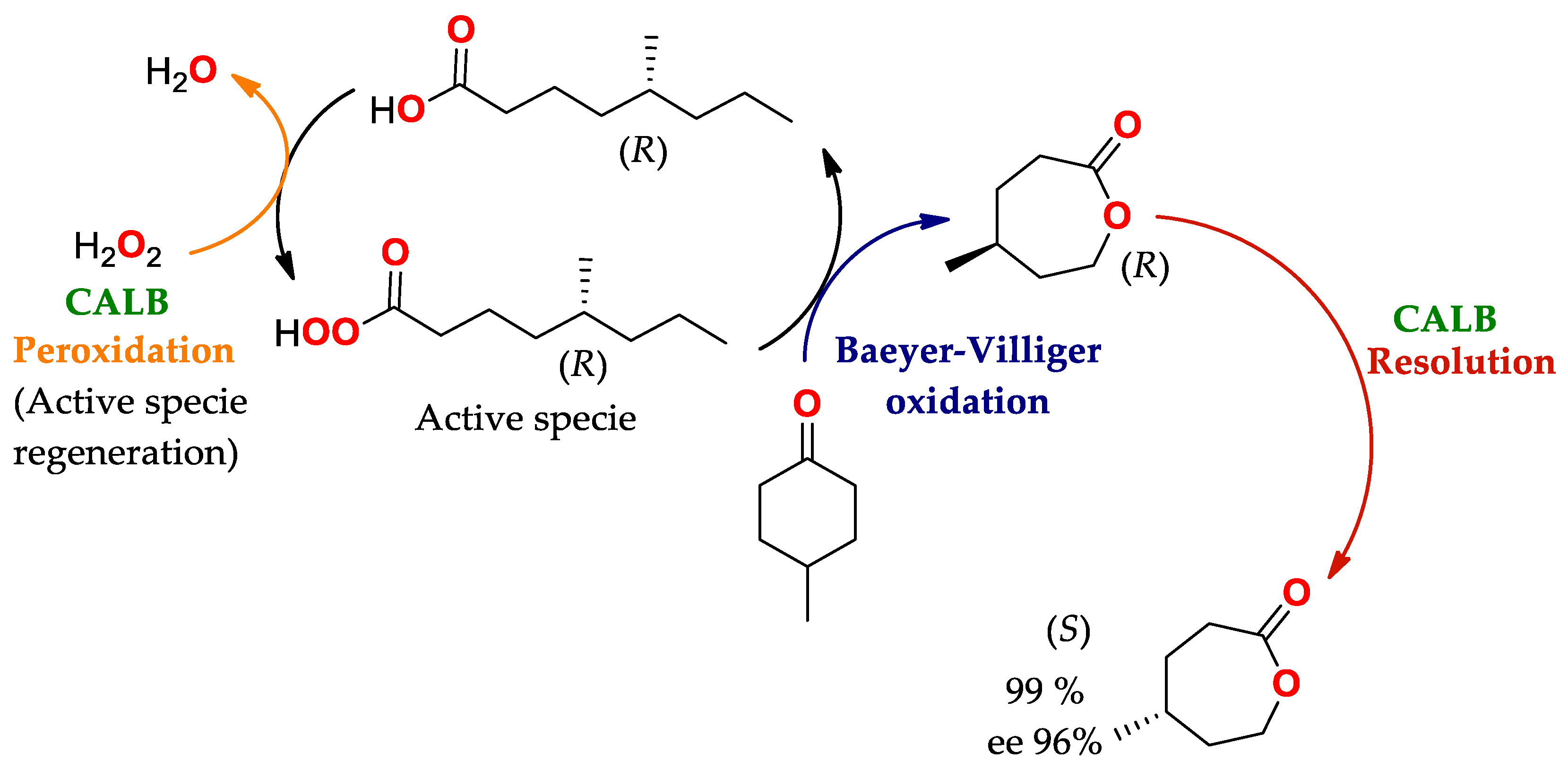
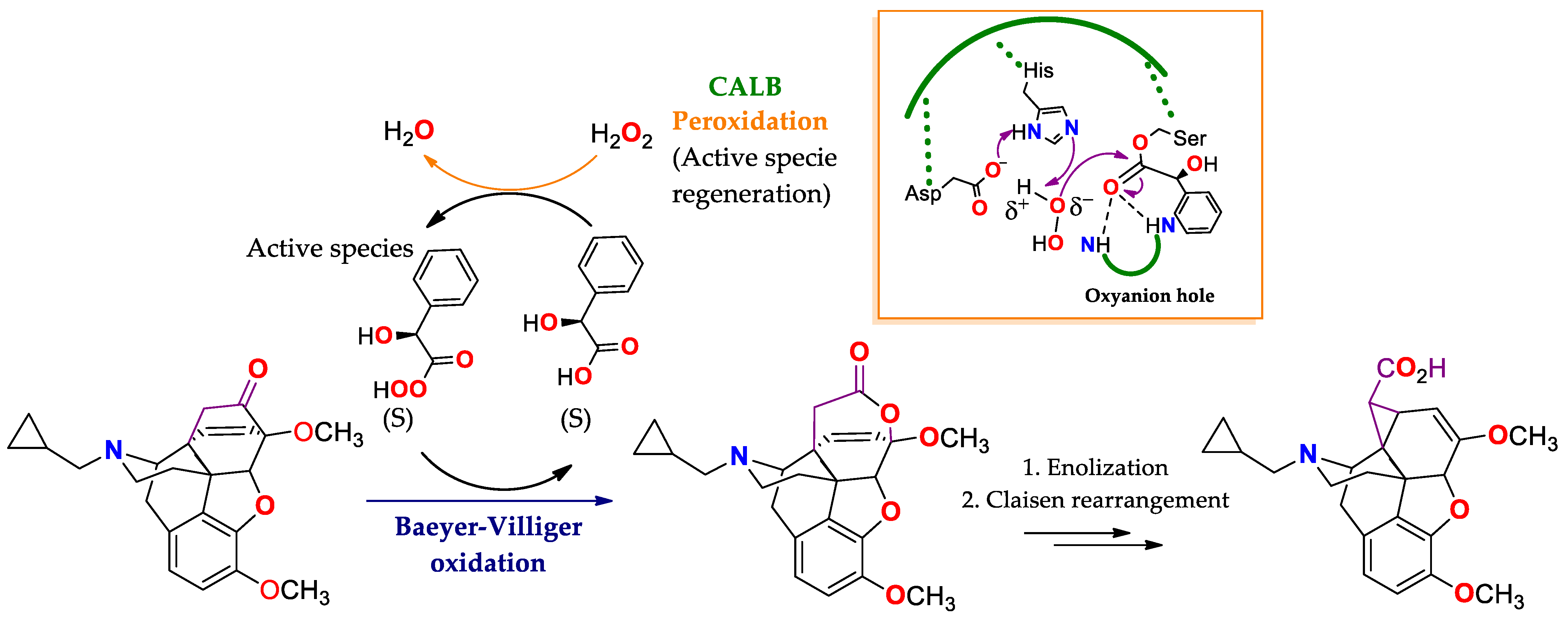
4.2. Oxidation of Baeyer–Villiger: Lipases Substituting Monooxygenases
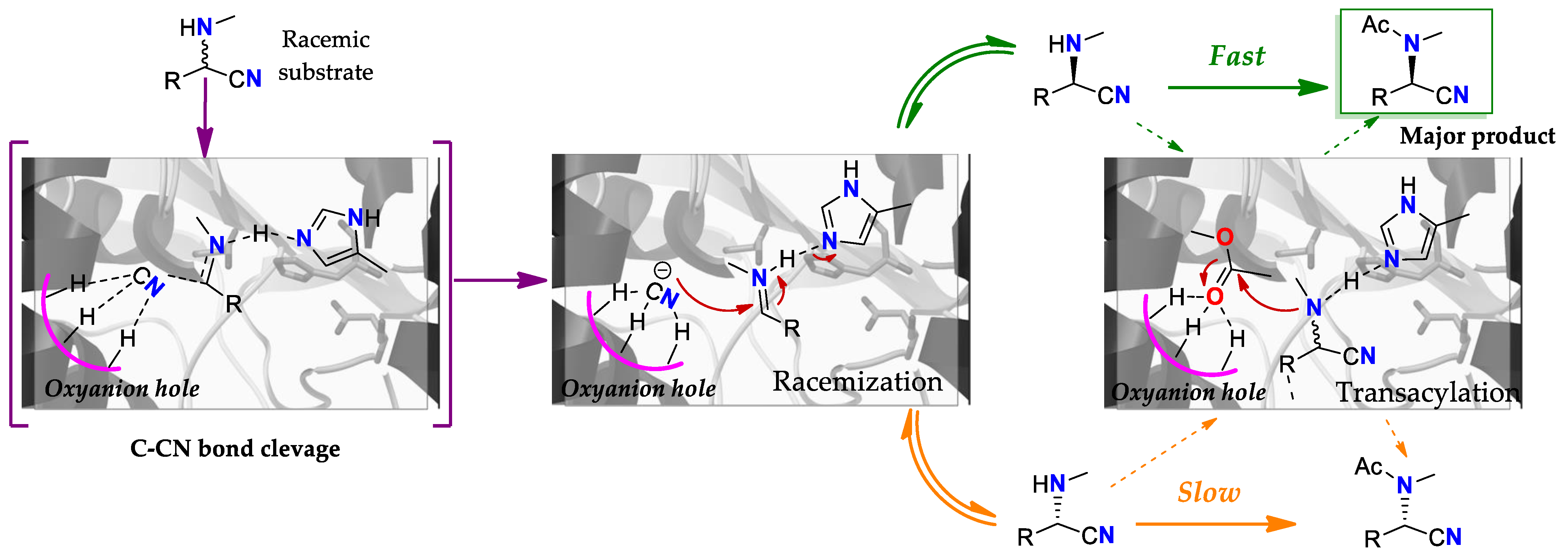
4.3. Racemization: A Lipase Substituting an Isomerase
5. Perspectives and Conclusions
- Studies on the environmental, economic and social costs (Life Cycle Assesment or LCA) with updated metrics that contrast the use of lipase-based biocatalysts against current synthesis strategies for specific APIs and the usual scale of production: of this there are examples in other industrial areas of application involving lipases and other enzymes [227,228]. Having well-defined cost–benefit ratios regarding the use of enzymatic processes in the industry is a solid starting point when overcoming obstacles to their implementation. This will allow us to prioritize what is worth investigating within what has not yet been done or take advantage of that which is still laboratory-scale or patent-protected but still unexploited.
- The development of processes for the synthesis of APIs with the use of immobilized lipases, with the appropriate studies of reuse or continuous use, and, if possible, in the absence of organic solvents: in comparative studies of ACL of biocatalysis vs. conventional process, those two aspects are some of the most critical when favoring the implementation of enzymatic processes over chemicals in the industry [227,229,230].
- The expansion of lipase promiscuity and the improvement of synthetic parameters such as reaction rates, performance and industrial-scale stereoselectivity: this requires the integrated use of tools such as directed evolution, enzymatic immobilization, enzyme chemical modification, QM/MM and, recently, Machine-Learning (ML) and Artificial Intelligence (AI) [231,232,233,234,235], applied to the old but also to the new microbial lipases that the required bioprospection can offer.
