
Delving into the Heterogeneity of Different Breast Cancer Subtypes and the Prognostic Models Utilizing scRNA-Seq and Bulk RNA-Seq


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Data Collection and Processing
2.2. Single-Cell Data Integration and Analysis
2.3. Identification of Cancer Cells by Calculating Cell Copy Number Variations
2.4. Identification and Functional Analysis of Differentially Expressed Genes
2.5. Inference of Gene Regulatory Networks (GRNs) in Tumor Cells
2.6. Construction of Prognostic Risk Model
2.7. Data Statistics and Visualization
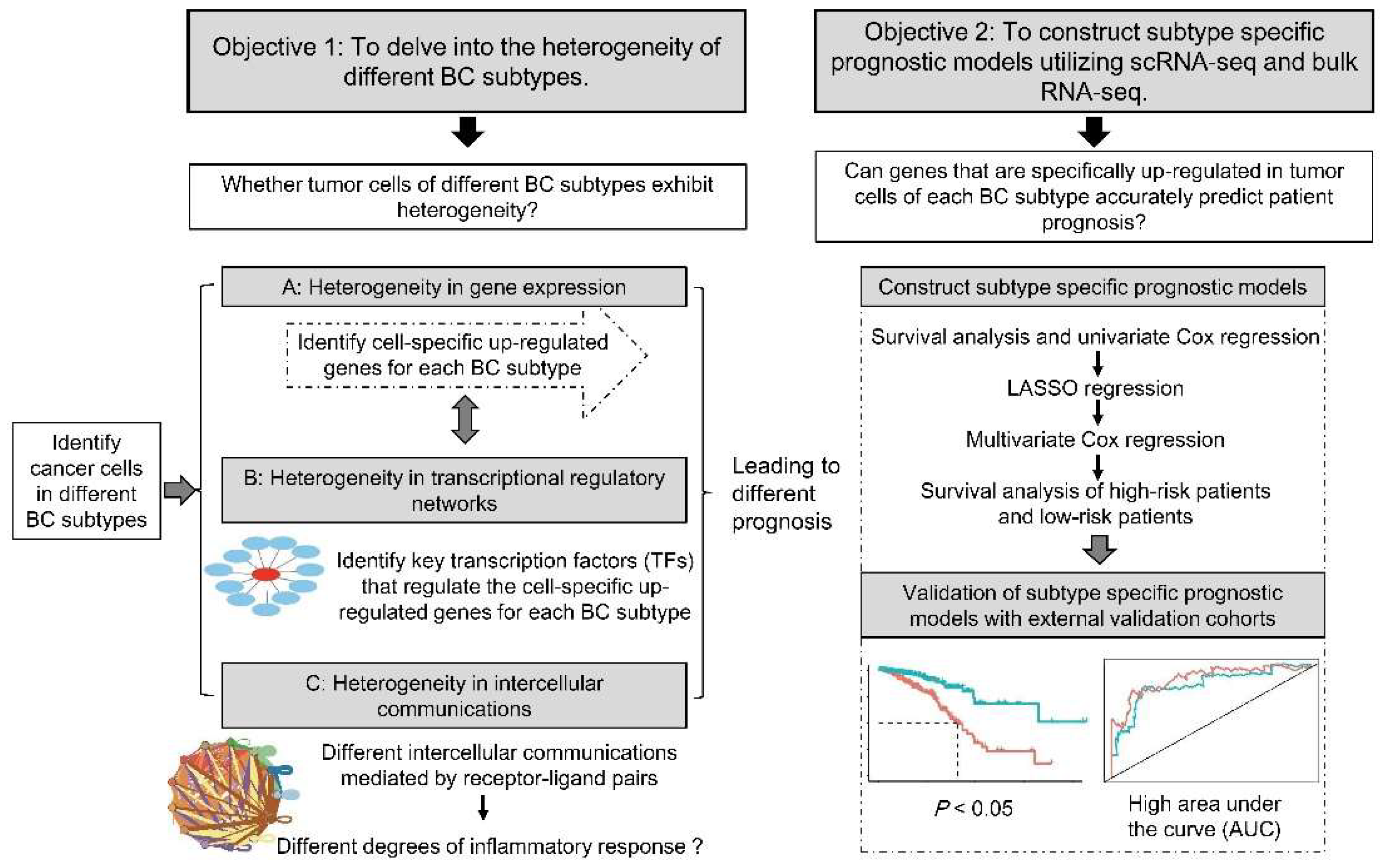
3. Results
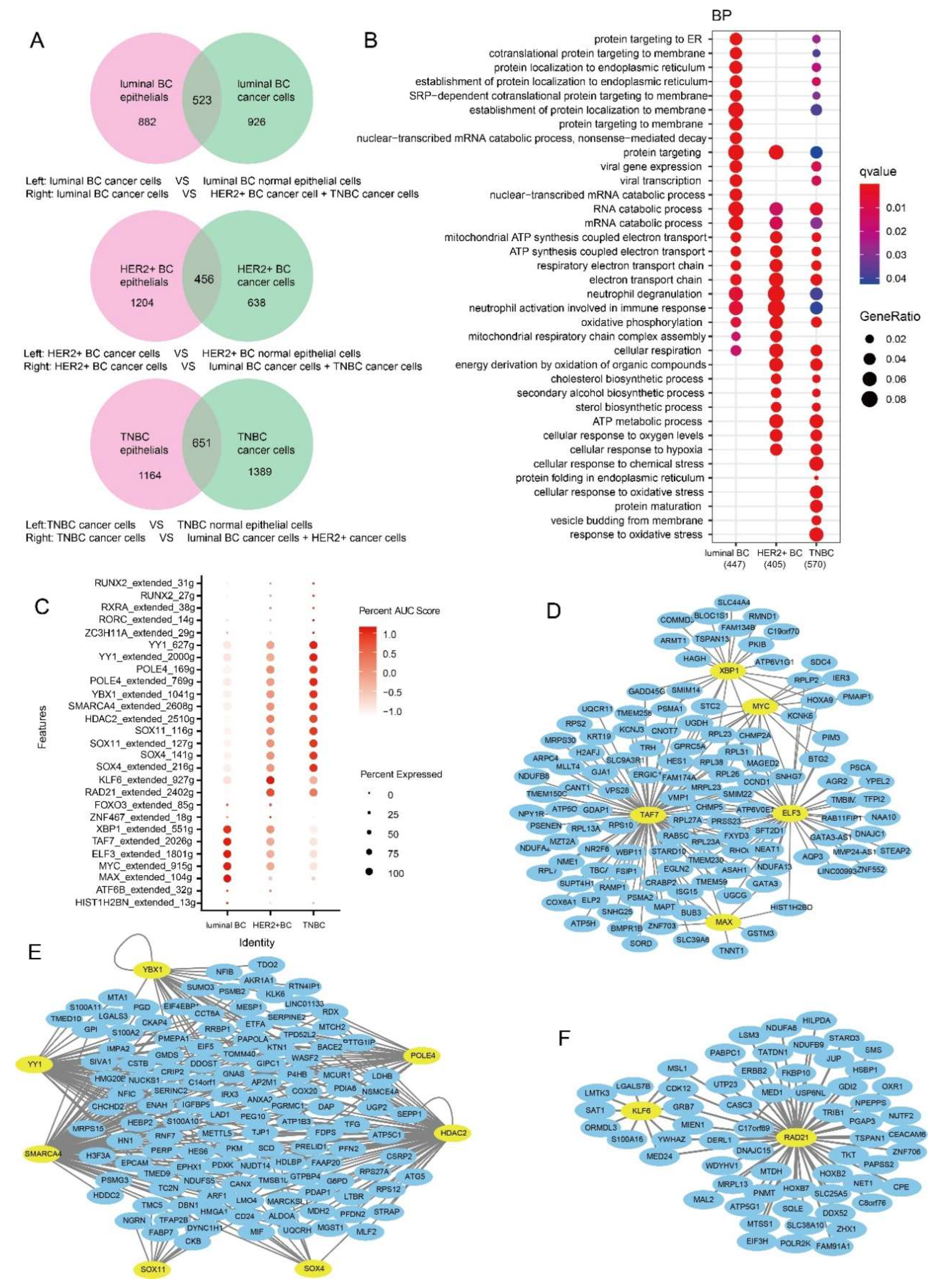
3.1. TNBC Has a Higher Proportion of Tumor Cells
3.2. Functions of Specifically Upregulated Genes and Variation of GRNs in Three BC Subtypes