- The experimental work will continue to be a source of knowledge in complex reaction systems such as those that normally involve immobilized lipases: an adequate experimental design for the generation of quality data will be indispensable to feed the improvement tools such as ML and AI.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Kokare, C. Microbial Enzymes of Use in Industry. In Biotechnology of Microbial Enzymes; Brahmachari, G., Ed.; Academic Press: Cambridge, MA, USA, 2017; Chapter 11; pp. 267–298. ISBN 978-0-12-803725-6. [Google Scholar]
- Javed, S.; Azeem, F.; Hussain, S.; Rasul, I.; Siddique, M.H.; Riaz, M.; Afzal, M.; Kouser, A.; Nadeem, H. Bacterial Lipases: A Review on Purification and Characterization. Prog. Biophys. Mol. Biol. 2018, 132, 23–34. [Google Scholar] [CrossRef]
- Olivecrona, G. Role of Lipoprotein Lipase in Lipid Metabolism. Curr. Opin. Lipidol. 2016, 27, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, H. Lysosomal Acid Lipase in Lipid Metabolism and Beyond. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 850–856. [Google Scholar] [CrossRef]
- Warakanont, J.; Li-Beisson, Y.; Benning, C. LIP4 Is Involved in Triacylglycerol Degradation in Chlamydomonas Reinhardtii. Plant Cell Physiol. 2019, 60, 1250–1259. [Google Scholar] [CrossRef]
- Singh, A.K.; Mukhopadhyay, M. Overview of Fungal Lipase: A Review. Appl. Biochem. Biotechnol. 2012, 166, 486–520. [Google Scholar] [CrossRef]
- Jawed, A.; Singh, G.; Kohli, S.; Sumera, A.; Haque, S.; Prasad, R.; Paul, D. Therapeutic Role of Lipases and Lipase Inhibitors Derived from Natural Resources for Remedies against Metabolic Disorders and Lifestyle Diseases. S. Afr. J. Bot. 2019, 120, 25–32. [Google Scholar] [CrossRef]
- Lee, H.-J.; Park, O.K. Lipases Associated with Plant Defense against Pathogens. Plant Sci. Int. J. Exp. Plant Biol. 2019, 279, 51–58. [Google Scholar] [CrossRef]
- Pascoal, A.; Estevinho, L.M.; Martins, I.M.; Choupina, A.B. REVIEW: Novel Sources and Functions of Microbial Lipases and Their Role in the Infection Mechanisms. Physiol. Mol. Plant Pathol. 2018, 104, 119–126. [Google Scholar] [CrossRef]
- Chandra, P.; Enespa; Singh, R.; Arora, P.K. Microbial Lipases and Their Industrial Applications: A Comprehensive Review. Microb. Cell Fact. 2020, 19, 1–42. [Google Scholar] [CrossRef]
- Guerrand, D. Lipases Industrial Applications: Focus on Food and Agroindustries. OCL 2017, 24, D403. [Google Scholar] [CrossRef]
- de María, P.D.; de Gonzalo, G.; Alcántara, A.R. Biocatalysis as Useful Tool in Asymmetric Synthesis: An Assessment of Recently Granted Patents (2014–2019). Catalysts 2019, 9, 802. [Google Scholar] [CrossRef]
- Bello, M.; Basilio-Antonio, L.; Fragoso-Vázquez, J.; Avalos-Soriano, A.; Correa-Basurto, J. Molecular Recognition between Pancreatic Lipase and Natural and Synthetic Inhibitors. Int. J. Biol. Macromol. 2017, 98, 855–868. [Google Scholar] [CrossRef]
- Verma, S.; Meghwanshi, G.K.; Kumar, R. Current Perspectives for Microbial Lipases from Extremophiles and Metagenomics. Biochimie 2021, 182, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Gao, Y.; Li, J.; Wang, K.; Zhang, Z.; Yan, S.; Ji, N.; Zhou, Y.; Lu, S. Directed Evolution of Pseudomonas Fluorescens Lipase Variants with Improved Thermostability Using Error-Prone PCR. Front. Bioeng. Biotechnol. 2020, 8, 1034. [Google Scholar] [CrossRef]
- Contesini, F.J.; Davanço, M.G.; Borin, G.P.; Vanegas, K.G.; Cirino, J.P.G.; de Melo, R.R.; Mortensen, U.H.; Hildén, K.; Campos, D.R.; de Oliveira Carvalho, P. Advances in Recombinant Lipases: Production, Engineering, Immobilization and Application in the Pharmaceutical Industry. Catalysts 2020, 10, 1032. [Google Scholar] [CrossRef]
- Arnold, F.H. Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed. Engl. 2018, 57, 4143–4148. [Google Scholar] [CrossRef]
- Cen, Y.; Singh, W.; Arkin, M.; Moody, T.S.; Huang, M.; Zhou, J.; Wu, Q.; Reetz, M.T. Artificial Cysteine-Lipases with High Activity and Altered Catalytic Mechanism Created by Laboratory Evolution. Nat. Commun. 2019, 10, 3198. [Google Scholar] [CrossRef]
- Drożdż, A.; Chrobok, A. Chemo-Enzymatic Baeyer–Villiger Oxidation of 4-Methylcyclohexanone via Kinetic Resolution of Racemic Carboxylic Acids: Direct Access to Enantioenriched Lactone. Chem. Commun. 2016, 52, 1230–1233. [Google Scholar] [CrossRef]
- Li, L.; Ji, F.; Wang, J.; Jiang, B.; Li, Y.; Bao, Y. Efficient Mono-Acylation of Fructose by Lipase-Catalyzed Esterification in Ionic Liquid Co-Solvents. Carbohydr. Res. 2015, 416, 51–58. [Google Scholar] [CrossRef]
- Bansode, S.R.; Rathod, V.K. An Intensified Technique for Lipase Catalysed Amide Synthesis. Chem. Eng. Process. Process Intensif. 2019, 143, 107605. [Google Scholar] [CrossRef]
- Dettori, L.; Jelsch, C.; Guiavarc’h, Y.; Delaunay, S.; Framboisier, X.; Chevalot, I.; Humeau, C. Molecular Rules for Selectivity in Lipase-Catalysed Acylation of Lysine. Process Biochem. 2018, 74, 50–60. [Google Scholar] [CrossRef]
- Sun, M.; Nie, K.; Wang, F.; Deng, L. Optimization of the Lipase-Catalyzed Selective Amidation of Phenylglycinol. Front. Bioeng. Biotechnol. 2020, 7, 486. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Shen, L.; Dong, Z.; Shen, J.; Du, L.; Luo, X. Enzymatic Synthesis of Thioesters from Thiols and Vinyl Esters in a Continuous-Flow Microreactor. Catalysts 2018, 8, 249. [Google Scholar] [CrossRef]
- Chang, P.; Zhang, Z.; Tang, S. Lipase-Catalyzed Synthesis of Sugar Ester in Mixed Biphasic System of Ionic Liquids and Supercritical Carbon Dioxide. J. Chin. Chem. Soc. 2018, 65, 452–458. [Google Scholar] [CrossRef]
- Kumar, A.; Dhar, K.; Kanwar, S.S.; Arora, P.K. Lipase Catalysis in Organic Solvents: Advantages and Applications. Biol. Proced. Online 2016, 18, 2. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.; Wang, C.; Shang, W.; Xiao, B.; Duan, S.; Li, F.; Wang, L.; Chen, P. Lipase-Catalyzed Aza-Michael Addition of Amines to Acrylates in Supercritical Carbon Dioxide. J. Chem. Technol. Biotechnol. 2019, 94, 3981–3986. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, H.; Li, X.; Li, L.-L.; Liang, Z.-H.; Liang, X.-Y.; Chen, X.-Y. MML-Catalyzed Direct Aldol Reaction in Green Solvents. Biomass Convers. Biorefin. 2020. [Google Scholar] [CrossRef]
- Li, K.; He, T.; Li, C.; Feng, X.-W.; Wang, N.; Yu, X.-Q. Lipase-Catalysed Direct Mannich Reaction in Water: Utilization of Biocatalytic Promiscuity for C–C Bond Formation in a “One-Pot” Synthesis. Green Chem. 2009, 11, 777–779. [Google Scholar] [CrossRef]
- Wang, C.; Wang, N.; Liu, X.; Wan, P.; He, X.; Shang, Y. Expanding Application of Immobilized Candida Antarctica Lipase B: A Green Enzyme Catalyst for Knoevenagel Condensation Reaction. Fibers Polym. 2018, 19, 1611–1617. [Google Scholar] [CrossRef]
- Kapoor, M.; Majumder, A.B.; Gupta, M.N. Promiscuous Lipase-Catalyzed C–C Bond Formation Reactions Between 4 Nitrobenzaldehyde and 2-Cyclohexen-1-One in Biphasic Medium: Aldol and Morita–Baylis–Hillman Adduct Formations. Catal. Lett. 2015, 145, 527–532. [Google Scholar] [CrossRef]
- Guezane lakoud, S.; Toffano, M.; Zouioueche-Aribi, L. Promiscuous Lipase Catalyzed a New P–C Bond Formation: Green and Efficient Protocol for One-Pot Synthesis of α-Aminophosphonates. Heteroat. Chem. 2017, 28, 21408. [Google Scholar] [CrossRef]
- Vongvilai, P.; Linder, M.; Sakulsombat, M.; Svedendahl Humble, M.; Berglund, P.; Brinck, T.; Ramström, O. Racemase Activity of B. Cepacia Lipase Leads to Dual-Function Asymmetric Dynamic Kinetic Resolution of α-Aminonitriles. Angew. Chem. Int. Ed. 2011, 50, 6592–6595. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, K.K.; Gupta, R. Synthesis of Chirally Pure Enantiomers by Lipase. J. Oleo Sci. 2017, 66, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, K.; Lütz, S. Recent Developments and Challenges of Biocatalytic Processes in the Pharmaceutical Industry. Curr. Opin. Green Sustain. Chem. 2018, 11, 58–64. [Google Scholar] [CrossRef]
- Sheldon, R.A. The E Factor 25 Years on: The Rise of Green Chemistry and Sustainability. Green Chem. 2017, 19, 18–43. [Google Scholar] [CrossRef]
- Domínguez de María, P. Biocatalysis, Sustainability, and Industrial Applications: Show Me the Metrics. Curr. Opin. Green Sustain. Chem. 2021, 31, 100514. [Google Scholar] [CrossRef]
- Rabbani, G.; Ahmad, E.; Khan, M.V.; Ashraf, M.T.; Bhat, R.; Khan, R.H. Impact of Structural Stability of Cold Adapted Candida Antarctica Lipase B (CaLB): In Relation to PH, Chemical and Thermal Denaturation. RSC Adv. 2015, 5, 20115–20131. [Google Scholar] [CrossRef]
- Lotti, M.; Pleiss, J.; Valero, F.; Ferrer, P. Effects of Methanol on Lipases: Molecular, Kinetic and Process Issues in the Production of Biodiesel. Biotechnol. J. 2015, 10, 22–30. [Google Scholar] [CrossRef]
- Wu, S.; Snajdrova, R.; Moore, J.C.; Baldenius, K.; Bornscheuer, U.T. Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem. Int. Ed. 2021, 60, 88–119. [Google Scholar] [CrossRef]
- Guisan, J.M.; López-Gallego, F.; Bolivar, J.M.; Rocha-Martín, J.; Fernandez-Lorente, G. The Science of Enzyme Immobilization. In Immobilization of Enzymes and Cells: Methods and Protocols; Guisan, J.M., Bolivar, J.M., López-Gallego, F., Rocha-Martín, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2020; pp. 1–26. ISBN 978-1-07-160215-7. [Google Scholar]
- Zhao, Z.; Zhou, M.-C.; Liu, R.-L. Recent Developments in Carriers and Non-Aqueous Solvents for Enzyme Immobilization. Catalysts 2019, 9, 647. [Google Scholar] [CrossRef]
- Hassan, M.E.; Yang, Q.; Xiao, Z.; Liu, L.; Wang, N.; Cui, X.; Yang, L. Impact of Immobilization Technology in Industrial and Pharmaceutical Applications. 3 Biotech 2019, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Mokhtar, N.F.; Abd Rahman, R.N.Z.R.; Muhd Noor, N.D.; Mohd Shariff, F.; Ali, M.S.M. The Immobilization of Lipases on Porous Support by Adsorption and Hydrophobic Interaction Method. Catalysts 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Almeida, F.L.C.; Castro, M.P.J.; Travália, B.M.; Forte, M.B.S. Trends in Lipase Immobilization: Bibliometric Review and Patent Analysis. Process Biochem. 2021, 110, 37–51. [Google Scholar] [CrossRef]
- Cesarini, S.; Infanzón, B.; Pastor, F.I.J.; Diaz, P. Fast and Economic Immobilization Methods Described for Non-Commercial Pseudomonaslipases. BMC Biotechnol. 2014, 14, 27. [Google Scholar] [CrossRef]
- Godoy, C.A.; de las Rivas, B.; Bezbradica, D.; Bolivar, J.M.; López-Gallego, F.; Fernandez-Lorente, G.; Guisan, J.M. Reactivation of a Thermostable Lipase by Solid Phase Unfolding/Refolding: Effect of Cysteine Residues on Refolding Efficiency. Enzyme Microb. Technol. 2011, 49, 388–394. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Bolivar, J.M.; Palau-Ors, A.; Volpato, G.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Guisan, J.M. Positive Effects of the Multipoint Covalent Immobilization in the Reactivation of Partially Inactivated Derivatives of Lipase from Thermomyces Lanuginosus. Enzym. Microb. Technol. 2009, 44, 386–393. [Google Scholar] [CrossRef]
- Rodrigues, R.C. Improved Reactivation of Immobilized-Stabilized Lipase from Thermomyces Lanuginosus by Its Coating with Highly Hydrophilic Polymers. J. Biotechnol. 2009, 144, 113–119. [Google Scholar] [CrossRef]
- Chen, J.-W.; Wu, W.-T. Regeneration of Immobilized Candida Antarctica Lipase for Transesterification. J. Biosci. Bioeng. 2003, 95, 466–469. [Google Scholar] [CrossRef]
- Basso, A.; Serban, S. Industrial Applications of Immobilized Enzymes—A Review. Mol. Catal. 2019, 479, 110607. [Google Scholar] [CrossRef]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The Lid Domain in Lipases: Structural and Functional Determinant of Enzymatic Properties. Front. Bioeng. Biotechnol. 2017, 5, 16. [Google Scholar] [CrossRef]
- Albayati, S.H.; Masomian, M.; Ishak, S.N.H.; bin Mohamad Ali, M.S.; Thean, A.L.; binti Mohd Shariff, F.; binti Muhd Noor, N.D.; Raja Abd Rahman, R.N.Z. Main Structural Targets for Engineering Lipase Substrate Specificity. Catalysts 2020, 10, 747. [Google Scholar] [CrossRef]
- Anobom, C.D.; Pinheiro, A.S.; De-Andrade, R.A.; Aguieiras, E.C.G.; Andrade, G.C.; Moura, M.V.; Almeida, R.V.; Freire, D.M. From Structure to Catalysis: Recent Developments in the Biotechnological Applications of Lipases. BioMed Res. Int. 2014, 2014, e684506. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.L.; Buchholz, P.C.F.; Pleiss, J. The Modular Structure of α/β-Hydrolases. FEBS J. 2020, 287, 1035–1053. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.Y.; Howles, P.N. Carboxyl Ester Lipase. J. Lipid Res. 2002, 43, 2017–2030. [Google Scholar] [CrossRef]
- Yasutake, Y.; Konishi, K.; Muramatsu, S.; Yoshida, K.; Aburatani, S.; Sakasegawa, S.-I.; Tamura, T. Bacterial Triacylglycerol Lipase Is a Potential Cholesterol Esterase: Identification of a Key Determinant for Sterol-Binding Specificity. Int. J. Biol. Macromol. 2021, 167, 578–586. [Google Scholar] [CrossRef]
- van Pouderoyen, G.; Eggert, T.; Jaeger, K.-E.; Dijkstra, B.W. The Crystal Structure of Bacillus Subtili Lipase: A Minimal α/β Hydrolase Fold Enzyme1†1Edited by R. Huber†This Paper Is Dedicated to the Memory of Professor Charles Colson, Louvain-La-Neuve, Who Initiated the Molecular Research on Bacillus Subtilis Lipase. J. Mol. Biol. 2001, 309, 215–226. [Google Scholar] [CrossRef]
- Akatsuka, H.; Kawai, E.; Omori, K.; Komatsubara, S.; Shibatani, T.; Tosa, T. The LipA Gene of Serratia Marcescens Which Encodes an Extracellular Lipase Having No N-Terminal Signal Peptide. J. Bacteriol. 1994, 176, 1949–1956. [Google Scholar] [CrossRef]
- Choi, W.-C.; Kim, M.H.; Ro, H.-S.; Ryu, S.R.; Oh, T.-K.; Lee, J.-K. Zinc in Lipase L1 from Geobacillus Stearothermophilus L1 and Structural Implications on Thermal Stability. FEBS Lett. 2005, 579, 3461–3466. [Google Scholar] [CrossRef]
- Timucin, E.; Sezerman, O.U. Zinc Modulates Self-Assembly of Bacillus Thermocatenulatus Lipase. Biochemistry 2015, 54, 3901–3910. [Google Scholar] [CrossRef]
- Arpigny, J.L.; Jaeger, K.E. Bacterial Lipolytic Enzymes: Classification and Properties. Biochem. J. 1999, 343 Pt 1, 177–183. [Google Scholar] [CrossRef]
- Zhang, J.-H.; Jiang, Y.-Y.; Lin, Y.; Sun, Y.-F.; Zheng, S.-P.; Han, S.-Y. Structure-Guided Modification of Rhizomucor Miehei Lipase for Production of Structured Lipids. PLoS ONE 2013, 8, e67892. [Google Scholar] [CrossRef]
- Domínguez de María, P.; Sánchez-Montero, J.M.; Sinisterra, J.V.; Alcántara, A.R. Understanding Candida Rugosa Lipases: An Overview. Biotechnol. Adv. 2006, 24, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Stauch, B.; Fisher, S.J.; Cianci, M. Open and Closed States of Candida Antarctica Lipase B: Protonation and the Mechanism of Interfacial Activation1. J. Lipid Res. 2015, 56, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-López, C. Activation of Bacterial Thermo Alkalophilic Lipases Is Spurred by Dramatic Structural Rearrangements. J. Biol. Chem. 2009, 284, 4365–4372. [Google Scholar] [CrossRef]
- Brzozowski, A.M. Structural Origins of the Interfacial Activation in Thermomyces (Humicola) Lanuginosa Lipase. Biochemistry 2000, 39, 15071–15082. [Google Scholar] [CrossRef]
- Jaeger, K.-E.; Dijkstra, B.W.; Reetz, M.T. Bacterial Biocatalysts: Molecular Biology, Three-Dimensional Structures, and Biotechnological Applications of Lipases. Annu. Rev. Microbiol. 1999, 53, 315–351. [Google Scholar] [CrossRef]
- Pleiss, J.; Fischer, M.; Schmid, R.D. Anatomy of Lipase Binding Sites: The Scissile Fatty Acid Binding Site. Chem. Phys. Lipids 1998, 93, 67–80. [Google Scholar] [CrossRef]
- Angkawidjaja, C.; Matsumura, H.; Koga, Y.; Takano, K.; Kanaya, S. X-ray Crystallographic and MD Simulation Studies on the Mechanism of Interfacial Activation of a Family I.3 Lipase with Two Lids. J. Mol. Biol. 2010, 400, 82–95. [Google Scholar] [CrossRef]
- Álvarez-Cao, M.-E.; González, R.; Pernas, M.A.; Rúa, M.L. Contribution of the Oligomeric State to the Thermostability of Isoenzyme 3 from Candida Rugosa. Microorganisms 2018, 6, 108. [Google Scholar] [CrossRef]
- Mateos-Diaz, E.; Amara, S.; Roussel, A.; Longhi, S.; Cambillau, C.; Carrière, F. Probing Conformational Changes and Interfacial Recognition Site of Lipases With Surfactants and Inhibitors. Methods Enzymol. 2017, 583, 279–307. [Google Scholar] [CrossRef]
- Skjold-Jørgensen, J.; Vind, J.; Svendsen, A.; Bjerrum, M.J. Understanding the Activation Mechanism of Thermomyces Lanuginosus Lipase Using Rational Design and Tryptophan-Induced Fluorescence Quenching. Eur. J. Lipid Sci. Technol. 2016, 118, 1644–1660. [Google Scholar] [CrossRef]
- Bbohr, S.; Lund, P.; Kallenbach, A.; Pinholt, H.; Thomsen, J.; Iversen, L.; Svendsen, A.; Christensen, S.; Hatzakis, N. Direct Observation of Thermomyces Lanuginosus Lipase Diffusional States by Single Particle Tracking and Their Remodeling by Mutations and Inhibition. Sci. Rep. 2019, 9, 16169. [Google Scholar] [CrossRef] [PubMed]
- Mathesh, M.; Luan, B.; Akanbi, T.O.; Weber, J.K.; Liu, J.; Barrow, C.J.; Zhou, R.; Yang, W. Opening Lids: Modulation of Lipase Immobilization by Graphene Oxides. ACS Catal. 2016, 6, 4760–4768. [Google Scholar] [CrossRef]
- Willems, N.; Lelimousin, M.; Skjold-Jørgensen, J.; Svendsen, A.; Sansom, M.S.P. The Effect of Mutations in the Lid Region of Thermomyces Lanuginosus Lipase on Interactions with Triglyceride Surfaces: A Multi-Scale Simulation Study. Chem. Phys. Lipids 2018, 211, 4–15. [Google Scholar] [CrossRef]
- Willems, N.; Lelimousin, M.; Koldsø, H.; Sansom, M.S.P. Interfacial Activation of M37 Lipase: A Multi-Scale Simulation Study. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 340–349. [Google Scholar] [CrossRef]
- Kapoor, M.; Gupta, M.N. Lipase Promiscuity and Its Biochemical Applications. Process Biochem. 2012, 47, 555–569. [Google Scholar] [CrossRef]
- Toro, E.C.; Rodríguez, D.F.; Morales, N.; García, L.M.; Godoy, C.A. Novel Combi-Lipase Systems for Fatty Acid Ethyl Esters Production. Catalysts 2019, 9, 546. [Google Scholar] [CrossRef]
- Zhao, D.; Peng, C.; Zhou, J. Lipase Adsorption on Different Nanomaterials: A Multi-Scale Simulation Study. Phys. Chem. Chem. Phys. 2014, 17, 840–850. [Google Scholar] [CrossRef]
- Berruex, L.G.; Freitag, R. Separation and Purification of Biochemicals. In Encyclopedia of Physical Science and Technology, 3rd ed.; Meyers, R.A., Ed.; Academic Press: New York, NY, USA, 2003; pp. 651–673. ISBN 978-0-12-227410-7. [Google Scholar]
- Sharma, S.; Berne, B.J.; Kumar, S.K. Thermal and Structural Stability of Adsorbed Proteins. Biophys. J. 2010, 99, 1157–1165. [Google Scholar] [CrossRef]
- Galmés, M.À.; García-Junceda, E.; Świderek, K.; Moliner, V. Exploring the Origin of Amidase Substrate Promiscuity in CALB by a Computational Approach. ACS Catal. 2020, 10, 1938–1946. [Google Scholar] [CrossRef]
- Świderek, K.; Martí, S.; Moliner, V. Theoretical Study of Primary Reaction of Pseudozyma Antarctica Lipase B as the Starting Point To Understand Its Promiscuity. ACS Catal. 2014, 4, 426–434. [Google Scholar] [CrossRef]
- Paiva, A.L.; Balcão, V.M.; Malcata, F.X. Kinetics and Mechanisms of Reactions Catalyzed by Immobilized Lipases. Enzym. Microb. Technol. 2000, 27, 187–204. [Google Scholar] [CrossRef]
- Gupta, P.; Chaubey, A.; Mahajan, N.; Anand, N. A Review on Arthrobacter Sp. Lipase: A Versatile Biocatalyst for the Kinetic Resolution to Access Enantiomerically Pure/Enriched Compounds. Chirality 2021, 33, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Caicedo, A.; Soto-Martínez, D.M.; Osorio, D.A.; Novoa, M.; Loaiza, A.E.; Jaramillo-Gómez, L.M. Synthesis of 1-Azaspiro[4.4]Nonane Derivatives Enabled by Domino Radical Bicyclization Involving Formation and Capture of Alkoxyaminyl Radicals. ACS Omega 2019, 4, 21100–21114. [Google Scholar] [CrossRef] [PubMed]
- Mnasri, T.; Hérault, J.; Gauvry, L.; Loiseau, C.; Poisson, L.; Ergan, F.; Pencréac’h, G. Lipase-Catalyzed Production of Lysophospholipids. OCL 2017, 24, D405. [Google Scholar] [CrossRef][Green Version]
- Drescher, S.; van Hoogevest, P. The Phospholipid Research Center: Current Research in Phospholipids and Their Use in Drug Delivery. Pharmaceutics 2020, 12, 1235. [Google Scholar] [CrossRef]
- Bond, P. Phosphatidic Acid: Biosynthesis, Pharmacokinetics, Mechanisms of Action and Effect on Strength and Body Composition in Resistance-Trained Individuals. Nutr. Metab. 2017, 14, 12. [Google Scholar] [CrossRef]
- Khan, M.S.Y.; Akhter, M. Glyceride Derivatives as Potential Prodrugs: Synthesis, Biological Activity and Kinetic Studies of Glyceride Derivatives of Mefenamic Acid. Die Pharm. 2005, 60, 110–114. [Google Scholar]
- Klein, R.R.; King, G.; Moreau, R.A.; Haas, M.J. Altered Acyl Chain Length Specificity of Rhizopus Delemar Lipase through Mutagenesis and Molecular Modeling. Lipids 1997, 32, 123–130. [Google Scholar] [CrossRef]
- Brundiek, H.; Padhi, S.K.; Kourist, R.; Evitt, A.; Bornscheuer, U.T. Altering the Scissile Fatty Acid Binding Site of Candida Antarctica Lipase A by Protein Engineering for the Selective Hydrolysis of Medium Chain Fatty Acids. Eur. J. Lipid Sci. Technol. 2012, 114, 1148–1153. [Google Scholar] [CrossRef]
- Quaglia, D.; Alejaldre, L.; Ouadhi, S.; Rousseau, O.; Pelletier, J.N. Holistic Engineering of Cal-A Lipase Chain-Length Selectivity Identifies Triglyceride Binding Hot-Spot. PLoS ONE 2019, 14, e0210100. [Google Scholar] [CrossRef]
- Borgdorf, R.; Warwel, S. Substrate Selectivity of Various Lipases in the Esterification of Cis- and Trans-9-Octadecenoic Acid. Appl. Microbiol. Biotechnol. 1999, 51, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Warwel, S.; Borgdorf, R. Substrate Selectivity of Lipases in the Esterification of Cis/Trans-Isomers and Positional Isomers of Conjugated Linoleic Acid (CLA). Biotechnol. Lett. 2000, 22, 1151–1155. [Google Scholar] [CrossRef]
- Brundiek, H.B.; Evitt, A.S.; Kourist, R.; Bornscheuer, U.T. Creation of a Lipase Highly Selective for Trans Fatty Acids by Protein Engineering. Angew. Chem. Int. Ed. 2012, 51, 412–414. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Zhao, G.; Holzmann, N.; Yuan, S.; Wang, J.; Wang, Y. Structure-Guided Rational Design of a Mono- and Diacylglycerol Lipase from Aspergillus Oryzae: A Single Residue Mutant Increases the Hydrolysis Ability. J. Agric. Food Chem. 2021, 69, 5344–5352. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Khan, F.I.; Zhao, Z.; Wang, W.; Yang, B.; Wang, Y. Diacylglycerol Production by Genetically Modified Lipase from Malassezia Globosa. J. Mol. Catal. B Enzym. 2016, 133, S204–S212. [Google Scholar] [CrossRef]
- Xu, T.; Liu, L.; Hou, S.; Xu, J.; Yang, B.; Wang, Y.; Liu, J. Crystal Structure of a Mono- and Diacylglycerol Lipase from Malassezia Globosa Reveals a Novel Lid Conformation and Insights into the Substrate Specificity. J. Struct. Biol. 2012, 178, 363–369. [Google Scholar] [CrossRef]
- Lan, D.; Popowicz, G.M.; Pavlidis, I.V.; Zhou, P.; Bornscheuer, U.T.; Wang, Y. Conversion of a Mono- and Diacylglycerol Lipase into a Triacylglycerol Lipase by Protein Engineering. ChemBioChem 2015, 16, 1431–1434. [Google Scholar] [CrossRef]
- Liu, L.; Lan, D.; Wang, Q.; Gao, C.; Li, Z.; Yang, B.; Wang, Y. A “Bridge-like” Structure Responsible for the Substrate Selectivity of Mono- and Diacylglycerol Lipase from Aspergillus Oryzae. J. Mol. Catal. B Enzym. 2013, 97, 144–149. [Google Scholar] [CrossRef]
- Wei, W.; Sun, C.; Wang, X.; Jin, Q.; Xu, X.; Akoh, C.C.; Wang, X. Lipase-Catalyzed Synthesis of Sn-2 Palmitate: A Review. Engineering 2020, 6, 406–414. [Google Scholar] [CrossRef]
- Sugasini, D.; Thomas, R.; Yalagala, P.C.R.; Tai, L.M.; Subbaiah, P.V. Dietary Docosahexaenoic Acid (DHA) as Lysophosphatidylcholine, but Not as Free Acid, Enriches Brain DHA and Improves Memory in Adult Mice. Sci. Rep. 2017, 7, 11263. [Google Scholar] [CrossRef]
- Duan, Z.-Q.; Du, W.; Liu, D.-H. The Mechanism of Solvent Effect on the Positional Selectivity of Candida Antarctica Lipase B during 1,3-Diolein Synthesis by Esterification. Bioresour. Technol. 2011, 102, 11048–11050. [Google Scholar] [CrossRef]
- He, Y.; Li, J.; Kodali, S.; Balle, T.; Chen, B.; Guo, Z. Liquid Lipases for Enzymatic Concentration of N-3 Polyunsaturated Fatty Acids in Monoacylglycerols via Ethanolysis: Catalytic Specificity and Parameterization. Bioresour. Technol. 2017, 224, 445–456. [Google Scholar] [CrossRef]
- Cha, H.-J.; Park, J.-B.; Park, S. Esterification of Secondary Alcohols and Multi-Hydroxyl Compounds by Candida Antarctica Lipase B and Subtilisin. Biotechnol. Bioprocess Eng. 2019, 24, 41–47. [Google Scholar] [CrossRef]
- Mao, J.; Hu, Z.; Hu, J.; Zhu, X.; Xiong, H. A Density Functional Theory (DFT) Study of the Acyl Migration Occurring during Lipase-Catalyzed Transesterifications. Int. J. Mol. Sci. 2019, 20, 3438. [Google Scholar] [CrossRef] [PubMed]
- Šinkūnienė, D.; Adlercreutz, P. Effects of Regioselectivity and Lipid Class Specificity of Lipases on Transesterification, Exemplified by Biodiesel Production. J. Am. Oil Chem. Soc. 2014, 91, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Mangas-Sánchez, J.; Feng, F.; Adlercreutz, P. Acyl Migration in Enzymatic Interesterification of Triacylglycerols: Effects of Lipases from Thermomyces Lanuginosus and Rhizopus Oryzae, Support Material, and Water Activity. Eur. J. Lipid Sci. Technol. 2016, 118, 1579–1587. [Google Scholar] [CrossRef]
- Iribarren, A.M.; Iglesias, L.E. An Update of Biocatalytic Selective Acylation and Deacylation of Monosaccharides. RSC Adv. 2016, 6, 16358–16386. [Google Scholar] [CrossRef]
- Terreni, M.; Salvetti, R.; Linati, L.; Fernandez-Lafuente, R.; Fernández-Lorente, G.; Bastida, A.; Guisan, J.M. Regioselective Enzymatic Hydrolysis of Acetylated Pyranoses and Pyranosides Using Immobilised Lipases. An Easy Chemoenzymatic Synthesis of α- and β-d-Glucopyranose Acetates Bearing a Free Secondary C-4 Hydroxyl Group. Carbohydr. Res. 2002, 337, 1615–1621. [Google Scholar] [CrossRef]
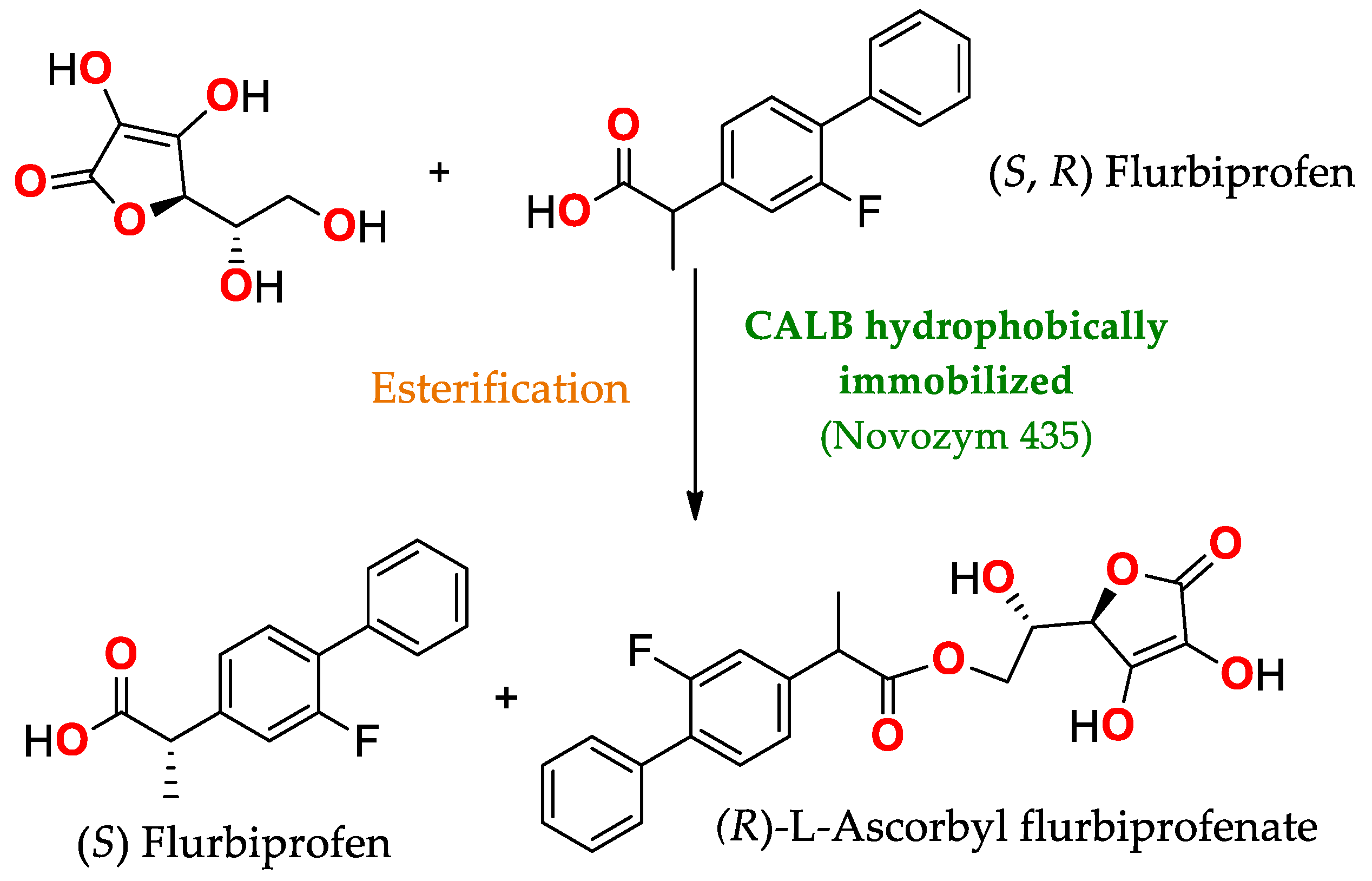
- Xin, J.; Sun, L.; Chen, S.; Wang, Y.; Xia, C. Synthesis of L-Ascorbyl Flurbiprofenate by Lipase-Catalyzed Esterification and Transesterification Reactions. BioMed Res. Int. 2017, 2017, e5751262. [Google Scholar] [CrossRef]
- de Melo Carvalho, A.C.L.; Fonseca, T.D.S.; de Mattos, M.C.; de Oliveira, M.D.C.F.; de Lemos, T.L.G.; Molinari, F.; Romano, D.; Serra, I. Recent Advances in Lipase-Mediated Preparation of Pharmaceuticals and Their Intermediates. Int. J. Mol. Sci. 2015, 16, 29682–29716. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. Compendium of Chemical Terminology, 2nd ed.; Blackwell Science Oxford: Oxford, UK, 1997; Volume 1669, ISBN 0-9678550-9-8. [Google Scholar]
- Gawley, R.E. Do the Terms “% Ee” and “% de” Make Sense as Expressions of Stereoisomer Composition or Stereoselectivity? J. Org. Chem. 2006, 71, 2411–2416. [Google Scholar] [CrossRef]
- Chen, H.; Meng, X.; Xu, X.; Liu, W.; Li, S. The Molecular Basis for Lipase Stereoselectivity. Appl. Microbiol. Biotechnol. 2018, 102, 3487–3495. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Park, S. Enhancing Enantioselectivity of Candida Antarctica Lipase B towards Chiral Sec-Alcohols Bearing Small Substituents through Hijacking Sequence of A Homolog. Tetrahedron Lett. 2021, 75, 153186. [Google Scholar] [CrossRef]
- Mezzetti, A.; Schrag, J.D.; Cheong, C.S.; Kazlauskas, R.J. Mirror-Image Packing in Enantiomer Discrimination: Molecular Basis for the Enantioselectivity of B.Cepacia Lipase toward 2-Methyl-3-Phenyl-1-Propanol. Chem. Biol. 2005, 12, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.R.; Alnoch, R.C.; de Almeida, J.M.; dos Santos, L.A.; Andretta, A.T.; del Pilar Cuaspa Ropaín, R.; de Souza, E.M.; Mitchell, D.A.; Krieger, N. Key Mutation Sites for Improvement of the Enantioselectivity of Lipases through Protein Engineering. Biochem. Eng. J. 2021, 172, 108047. [Google Scholar] [CrossRef]
- Li, D.-Y.; Lou, Y.-J.; Xu, J.; Chen, X.-Y.; Lin, X.-F.; Wu, Q. Electronic Effect-Guided Rational Design of Candida Antarctica Lipase B for Kinetic Resolution Towards Diarylmethanols. Adv. Synth. Catal. 2021, 363, 1867–1872. [Google Scholar] [CrossRef]
- Holmquist, M.; Haeffner, F.; Norin, T.; Hult, K. A Structural Basis for Enantioselective Inhibition of Candida Rugosa Lipase by Long-Chain Aliphatic Alcohols. Protein Sci. Publ. Protein Soc. 1996, 5, 83–88. [Google Scholar] [CrossRef][Green Version]
- Park, A.; Kim, S.; Park, J.; Joe, S.; Min, B.; Oh, J.; Song, J.; Park, S.; Park, S.; Lee, H. Structural and Experimental Evidence for the Enantiomeric Recognition toward a Bulky Sec-Alcohol by Candida Antarctica Lipase B. ACS Catal. 2016, 6, 7458–7465. [Google Scholar] [CrossRef]
- Kitamoto, Y.; Kuruma, Y.; Suzuki, K.; Hattori, T. Effect of Solvent Polarity on Enantioselectivity in Candida Antarctica Lipase B Catalyzed Kinetic Resolution of Primary and Secondary Alcohols. J. Org. Chem. 2015, 80, 521–527. [Google Scholar] [CrossRef]
- Otsu, M.; Suzuki, Y.; Koesoema, A.A.; Hoang, H.N.; Tamura, M.; Matsuda, T. CO2-Expanded Liquids as Solvents to Enhance Activity of Pseudozyma Antarctica Lipase B towards Ortho-Substituted 1-Phenylethanols. Tetrahedron Lett. 2020, 61, 152424. [Google Scholar] [CrossRef]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of Conventional Anti-Cancer Drugs as New Tools against Multidrug Resistant Tumors. Drug Resist. Updat. 2020, 50. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Paterson, D.L. Antibiotics in the Clinical Pipeline in October 2019. J. Antibiot. 2020, 73, 329–364. [Google Scholar] [CrossRef] [PubMed]
- Branneby, C.; Carlqvist, P.; Magnusson, A.; Hult, K.; Brinck, T.; Berglund, P. Carbon−Carbon Bonds by Hydrolytic Enzymes. J. Am. Chem. Soc. 2003, 125, 874–875. [Google Scholar] [CrossRef]
- Kazlauskas, R.J. Enhancing Catalytic Promiscuity for Biocatalysis. Curr. Opin. Chem. Biol. 2005, 9, 195–201. [Google Scholar] [CrossRef]
- Yang, F.; Wang, H.; Jiang, L.; Yue, H.; Zhang, H.; Wang, Z.; Wang, L. A Green and One-Pot Synthesis of Benzo[g]Chromene Derivatives through a Multi-Component Reaction Catalyzed by Lipase. RSC Adv. 2014, 5, 5213–5216. [Google Scholar] [CrossRef]
- Dwivedee, B.P.; Soni, S.; Sharma, M.; Bhaumik, J.; Laha, J.K.; Banerjee, U.C. Promiscuity of Lipase-Catalyzed Reactions for Organic Synthesis: A Recent Update. ChemistrySelect 2018, 3, 2441–2466. [Google Scholar] [CrossRef]
- Patti, A.; Sanfilippo, C. Stereoselective Promiscuous Reactions Catalyzed by Lipases. Int. J. Mol. Sci. 2022, 23, 2675. [Google Scholar] [CrossRef]
- Hult, K.; Berglund, P. Enzyme Promiscuity: Mechanism and Applications. Trends Biotechnol. 2007, 25, 231–238. [Google Scholar] [CrossRef]
- Atkins, W.M. Biological Messiness vs. Biological Genius: Mechanistic Aspects and Roles of Protein Promiscuity. J. Steroid Biochem. Mol. Biol. 2015, 151, 3–11. [Google Scholar] [CrossRef]
- Atkins, W.M. Mechanisms of Promiscuity among Drug Metabolizing Enzymes and Drug Transporters. FEBS J. 2020, 287, 1306–1322. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.N.; dos Anjos, C.S.; Orozco, E.V.M.; Porto, A.L.M. Versatility of Candida Antarctica Lipase in the Amide Bond Formation Applied in Organic Synthesis and Biotechnological Processes. Mol. Catal. 2019, 466, 75–105. [Google Scholar] [CrossRef]
- Mohammadi Ziarani, G.; Gholamzadeh, P.; Asadiatouei, P.; Lashgari, N. The Role of Pseudomonas Cepacia Lipase in the Asymmetric Synthesis of Heterocyclic Based Compounds. J. Mol. Catal. B Enzym. 2015, 122, 93–116. [Google Scholar] [CrossRef]
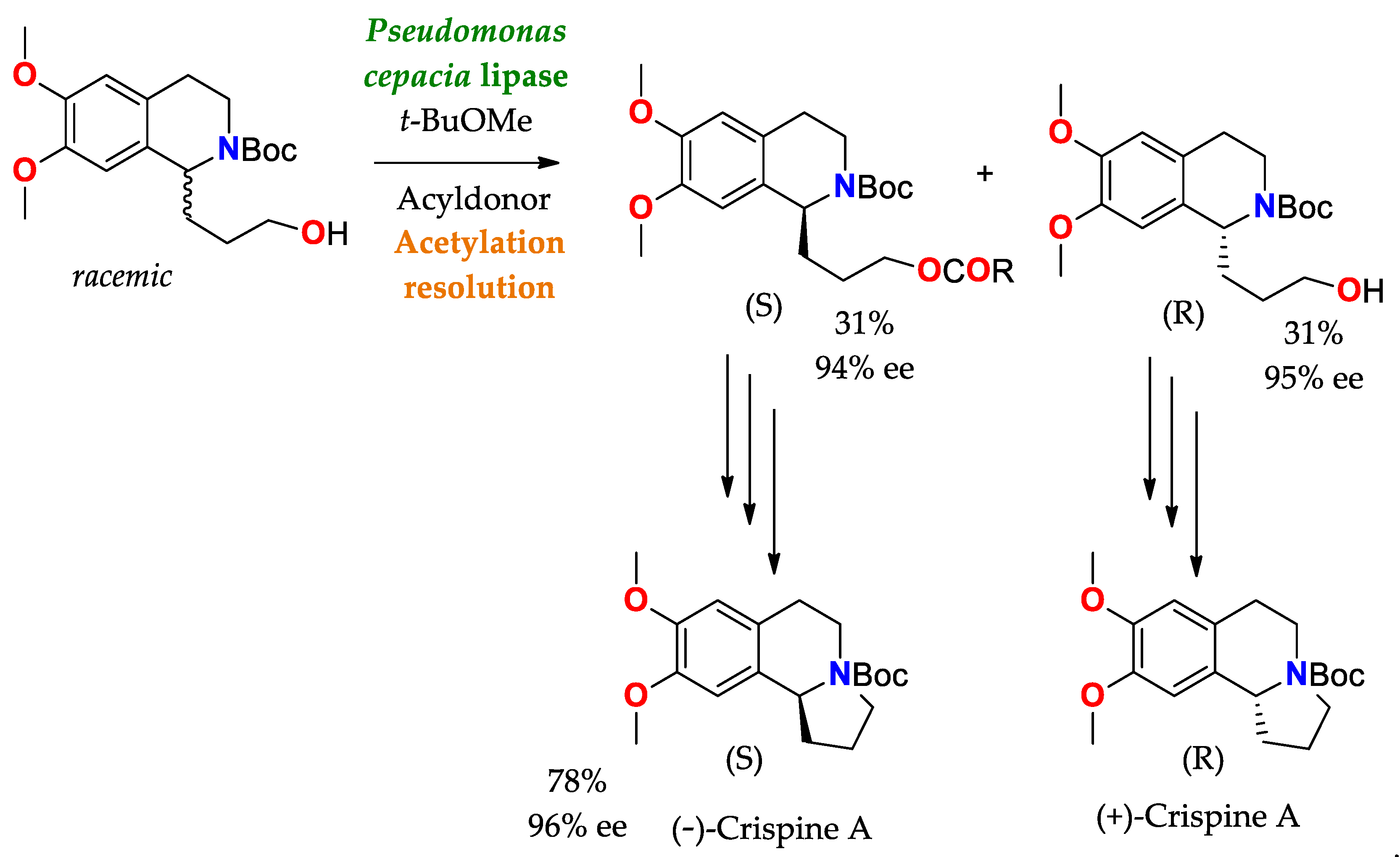
- Forró, E.; Schönstein, L.; Fülöp, F. Total Synthesis of Crispine A Enantiomers through a Burkholderia Cepacia Lipase-Catalysed Kinetic Resolution. Tetrahedron Asymmetry 2011, 22, 1255–1260. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Woodley, J.M.; Fernandez-Lafuente, R. Is Enzyme Immobilization a Mature Discipline? Some Critical Considerations to Capitalize on the Benefits of Immobilization. Chem. Soc. Rev. 2022, 51, 6251–6290. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; Dos Santos, J.C.S.; Rodrigues, R.C.; Berenguer-Murcia, Á.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “Perfect” Lipase Immobilized Biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef]
- Lv, L.; Dai, L.; Du, W.; Liu, D. Progress in Enzymatic Biodiesel Production and Commercialization. Processes 2021, 9, 355. [Google Scholar] [CrossRef]
- da Silva Dutra, L.; Costa Cerqueira Pinto, M.; Cipolatti, E.P.; Aguieiras, E.C.G.; Manoel, E.A.; Greco-Duarte, J.; Guimarães Freire, D.M.; Pinto, J.C. How the Biodiesel from Immobilized Enzymes Production Is Going on: An Advanced Bibliometric Evaluation of Global Research. Renew. Sustain. Energy Rev. 2022, 153, 111765. [Google Scholar] [CrossRef]
- Adlercreutz, P. Immobilisation and Application of Lipases in Organic Media. Chem. Soc. Rev. 2013, 42, 6406–6436. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Rios, N.S.; Carballares, D.; Mendez-Sanchez, C.; Lokha, Y.; Gonçalves, L.R.B.; Fernandez-Lafuente, R. Effects of Enzyme Loading and Immobilization Conditions on the Catalytic Features of Lipase from Pseudomonas Fluorescens Immobilized on Octyl-Agarose Beads. Front. Bioeng. Biotechnol. 2020, 8, 1–13. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Virgen-Ortíz, J.J.; dos Santos, J.C.S.; Berenguer-Murcia, Á.; Alcantara, A.R.; Barbosa, O.; Ortiz, C.; Fernandez-Lafuente, R. Immobilization of Lipases on Hydrophobic Supports: Immobilization Mechanism, Advantages, Problems, and Solutions. Biotechnol. Adv. 2019, 37, 746–770. [Google Scholar] [CrossRef]
- Zhuang, W.; Quan, X.; Wang, Z.; Zhou, W.; Yang, P.; Ge, L.; Villacorta Hernandez, B.; Wu, J.; Li, M.; Zhou, J.; et al. Interfacial Microenvironment for Lipase Immobilization: Regulating the Heterogeneity of Graphene Oxide. Chem. Eng. J. 2020, 394, 125038. [Google Scholar] [CrossRef]
- Cabrera, Z.; Fernandez-Lorente, G.; Fernandez-Lafuente, R.; Palomo, J.M.; Guisan, J.M. Novozym 435 Displays Very Different Selectivity Compared to Lipase from Candida Antarctica B Adsorbed on Other Hydrophobic Supports. J. Mol. Catal. B Enzym. 2009, 57, 171–176. [Google Scholar] [CrossRef]
- Bastida, A. A Single Step Purification, Immobilization, and Hyperactivation of Lipases via Interfacial Adsorption on Strongly Hydrophobic Supports. Biotechnol. Bioeng. 1998, 58, 486–493. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Cabrera, Z.; Godoy, C.; Fernandez-Lafuente, R.; Palomo, J.M.; Guisan, J.M. Interfacially Activated Lipases against Hydrophobic Supports: Effect of the Support Nature on the Biocatalytic Properties. Process Biochem. 2008, 43, 1061–1067. [Google Scholar] [CrossRef]
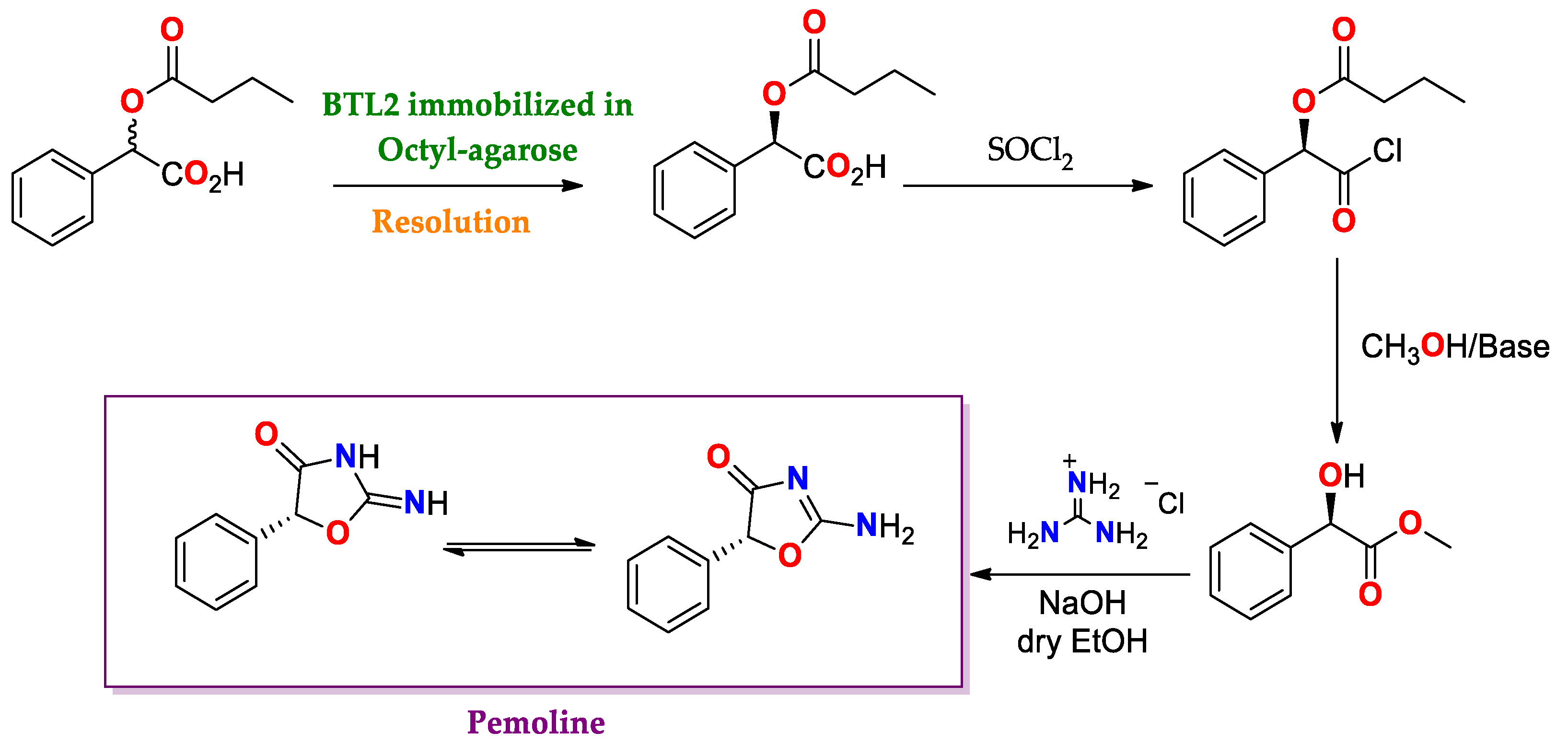
- Poterała, M.; Dranka, M.; Borowiecki, P. Chemoenzymatic Preparation of Enantiomerically Enriched (R)-(–)-Mandelic Acid Derivatives: Application in the Synthesis of the Active Agent Pemoline. Eur. J. Org. Chem. 2017, 2017, 2290–2304. [Google Scholar] [CrossRef]
- Verma, S.; Choudhary, R.N.; Kanadje, A.P.; Banerjee, U.C. Diversifying Arena of Drug Synthesis: In the Realm of Lipase Mediated Waves of Biocatalysis. Catalysts 2021, 11, 1328. [Google Scholar] [CrossRef]
- Muthuraja, P.; Usman, R.; Sajeev, R.; Gopinath, P. Controlled Meta-Selective C–H Mono- and Di-Olefination of Mandelic Acid Derivatives. Org. Lett. 2021, 23, 6014–6018. [Google Scholar] [CrossRef]
- Faber, K. Biocatalytic Applications. In Biotransformations in Organic Chemistry: A Textbook; Faber, K., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 31–313. ISBN 978-3-642-17393-6. [Google Scholar]
- Filice, M.; Guisan, J.M.; Terreni, M.; Palomo, J.M. Regioselective Monodeprotection of Peracetylated Carbohydrates. Nat. Protoc. 2012, 7, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, K.S.; Cavicchioli, R. Improved Thermal Stability and Activity in the Cold-Adapted Lipase B from Candida Antarctica Following Chemical Modification with Oxidized Polysaccharides. Extremophiles 2005, 9, 471–476. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, J.C.S.; Bonazza, H.L.; de Matos, L.J.B.L.; Carneiro, E.A.; Barbosa, O.; Fernandez-Lafuente, R.; Gonçalves, L.R.B.; de Sant’ Ana, H.B.; Santiago-Aguiar, R.S. Immobilization of CALB on Activated Chitosan: Application to Enzymatic Synthesis in Supercritical and near-Critical Carbon Dioxide. Biotechnol. Rep. 2017, 14, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Palomo, J.M.; Filice, M.; Romero, O.; Guisan, J. Improving Lipase Activity by Immobilization and Post-Immobilization Strategies. In Immobilization of Enzymes and Cells; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1051, pp. 255–273. [Google Scholar] [CrossRef]
- Wiemann, L.O.; Nieguth, R.; Eckstein, M.; Naumann, M.; Thum, O.; Ansorge-Schumacher, M.B. Composite Particles of Novozyme 435 and Silicone: Advancing Technical Applicability of Macroporous Enzyme Carriers. ChemCatChem 2009, 1, 455–462. [Google Scholar] [CrossRef]
- Orrego, A.H.; Ghobadi, R.; Moreno-Perez, S.; Mendoza, A.J.; Fernandez-Lorente, G.; Guisan, J.M.; Rocha-Martin, J. Stabilization of Immobilized Lipases by Intense Intramolecular Cross-Linking of Their Surfaces by Using Aldehyde-Dextran Polymers. Int. J. Mol. Sci. 2018, 19, 553. [Google Scholar] [CrossRef] [PubMed]
- Virgen-Ortíz, J.J.; Pedrero, S.G.; Fernandez-Lopez, L.; Lopez-Carrobles, N.; Gorines, B.C.; Otero, C.; Fernandez-Lafuente, R. Desorption of Lipases Immobilized on Octyl-Agarose Beads and Coated with Ionic Polymers after Thermal Inactivation. Stronger Adsorption of Polymers/Unfolded Protein Composites. Molecules 2017, 22, 91. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.C.P.; Amaral, J.C.; Lopes, L.A.; Fernandez-Lafuente, R.; Tardioli, P.W. Stabilization and Operational Selectivity Alteration of Lipozyme 435 by Its Coating with Polyethyleneimine: Comparison of the Biocatalyst Performance in the Synthesis of Xylose Fatty Esters. Int. J. Biol. Macromol. 2021, 192, 665–674. [Google Scholar] [CrossRef]
- Mao, Y.; Cai, Z.; Zhou, C.; Lan, H.; Ye, X. Characteristics of Crosslinking Polymers Play Major Roles in Improving the Stability and Catalytic Properties of Immobilized Thermomyces Lanuginosus Lipase. Int. J. Mol. Sci. 2022, 23, 2917. [Google Scholar] [CrossRef]
- Aasim, M.; Khan, M.H.; Bibi, N.S.; Fernandez-Lahore, M. Understanding the Interaction of Proteins to Ion Exchange Chromatographic Supports: A Surface Energetics Approach. Biotechnol. Prog. 2022, 38, e3232. [Google Scholar] [CrossRef]
- Vaz, R.P.; Filho, E.X.F. Ion Exchange Chromatography for Enzyme Immobilization. In Applications of Ion Exchange Materials in Biomedical Industries; Inamuddin, Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 13–27. ISBN 978-3-030-06082-4. [Google Scholar]
- Benamia, F.; Benouis, S.; Belafriekh, A.; Semache, N.; Rebbani, N.; Djeghaba, Z. Efficient Candida Rugosa Lipase Immobilization on Maghnite Clay and Application for the Production of (1R)-(−)-Menthyl Acetate. Chem. Pap. 2017, 71, 785–793. [Google Scholar] [CrossRef]
- Wang, D.-L.; Nag, A.; Lee, G.-C.; Shaw, J.-F. Factors Affecting the Resolution of Dl-Menthol by Immobilized Lipase-Catalyzed Esterification in Organic Solvent. J. Agric. Food Chem. 2002, 50, 262–265. [Google Scholar] [CrossRef]
- Prakash, C.; Jajoo, H.K.; Blair, I.A.; Mayol, R.F. Resolution of Enantiomers of the Antiarrhythmic Drug Encainide and Its Major Metabolites by Chiral Derivatization and High-Performance Liquid Chromatography. J. Chromatogr. B. Biomed. Sci. App. 1989, 493, 325–335. [Google Scholar] [CrossRef]
- Torres, R.; Ortiz, C.; Pessela, B.C.C.; Palomo, J.M.; Mateo, C.; Guisán, J.M.; Fernández-Lafuente, R. Improvement of the Enantioselectivity of Lipase (Fraction B) from Candida Antarctica via Adsorpiton on Polyethylenimine-Agarose under Different Experimental Conditions. Enzym. Microb. Technol. 2006, 39, 167–171. [Google Scholar] [CrossRef]
- Ren, M.-Y.; Bai, S.; Zhang, D.-H.; Sun, Y. PH Memory of Immobilized Lipase for (±)-Menthol Resolution in Ionic Liquid. J. Agric. Food Chem. 2008, 56, 2388–2391. [Google Scholar] [CrossRef] [PubMed]
- Marciello, M.; Filice, M.; Palomo, J.M. Different Strategies to Enhance the Activity of Lipase Catalysts. Catal. Sci. Technol. 2012, 2, 1531–1543. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W. Surfactant Imprinting Hyperactivated Immobilized Lipase as Efficient Biocatalyst for Biodiesel Production from Waste Cooking Oil. Catalysts 2019, 9, 914. [Google Scholar] [CrossRef]
- Cao, L. Carrier-Bound Immobilized Enzymes: Principles, Application and Design; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 978-3-527-60708-2. [Google Scholar]
- Guisan, J.M. Immobilization of Enzymes and Cells; Humana Press: Totowa, NJ, USA, 2006; ISBN 978-1-58829-290-2. [Google Scholar]
- Zhao, X.; Fan, P.-R.; Mo, C.-E.; Huang, Y.-P.; Liu, Z.-S. Green Synthesis of Monolithic Enzyme Microreactor Based on Thiol-Ene Click Reaction for Enzymatic Hydrolysis of Protein. J. Chromatogr. A 2020, 1611, 460618. [Google Scholar] [CrossRef] [PubMed]
- Mateo, C.; Abian, O.; Fernandez-Lorente, G.; Pessela, B.C.C.; Grazu, V.; Guisan, J.M.; Fernandez-Lafuente, R. Multi-Point Covalent Immobilization of Enzymes on Supports Activated with Epoxy Groups: Stabilization of Industrial Enzymes. In Immobilization of Enzymes and Cells; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2020; Volume 2100, pp. 109–117. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Carballares, D.; Morellon-Sterling, R.; Rocha-Martin, J.; Fernandez-Lafuente, R. The Combination of Covalent and Ionic Exchange Immobilizations Enables the Coimmobilization on Vinyl Sulfone Activated Supports and the Reuse of the Most Stable Immobilized Enzyme. Int. J. Biol. Macromol. 2022, 199, 51–60. [Google Scholar] [CrossRef]
- Godoy, C.A. New Strategy for the Immobilization of Lipases on Glyoxyl–Agarose Supports: Production of Robust Biocatalysts for Natural Oil Transformation. Int. J. Mol. Sci. 2017, 18, 2130. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G. Immobilization of Proteins on Glyoxyl Activated Supports: Dramatic Stabilization of Enzymes by Multipoint Covalent Attachment on Pre-Existing Supports. Curr. Org. Chem. 2015, 19, 1–13. [Google Scholar] [CrossRef]
- Steen Redeker, E.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein Engineering for Directed Immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef]
- de Melo, R.R.; Alnoch, R.C.; Vilela, A.F.L.; de Souza, E.M.; Krieger, N.; Ruller, R.; Sato, H.H.; Mateo, C. New Heterofunctional Supports Based on Glutaraldehyde-Activation: A Tool for Enzyme Immobilization at Neutral PH. Molecules 2017, 22, 1088. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Mateo, C.; Godoy, C.; Pessela, B.C.C.; Rodrigues, D.S.; Giordano, R.L.C.; Fernandez-Lafuente, R.; Guisan, J.M. The Co-Operative Effect of Physical and Covalent Protein Adsorption on Heterofunctional Supports. Process Biochem. 2009, 44, 757–763. [Google Scholar] [CrossRef]
- Mateo, C.; Grazu, V.; Guisan, J.M. Immobilization of Enzymes on Monofunctional and Heterofunctional Epoxy-Activated Supports. In Immobilization of Enzymes and Cells; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 1051, pp. 43–57. [Google Scholar] [CrossRef]
- Godoy, C.A.; de las Rivas, B.; Grazú, V.; Montes, T.; Guisán, J.M.; López-Gallego, F. Glyoxyl-Disulfide Agarose: A Tailor-Made Support for Site-Directed Rigidification of Proteins. Biomacromolecules 2011, 12, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Godoy, C.A.; de las Rivas, B.; Guisán, J.M. Site-Directing an Intense Multipoint Covalent Attachment (MCA) of Mutants of the Geobacillus Thermocatenulatus Lipase 2 (BTL2): Genetic and Chemical Amination plus Immobilization on a Tailor-Made Support. Process Biochem. 2014, 49, 1324–1331. [Google Scholar] [CrossRef]
- Abaházi, E.; Lestál, D.; Boros, Z.; Poppe, L. Tailoring the Spacer Arm for Covalent Immobilization of Candida Antarctica Lipase B—Thermal Stabilization by Bisepoxide-Activated Aminoalkyl Resins in Continuous-Flow Reactors. Molecules 2016, 21, 767. [Google Scholar] [CrossRef]
- Mateo, C.; Fernandez-Lorente, G.; Rocha-Martin, J.; Bolivar, J.M.; Guisan, J.M. Oriented Covalent Immobilization of Enzymes on Heterofunctional-Glyoxyl Supports. In Immobilization of Enzymes and Cells; Guisan, J.M., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; pp. 73–88. ISBN 978-1-62703-549-1. [Google Scholar]
- Pereira, M.G.; Facchini, F.D.A.; Filó, L.E.C.; Polizeli, A.M.; Vici, A.C.; Jorge, J.A.; Fernandez-Lorente, G.; Pessela, B.C.; Guisan, J.M.; de Moraes Polizeli, M.d.L.T. Immobilized Lipase from Hypocrea Pseudokoningii on Hydrophobic and Ionic Supports: Determination of Thermal and Organic Solvent Stabilities for Applications in the Oleochemical Industry. Process Biochem. 2015, 50, 561–570. [Google Scholar] [CrossRef]
- Ruiz, M.; Plata, E.; Castillo, J.J.; Ortiz, C.C.; López, G.; Baena, S.; Torres, R.; Fernandez-Lafuente, R. Modulation of the Biocatalytic Properties of a Novel Lipase from Psychrophilic Serratia Sp. (USBA-GBX-513) by Different Immobilization Strategies. Molecules 2021, 26, 1574. [Google Scholar] [CrossRef] [PubMed]
- Rueda, N.; dos Santos, J.C.S.; Torres, R.; Ortiz, C.; Barbosa, O.; Fernandez-Lafuente, R. Improved Performance of Lipases Immobilized on Heterofunctional Octyl-Glyoxyl Agarose Beads. RSC Adv. 2015, 5, 11212–11222. [Google Scholar] [CrossRef]
- Fernandez-Lorente, G.; Godoy, C.A.; Mendes, A.A.; Lopez-Gallego, F.; Grazu, V.; de Las Rivas, B.; Palomo, J.M.; Hermoso, J.; Fernandez-Lafuente, R.; Guisan, J.M. Solid-Phase Chemical Amination of a Lipase from Bacillus Thermocatenulatus to Improve Its Stabilization via Covalent Immobilization on Highly Activated Glyoxyl-Agarose. Biomacromolecules 2008, 9, 2553–2561. [Google Scholar] [CrossRef]
- Chen, B.; Miller, M.E.; Gross, R.A. Effects of Porous Polystyrene Resin Parameters on Candida Antarctica Lipase B Adsorption, Distribution, and Polyester Synthesis Activity. Langmuir ACS J. Surf. Colloids 2007, 23, 6467–6474. [Google Scholar] [CrossRef]
- Palanirajan, S.K.; Govindasamy, P.; Gummadi, S.N. Polystyrene Adsorbents: Rapid and Efficient Surrogate for Dialysis in Membrane Protein Purification. Sci. Rep. 2020, 10, 16334. [Google Scholar] [CrossRef]
- Modenez, I.A.; Sastre, D.E.; Moraes, F.C.; Marques Netto, C.G.C. Influence of Glutaraldehyde Cross-Linking Modes on the Recyclability of Immobilized Lipase B from Candida Antarctica for Transesterification of Soy Bean Oil. Mol. J. Synth. Chem. Nat. Prod. Chem. 2018, 23, 2230. [Google Scholar] [CrossRef]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in Bio-Catalysts Design: A Useful Crosslinker and a Versatile Tool in Enzyme Immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Ye, P.; Wan, R.-B.; Wang, X.-P. Quantitative Enzyme Immobilization: Control of the Carboxyl Group Density on Support Surface. J. Mol. Catal. B Enzym. 2009, 61, 296–302. [Google Scholar] [CrossRef]
- Smith, S.; Goodge, K.; Delaney, M.; Struzyk, A.; Tansey, N.; Frey, M. A Comprehensive Review of the Covalent Immobilization of Biomolecules onto Electrospun Nanofibers. Nanomaterials 2020, 10, 2142. [Google Scholar] [CrossRef]
- Palomo, J.M.; Mateo, C.; Fernández-Lorente, G.; Solares, L.F.; Diaz, M.; Sánchez, V.M.; Bayod, M.; Gotor, V.; Guisan, J.M.; Fernandez-Lafuente, R. Resolution of (±)-5-Substituted-6-(5-Chloropyridin-2-Yl)-7-Oxo-5,6-Dihydropyrrolo[3,4b]Pyrazine Derivatives-Precursors of (S)-(+)-Zopiclone, Catalyzed by Immobilized Candida Antarctica B Lipase in Aqueous Media. Tetrahedron Asymmetry 2003, 14, 429–438. [Google Scholar] [CrossRef]
- Palomo, J.M.; Fernández-Lorente, G.; Mateo, C.; Fuentes, M.; Fernández-Lafuente, R.; Guisan, J.M. Modulation of the Enantioselectivity of Candida Antarctica B Lipase via Conformational Engineering. Kinetic Resolution of (±)-α-Hydroxy-Phenylacetic Acid Derivatives. Tetrahedron Asymmetry 2002, 13, 1337–1345. [Google Scholar] [CrossRef]
- Rodrigues, D.S.; Mendes, A.A.; Filice, M.; Fernandez-Lafuente, R.; Guisan, J.M.; Palomo, J.M. Different Derivatives of a Lipase Display Different Regioselectivity in the Monohydrolysis of Per-O-Acetylated 1-O-Substituted-β-Galactopyranosides. J. Mol. Catal. B Enzym. 2009, 58, 36–40. [Google Scholar] [CrossRef]
- Filice, M.; Palomo, J.M. Monosaccharide Derivatives as Central Scaffolds in the Synthesis of Glycosylated Drugs. RSC Adv. 2012, 2, 1729–1742. [Google Scholar] [CrossRef]
- Martins, A.B.; Friedrich, J.L.R.; Cavalheiro, J.C.; Garcia-Galan, C.; Barbosa, O.; Ayub, M.A.Z.; Fernandez-Lafuente, R.; Rodrigues, R.C. Improved Production of Butyl Butyrate with Lipase from Thermomyces Lanuginosus Immobilized on Styrene–Divinylbenzene Beads. Bioresour. Technol. 2013, 134, 417–422. [Google Scholar] [CrossRef]
- Amano Lipase PS (Immobilized on Diatomite)|Sigma-Aldrich. Available online: http://www.sigmaaldrich.com/ (accessed on 4 March 2022).
- Sawicka, A.K.; Renzi, G.; Olek, R.A. The Bright and the Dark Sides of L-Carnitine Supplementation: A Systematic Review. J. Int. Soc. Sports Nutr. 2020, 17, 49. [Google Scholar] [CrossRef]
- Nichols, D.E.; Fantegrossi, W.E. Chapter Nineteen—Emerging Designer Drugs. In The Effects of Drug Abuse on the Human Nervous System; Madras, B., Kuhar, M., Eds.; Academic Press: Boston, MA, USA, 2014; pp. 575–596. ISBN 978-0-12-418679-8. [Google Scholar]
- Tufiño, C.; Bernal, C.; Ottone, C.; Romero, O.; Illanes, A.; Wilson, L. Synthesis with Immobilized Lipases and Downstream Processing of Ascorbyl Palmitate. Molecules 2019, 24, 3227. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, R.; Yu, X.; Xu, X.; Guo, Z.; Tan, T.; Fedosov, S.N. Lipase-Catalyzed Knoevenagel Condensation in Water–Ethanol Solvent System. Does the Enzyme Possess the Substrate Promiscuity? Biochem. Eng. J. 2015, 101, 99–107. [Google Scholar] [CrossRef]
- Monsalve, L.N.; Gillanders, F.; Baldessari, A. Promiscuous Behavior of Rhizomucor Miehei Lipase in the Synthesis of N-Substituted β-Amino Esters. Eur. J. Org. Chem. 2012, 2012, 1164–1170. [Google Scholar] [CrossRef]
- Xu, F.; Wu, Q.; Chen, X.; Lin, X.; Wu, Q. A Single Lipase-Catalysed One-Pot Protocol Combining Aminolysis Resolution and Aza-Michael Addition: An Easy and Efficient Way to Synthesise β-Amino Acid Esters. Eur. J. Org. Chem. 2015, 2015, 5393–5401. [Google Scholar] [CrossRef]
- Ham, J.R.; Choi, R.-Y.; Lee, H.-I.; Lee, M.-K. Methoxsalen and Bergapten Prevent Diabetes-Induced Osteoporosis by the Suppression of Osteoclastogenic Gene Expression in Mice. Int. J. Mol. Sci. 2019, 20, 1298. [Google Scholar] [CrossRef]
- Fongemie, J.; Felix-Getzik, E. A Review of Nebivolol Pharmacology and Clinical Evidence. Drugs 2015, 75, 1349–1371. [Google Scholar] [CrossRef]
- Beutler, U.; Fuenfschilling, P.C.; Steinkemper, A. An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part II. Org. Process Res. Dev. 2007, 11, 341–345. [Google Scholar] [CrossRef]
- Carrasco-Sanchez, V.; Simirgiotis, M.J.; Santos, L.S. The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry. Molecules 2009, 14, 3989–4021. [Google Scholar] [CrossRef]
- Basavaiah, D.; Reddy, B.S.; Badsara, S.S. Recent Contributions from the Baylis−Hillman Reaction to Organic Chemistry. Chem. Rev. 2010, 110, 5447–5674. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, S.; Zheng, L. First Novozym 435 Lipase-Catalyzed Morita–Baylis–Hillman Reaction in the Presence of Amides. Enzyme Microb. Technol. 2016, 84, 32–40. [Google Scholar] [CrossRef]
- Burk, M.J.; de Koning, P.D.; Grote, T.M.; Hoekstra, M.S.; Hoge, G.; Jennings, R.A.; Kissel, W.S.; Le, T.V.; Lennon, I.C.; Mulhern, T.A.; et al. An Enantioselective Synthesis of (S)-(+)-3-Aminomethyl-5-Methylhexanoic Acid via Asymmetric Hydrogenation. J. Org. Chem. 2003, 68, 5731–5734. [Google Scholar] [CrossRef] [PubMed]
- Koszelewski, D.; Kowalczyk, P.; Śmigielski, P.; Samsonowicz-Górski, J.; Kramkowski, K.; Wypych, A.; Szymczak, M.; Ostaszewski, R. Relationship between Structure and Antibacterial Activity of α-Aminophosphonate Derivatives Obtained via Lipase-Catalyzed Kabachnik−Fields Reaction. Materials 2022, 15, 3846. [Google Scholar] [CrossRef] [PubMed]
- Aissa, R.; Guezane-Lakoud, S.; Kolodziej, E.; Toffano, M.; Aribi-Zouioueche, L. Diastereoselective Synthesis of Bis(α-Aminophosphonates) by Lipase Catalytic Promiscuity. New J. Chem. 2019, 43, 8153–8159. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields Reaction: Mechanism and Synthetic Use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef]
- Varga, P.R.; Keglevich, G. Synthesis of α-Aminophosphonates and Related Derivatives; The Last Decade of the Kabachnik–Fields Reaction. Molecules 2021, 26, 2511. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Koszelewski, D.; Gawdzik, B.; Samsonowicz-Górski, J.; Kramkowski, K.; Wypych, A.; Lizut, R.; Ostaszewski, R. Promiscuous Lipase-Catalyzed Markovnikov Addition of H-Phosphites to Vinyl Esters for the Synthesis of Cytotoxic α-Acyloxy Phosphonate Derivatives. Materials 2022, 15, 1975. [Google Scholar] [CrossRef]
- Bal, A.M.; Capoor, M.R. 7.04—Fosfomycin. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Oxford, UK, 2022; pp. 100–105. ISBN 978-0-12-820876-2. [Google Scholar]
- Mansouri, H.R.; Mihovilovic, M.D.; Rudroff, F. Investigation of a New Type I Baeyer–Villiger Monooxygenase from Amycolatopsis Thermoflava Revealed High Thermodynamic but Limited Kinetic Stability. Chembiochem 2020, 21, 971–977. [Google Scholar] [CrossRef]
- Alavijeh, M.K.; Amini, M.M. Tin and Copper Species Dispersed on a Metal-Organic Framework as a New Catalyst in Aerobic Baeyer-Villiger Oxidation: An Insight into the Mechanism. Catal. Commun. 2020, 139, 105985. [Google Scholar] [CrossRef]
- Hino, T.; Kutsumura, N.; Saitoh, T.; Yamamoto, N.; Nagumo, Y.; Mogi, Y.; Watanabe, Y.; Nagase, H. Novel Baeyer–Villiger-Type Oxidation of 4,5-Epoxymorphinan Derivatives. Tetrahedron Lett. 2021, 63, 152714. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The Market of Chiral Drugs: Chiral Switches versus de Novo Enantiomerically Pure Compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Stradomska, D.; Heba, M.; Czernek, A.; Kuźnik, N.; Gillner, D.; Maresz, K.; Pudło, W.; Jarzębski, A.; Szymańska, K. Lipase Immobilized on MCFs as Biocatalysts for Kinetic and Dynamic Kinetic Resolution of Sec-Alcohols. Catalysts 2021, 11, 518. [Google Scholar] [CrossRef]
- Delgove, M.A.F.; Laurent, A.-B.; Woodley, J.M.; De Wildeman, S.M.A.; Bernaerts, K.V.; van der Meer, Y. A Prospective Life Cycle Assessment (LCA) of Monomer Synthesis: Comparison of Biocatalytic and Oxidative Chemistry. ChemSusChem 2019, 12, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Jegannathan, K.R.; Nielsen, P.H. Environmental Assessment of Enzyme Use in Industrial Production—A Literature Review. J. Clean. Prod. 2013, 42, 228–240. [Google Scholar] [CrossRef]
- Santi, M.; Sancineto, L.; Nascimento, V.; Braun Azeredo, J.; Orozco, E.V.M.; Andrade, L.H.; Gröger, H.; Santi, C. Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. Int. J. Mol. Sci. 2021, 22, 990. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Woodley, J.M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. [Google Scholar] [CrossRef]
- Jang, W.D.; Kim, G.B.; Kim, Y.; Lee, S.Y. Applications of Artificial Intelligence to Enzyme and Pathway Design for Metabolic Engineering. Curr. Opin. Biotechnol. 2022, 73, 101–107. [Google Scholar] [CrossRef]
- Repecka, D.; Jauniskis, V.; Karpus, L.; Rembeza, E.; Rokaitis, I.; Zrimec, J.; Poviloniene, S.; Laurynenas, A.; Viknander, S.; Abuajwa, W.; et al. Expanding Functional Protein Sequence Spaces Using Generative Adversarial Networks. Nat. Mach. Intell. 2021, 3, 324–333. [Google Scholar] [CrossRef]
- Singh, N.; Malik, S.; Gupta, A.; Srivastava, K.R. Revolutionizing Enzyme Engineering through Artificial Intelligence and Machine Learning. Emerg. Top. Life Sci. 2021, 5, 113–125. [Google Scholar] [CrossRef]
- Lu, H.; Diaz, D.J.; Czarnecki, N.J.; Zhu, C.; Kim, W.; Shroff, R.; Acosta, D.J.; Alexander, B.R.; Cole, H.O.; Zhang, Y.; et al. Machine Learning-Aided Engineering of Hydrolases for PET Depolymerization. Nature 2022, 604, 662–667. [Google Scholar] [CrossRef]
- Yang, M.; Fehl, C.; Lees, K.V.; Lim, E.-K.; Offen, W.A.; Davies, G.J.; Bowles, D.J.; Davidson, M.G.; Roberts, S.J.; Davis, B.G. Functional and Informatics Analysis Enables Glycosyltransferase Activity Prediction. Nat. Chem. Biol. 2018, 14, 1109–1117. [Google Scholar] [CrossRef]
- Benítez-Mateos, A.I.; Roura Padrosa, D.; Paradisi, F. Multistep Enzyme Cascades as a Route towards Green and Sustainable Pharmaceutical Syntheses. Nat. Chem. 2022, 14, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Del Arco, J.; Acosta, J.; Fernández-Lucas, J. New Trends in the Biocatalytic Production of Nucleosidic Active Pharmaceutical Ingredients Using 2’-Deoxyribosyltransferases. Biotechnol. Adv. 2021, 51, 107701. [Google Scholar] [CrossRef] [PubMed]
- Nazor, J.; Liu, J.; Huisman, G. Enzyme Evolution for Industrial Biocatalytic Cascades. Curr. Opin. Biotechnol. 2021, 69, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Nerke, P.; Siedentop, R.; Steinmetz, T.; Classen, T.; Rosenthal, K.; Nett, M.; Pietruszka, J.; Lütz, S. Recent Advances in Biocatalysis for Drug Synthesis. Biomedicines 2022, 10, 964. [Google Scholar] [CrossRef] [PubMed]
- Siedentop, R.; Rosenthal, K. Industrially Relevant Enzyme Cascades for Drug Synthesis and Their Ecological Assessment. Int. J. Mol. Sci. 2022, 23, 3605. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enantioselectivity Origins | Lipase | Reaction Type/Conditions | Substrate | Product-Favored | Activity | ee%/(E) | Reference |
|---|---|---|---|---|---|---|---|
| Electronic effects: the size and polarity of the stereoselectivity pocket favor interactions with heterocyclic rings. | CAL B W104T | Hydrolysis/Phosphate buffered-saline or PBS (50 mM, pH = 7.5) at 37 °C. | rac phenyl(pyridin-4-yl) methyl acetate Related API: (S)-Bepotastine | (S) | No reported (N.R.) | 98 (E > 200) | [121] |
| Complementary structure of the acyl site, hydrophobic pocket or oxyanion hole (KRIE): selectivity towards derivatives of chiral acids is increased by replacing large residues with small ones. | CAL B and mutants | Hydrolysis/PBS (50 mM, pH = 7.5) and 1% Triton X-100) at 30 °C. | rac p-nitrophenyl 2-phenylpropanoate Related API: Ipatropium | (S) | N.R. | (E = 1.2–72) | [120] |
| H-bonds: the (S) enantiomer is properly oriented to form H-bonds between N(ε) of His and its O alkoxy. | CRL | Esterification/1 volume of cyclohexane with 0.15 M acid and 1 volume 0.11 M decanol aqueous saline solution (aw = 0.16) at RT. | rac 2- methyldecanoic acid Related API: Polymyxins | (S) | 1.52 µmol.g−1·min−1 | 97% (E = 90) | [122] |
| H-bonds as a function of dihedral angle C carbonyl-O alkoxy- Cα-Cβ alkoxy: the shorter the acyl residue (C2), the dihedral will be closer to 60° which will facilitate the building of an H-bond between O beta as acceptor and the -OH of Ty29 as donor. | P.cepacia lipase (PCL) | Hydrolysis/PBS pH 7.0 (100 mM and 10% isopropanol v/v) at 25 °C, 200 rpm. | rac acyl esters of 2-phenoxypropan-1-ol (acyl n = C2 to C12) Related API: Metoprolol related β-blockers | (S) | 0.011 (C12)–0.52 (C2) µmol·min−1 | E values 1,1 (C12) to 33 (C2). | [122] |
| Steric hindrance or Kazlauskas rule | CAL B | Transesterification/in t-BuOMe at 30 °C, 200 rpm with 0.05 mmol substrate and 0.15 mmol vinyl butanoate. | rac-1-phenyl-1-ethanol Related API: Norephedrine | (R) | 28% conversion in 12 h | >99 (E > 200) | [123] |
| Dynamics of the catalytic cavity “contradicts” the Kazlauskas rule: a rotation of leu278 was presented to give more space to the substrate with the bulky substituent; additional H-bond in the case of the (S) with the oxyanion hole. | CAL B | Transesterification/in t-BuOMe at 30 °C, 200 rpm with 0.05 mmol substrate and 0.15 mmol vinyl butanoate. | rac-2-amino-1-phenylethanol N-Boc-aminomethylene Related API: Denopamine | (S) | 10% conversion in 48 h. | >99 (E > 200) | [123] |
| Mirror-image-packing of the substrate: favored when a stereocenter of the substrate has an H as substituent, explaining a packing of the less favored enantiomer different from that assumed by the Kazlauscas rule (Figure 5). | Burkholderia cepacia lipase (BCL) | Hydrolysis/PBS pH 7.0 in 30% n-propanol with 4.5 mmol substrate. | rac 2-methyl-3-phenyl-1-propyl heptanoate Related API: Squalestatin | (S) | kcat = 0.4 min−1 | 98 (E > 190) | [119] |
| Substrate structure/presence of inhibitor: by acquiring the hair-pin conformation, the (R)-enantiomer binds to N(ε) of His through its O alkoxy induced by the presence of coordinated heptanol as inhibitor. | CRL | Esterification/1 volume of cyclohexane with 0.15 M acid and 1 volume 0.11 M decanol aqueous saline solution (aw = 0.16) at RT in presence of 900 mM heptanol. | rac 2- methyldecanoic acid Related API: Polymyxins | Relative increase (but not absolute) towards the (R) enantiomer. | N.R. | E = 37 | [122] |
| Solvation effects/Kazlauskas rule: the reaction medium affects the interaction of the racemic substrate with the solvent, its peers and the enzyme, with the latter, enzyme-substrate diastereomeric complexes with different km values are formed. The one with the largest km will be the one most affected by the permittivity of the medium depending on the solvent. | CAL B | Transesterification/alcohol 2 mmol and vinyl acetate 8 mmol in toluene−acetonitrile (variable proportion to change polarity, ε) at 30 °C. | rac 1-(naphth-2-yl) ethanol or rac 2-methoxy-2-phenylethanol Related API: Nifedipine | (R) (R) | 100 mol·h−1·mgenz−1 220 mol·h−1·mgenz−1 | E values from 30–120 (E 120 with ε = 12) E values from 60–200 (E 60 with ε = 14) | [124] |
| Kazlauskas rule/Higher structural flexibility: in the lid and the entrance to the catalytic cavity (helix α-5) induced by the reaction medium (solvent expanded by CO2), it allows better accommodation of bulky substrates, accelerating the reaction. | CAL B | Transesterification/alcohol 0.1 mmol and vinyl acetate 0.2 mmol in 10 mL hexane or CO2-hexane (5%, 6 MPa) at 50 °C, 30 min. | rac ortho-substituted 1-phenylethanols Related API: Crizotinib | (R) | Conversion rates increased up to 2.5 times in presence of CO2-hexane. | >99 | [125] |
| Derivative | Support Used | Type of Reaction/Conditions | Substrate | Product | Activity (µmol·min−1) | ee(%)/(E) | Uses Related to API |
|---|---|---|---|---|---|---|---|
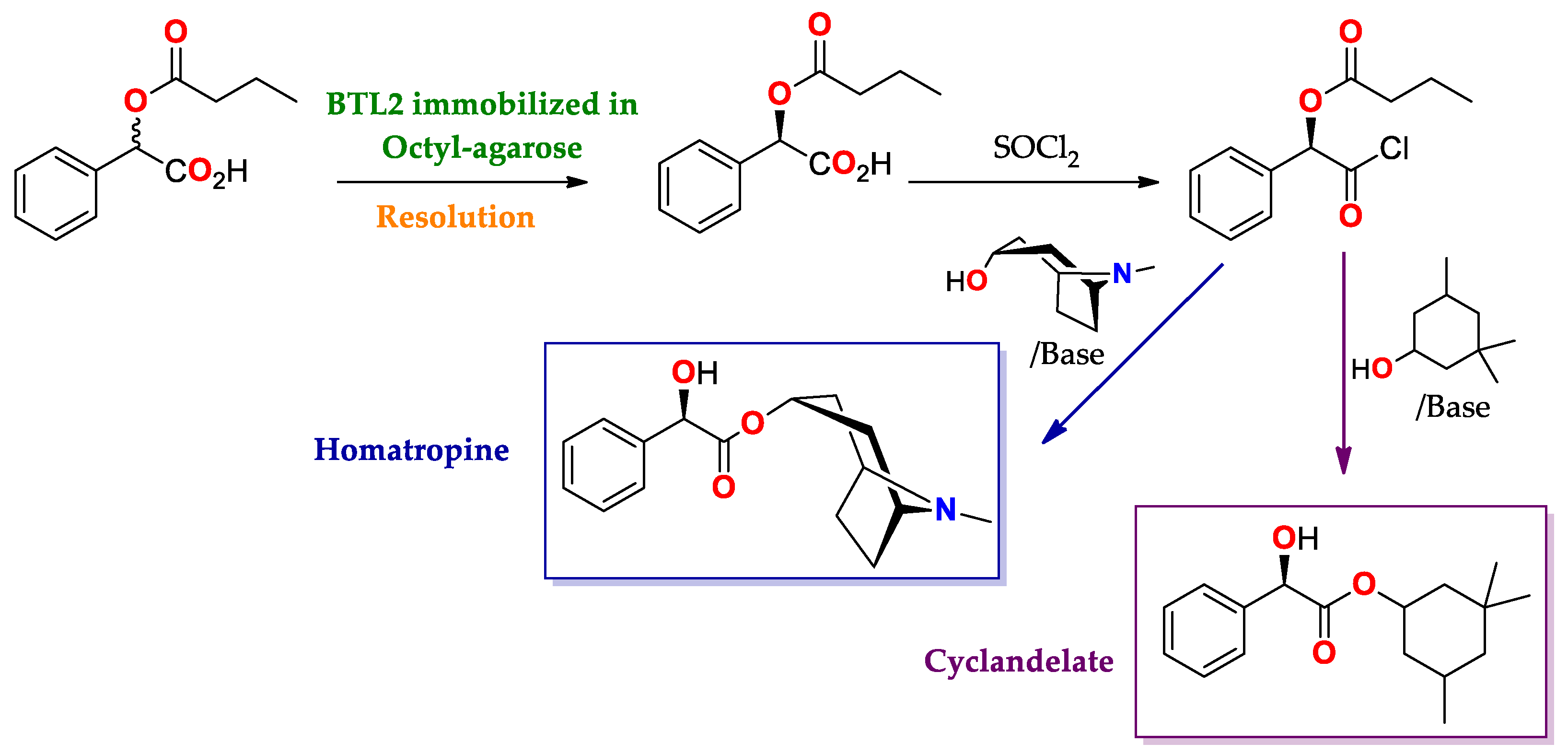
| BTL2 immobilized by hydrophobic interaction [149] | Octyl-agarose | Hydrolysis/Acetate buffered-saline or ABS (25 mM, pH = 5.0) at 25 °C. | rac-2-O-Butyryl-2-phenylacetic acid | (R)-O-Butyryl-2-phenylacetic acid | 0.25 | >99/E > 100 | Precursor of mandelic acid derivatives APIs: pemoline ([150], Scheme 7), homatropine, cyclandelate (Scheme 8), cephalosporin, antineoplastics, antiobesity and anti-thrombotic agents [150,151,152] |
| CAL-B immobilized by hydrophobic interaction [147] | Novozym 435 Octyl-agarose Octadecyl-Sepabeads Butyl-agarose | Hydrolysis/Acetate buffered-saline or ABS (25 mM, pH = 5.0) at 25 °C. | rac-2-O-butyryl-2-phenylacetic acid | (S)-2-hydroxy-2-phenylacetic acid (R)-2-hydroxy-2-phenylacetic acid (R)-2-hydroxy-2-phenylacetic acid (R)-2-hydroxy-2-phenylacetic acid | 0.75 0.19 0.35 0.16 | >99/E > 100 95/E = 49 90/E = 23 72/E = 7.5 | Precursor of mandelic acid derivatives APIs: pemoline ([150], Scheme 7), homatropine, cyclandelate (Scheme 8), cephalosporin, antineoplastics, antiobesity and anti-thrombotic agents [150,151,152]. |
| TLL immobilized by hydrophobic interaction [154] | octyl-Sepharose® | Hydrolysis/Acetate buffered-saline or ABS (25 mM, pH = 5.0) at 25 °C. | β -D-Galactose pentaacetate | C–6 deprotected β -D-Galactose tetraacetylated 99% | N.R. | N/A | Lacto-N-neotetraose in turn precursor of the tumor-associated carbohydrate antigen T and the antitumoral drug [154]. |
| CRL immobilized by ion exchange [165] | Amberjet®1200-H (Cation exchanger) Amberjet®4200-Cl (Anion ex-changer) | Transesterification/Rac-Menthol with vinyl acetate 1:2 in anhydrous solvent at 30 °C. | rac-Menthol | (R)-(-)- Menthyl acetate | N.R. | >99/ E = 208 >99/ E = 213 | Chiral additive in the resolution of enantiomers of the antiarrhythmic drug encainide and its major metabolites [167]. |
| CRL immobilized by ion exchange [166] | Sephadex® G-25 Sephadex® G-50 | Transesterification/Rac-Menthol with valeric acetate 1:2 in anhydrous solvent at 30 °C. | rac-Menthol | (R)-(-)- Menthyl acetate | N.R. | 92/ E 70.7 90%/ E = 33.1 | Chiral additive in the resolution of enantiomers of the antiarrhythmic drug encainide and its major metabolites [167]. |
| CAL-B immobilized by ion exchange [168] | PEI-Agarose at 4 °C PEI-Agarose at 25 °C PEI-Agarose at 37 °C | Hydrolysis/Acetate buffered-saline or ABS (25 mM, pH = 5.0) at 25 °C | rac- mandelic acid methyl ester | (R)-2-hydroxy-2-phenylacetic acid | 0.21 0.21 0.39 | E = 25 E = 16 E = 10.5 | Precursor of mandelic acid derivatives APIs: pemoline ([150], Scheme 7), homatropine, cyclandelate (Scheme 8), cephalosporin, antineoplastics, antiobesity and anti-thrombotic agents [150,151,152]. |
| CRL immobilized by ion exchange [169] | DEAE-glycidyl methacrylate (GMA)- ethylene dimethacrylate (EDMA) activated | Transesterifica-tion/Rac-Menthol with propionic anhydride 1:1 in anhydrous solvent at 30 °C. | rac-Menthol | (R)-(-)- Menthyl acetate | N.R. | >90 | Chiral additive in the resolution of enantiomers of the antiarrhythmic drug encainide and its major metabolites [167]. |
| Derivative (Agarose Matrix) | Type of Covalent Union/Immobilization Conditions | Thermal Stability at 70 °C t½ (min) | Resolution of ee-2-O-butyryl-2-phenylacetic Acid (“a”). ee(R) (%)/Activity (µmol·min−1 mglipase−1) | Desymmetrization of Phenyl Glutaric Acid Methyl Diester (“b”) ee(S) (%)/Activity (µmol·min−1 mglipase−1) |
|---|---|---|---|---|
| CNBr (Cyanate ester) | Monopoint (via N-terminal)/pH 7 in 50 mM PBS at 4 °C. | 26 | 24/(0.71) | 94/(0.51) |
| Disulfide (-S-S-) | Monopoint through the introduced cysteine (Q40C C65S, C296S BTL2)/at pH 7 in PBS 50 mM at 4 °C. | 21 | 45/(0.56) | 91/(0.47) |
| Glyoxyl (aldehyde) | Non oriented multipoint through Lys/N-terminal residues/At pH 10 in sodium carbonate 100 mM (25 °C). | 602 | 80/(0.68) | >99/(0.44) |
| Glyoxyl + disulfide | Oriented through cysteine (Q40C) plus soft multipoint union through adjacent native ε-amino groups./at pH 7 in PBS 50 mM and then pH increased to 10 in sodium carbonate 100 mM (25 °C). | 510 | >99/(0.59) | 96/(0.48) |
| Glyoxyl + disulfide | Oriented through cysteine (Q40C) plus intense multipoint union through adjacent native and other amino groups introduced chemically/at pH 7 in PBS 50 mM and then pH increased to 10 in sodium carbonate 100 mM (25 °C). | 1010 | >99/(0.29) | 81.8/(0.86) |
| Glyoxyl + disulfide | Oriented through cysteine (Q40C) plus intense multipoint union through adjacent native and other amino groups introduced chemically and genetically (Arg→Lys in positions 374, 379, 48, 35, 22 and 5)/at pH 7 in PBS 50 mM and then pH increased to 10 in sodium carbonate 100 mM (25 °C). | 380 | >99/(0.09) | >99/(0.11) |
| Derivative/Immobilization Conditions | Commercial Name | Type of Reaction/Conditions | Substrate | Product/Yield, ee%/(E) | Uses Related to API |
|---|---|---|---|---|---|
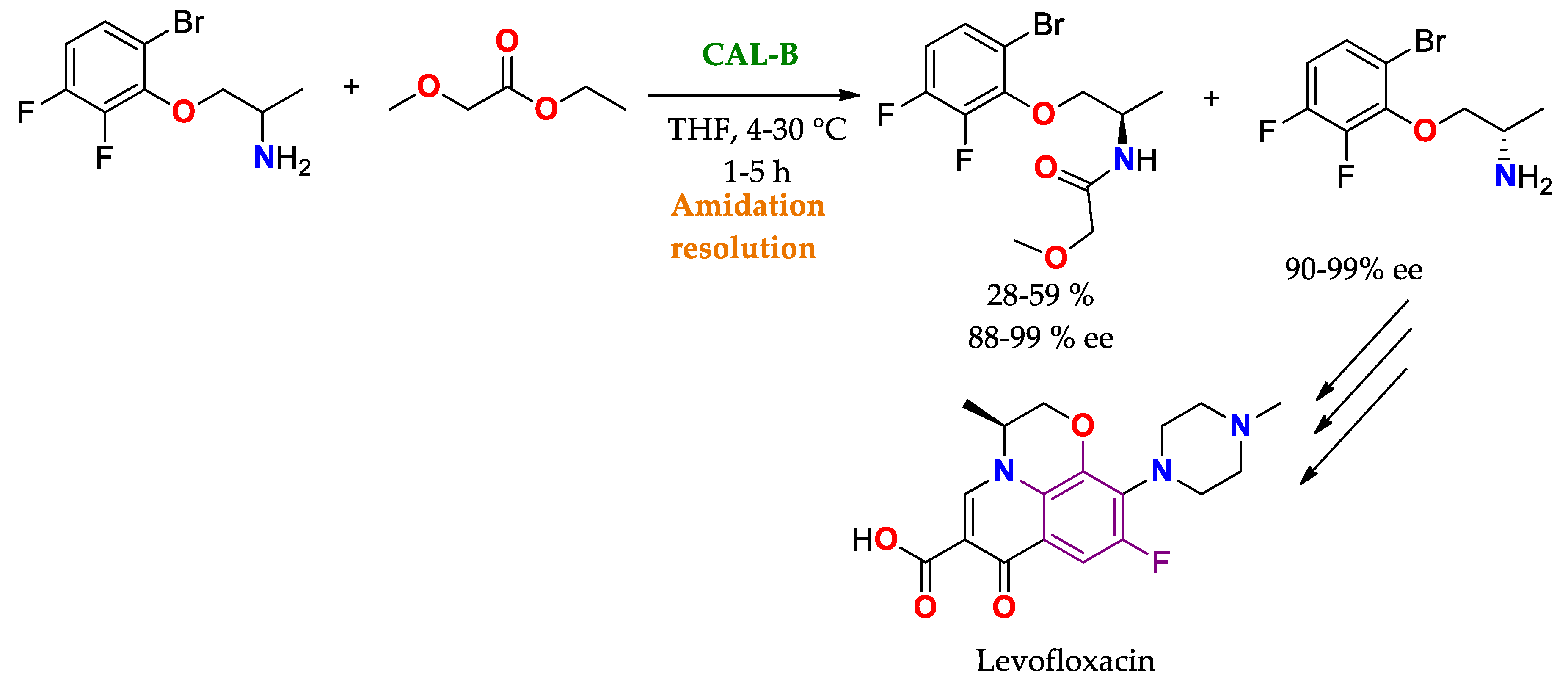
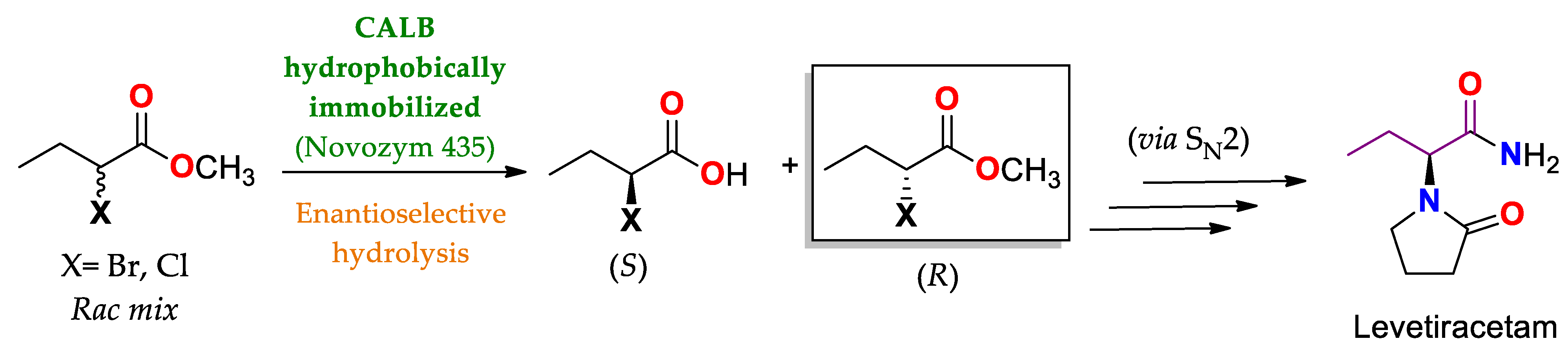
| CAL B immobilized by hydrophobic interaction on Lewatit® VP OC 1600/conditions non specified | Novozym® 435 | Hydrolysis Amidation/PBS buffer pH 6.5–7.5 at 30–35 °C (24–48 h). | Methyl rac-2-bromobutyrate | (R)-methyl 2-bromobutanoate/No reported (N.R.) | Production of anti-epileptic drug levetiracetam (Elepsia® o Keppra®) [12] |
| CAL B immobilized/type or conditions non specified | Chirazyme L2TM | Hydrolysis resolution/Buffer pH 7.0/MTBE(5:1, v/v) at 20 °C. | rac-1-({[(1,1,1,3,3,3-hexafluoropropan-2-yl)oxy]carbonyl}oxy)ethyl 2-methylpropanoate | (R)-1-((((1,1,1,3,3,3-hexafluoropropan-2-yl)oxy)carbonyl)oxy)ethyl isobutyrate and (S)-1,1,1,3,3,3-hexafluoropropan-2-yl (1-hydroxyethyl) carbonate/Yield 44%, ee 99%. | Production of a prodrug type carbamate with inhibitory activity of TAFIa (thrombin-activatable fibrinolysis inhibitor) for the treatment of thromboembolic disorders [12] |
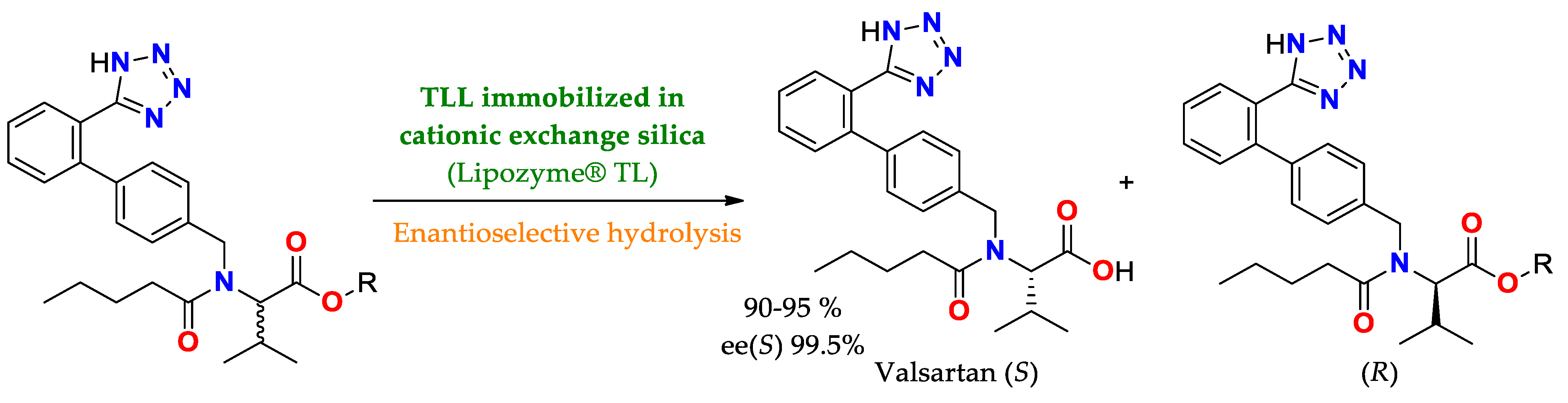
| TLL immobilized by cationic exchange in silica/conditions non specified | Lipozyme® TL IM Lipozyme® RM IM Lipase PS IM | Enatioselective hydrolysis | Precursor esters of valsartan | Valsartan/Yield of 90–95%ee(S) = 99.5 | Enatioselective hydrolysis of esters of valsartan (Diovan®, a drug that modulates angiotensin-renin system to treat hypertension [201,202] |
| CAL B immobilized by hydrophobic interaction on Lewatit® VP OC 1600/conditions non specified | Novozym® 435 | Chemoselective hydrolysis of esters/PBS pH 8.0 and organic solvent (1:1 v/v) | N-Boc-(1R,3S,4S)-2-azabicyclo[2.2.1]heptane-3-carboxylate | Carboxylic derivative/Yield > 70%, diastereomeric excess values of 99.7% | Production of a building block required for the preparation of ledispavir (Harvoni, Gilead, along with sofosbuvir for the treatment of hepatitis C) [12] |
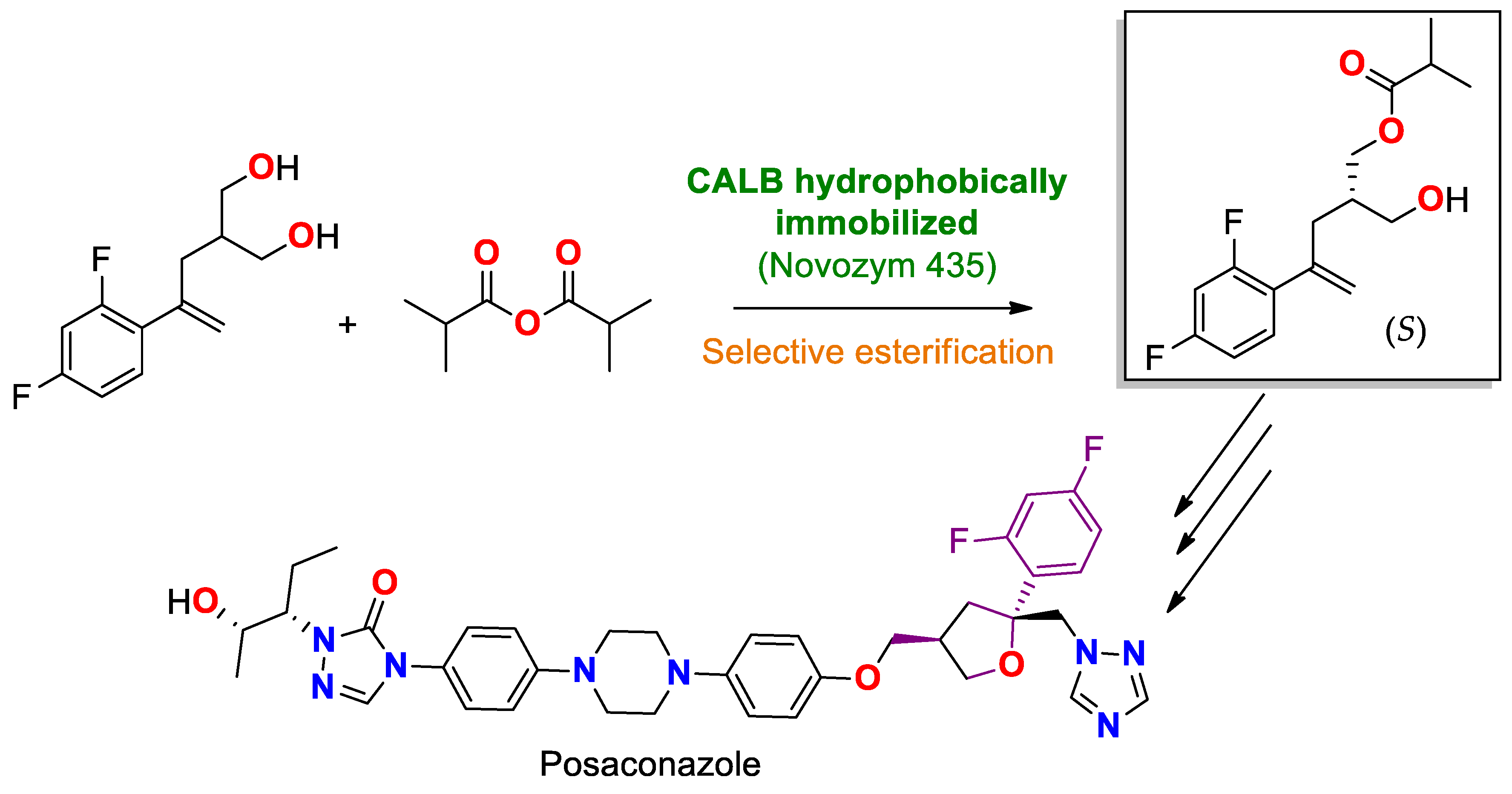
| CAL B immobilized by hydrophobic interaction on Lewatit® VP OC 1600/conditions non specified | Novozyme® 435 | Stereoselective acylation/with isopropanoic anhydride in a mixture of NaHCO3/toluene with vinyl acetate at 15 °C during 24 h. | prochiral 2-(2-(2,4-difluorophenyl)allyl)propane-1,3-diol | (S)-4-(2,4-difluorophenyl)-2-(hydroxymethyl)pent-4-en-1-yl isobutyrate/Yield 75% | Production of a precursor of the antifungal posaconazole (Posanol®), Noxafil®) [203] |
| CAL B immobilized by hydrophobic interaction on Lewatit® VP OC 1600/conditions non specified | Novozyme® 435 | Transesterification/ tolunene with perchlorophenyl acetate at 50–60 °C. | rac-3-cyano-2-hydroxy-N,N,N-trimethylpropan-1-aminium | (R)-2-acetoxy-3-cyano-N,N,N-trimethylpropan-1-aminium | Obtaining acetate (2R) as a precursor of L-carnitine, key in lipid metabolism and sold as a supplement [203] |
| CAL B immobilized by hydrophobic interaction on Lewatit® VP OC 1600/conditions non specified | Novozyme® 435 | Hydrolysis of a uracil nucleoside/methoxypropanol/water at 60 °C, 48 h. | 2-methyl-2-fluoro-deoxyribose peracetylate | deacetylated derivative selectively in position C-5′/yield > 99% | obtaining a precursor of Sofosbuvir, a drug used in the treatment of chronic hepatitis [203] |
| CAL B immobilized by hydrophobic interaction on Lewatit® VP OC 1600/conditions non specified | Novozym®435 | Dynamic nantioselective acetylation/Toluene with (R)-O-acetyl mandelic acid, H2/nickel catalyst (KT-02), 1 MPa H2, at 70 °C, 24 h. | rac-2-aminotetraline | (R)-2-aminotetraline/ee > 99% | Obtention of a pharmacophore based stimulant drugs with action on dopamine receptor D3 [204] |
| CAL B immobilized by hydrophobic interaction/conditions non specified | Novozym®435 | Monoacetylation in C-6/THF, 40 °C, 60 h. | β-D-glucosides polydatin and geniposide | Acylated derivatives of β-D-glucosides polydatin and geniposide/Yield 78% | Acylated derivatives of β-D-glucosides polydatin (anti-inflammatory, anti-oxidant and in anti-angiogenesis chemotherapy) and geniposide (used in the prevention and therapy of hepatic injury-HI) [12] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godoy, C.A.; Pardo-Tamayo, J.S.; Barbosa, O. Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks. Int. J. Mol. Sci. 2022, 23, 9933. https://doi.org/10.3390/ijms23179933
Godoy CA, Pardo-Tamayo JS, Barbosa O. Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks. International Journal of Molecular Sciences. 2022; 23(17):9933. https://doi.org/10.3390/ijms23179933
Chicago/Turabian StyleGodoy, César A., Juan S. Pardo-Tamayo, and Oveimar Barbosa. 2022. "Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks" International Journal of Molecular Sciences 23, no. 17: 9933. https://doi.org/10.3390/ijms23179933
APA StyleGodoy, C. A., Pardo-Tamayo, J. S., & Barbosa, O. (2022). Microbial Lipases and Their Potential in the Production of Pharmaceutical Building Blocks. International Journal of Molecular Sciences, 23(17), 9933. https://doi.org/10.3390/ijms23179933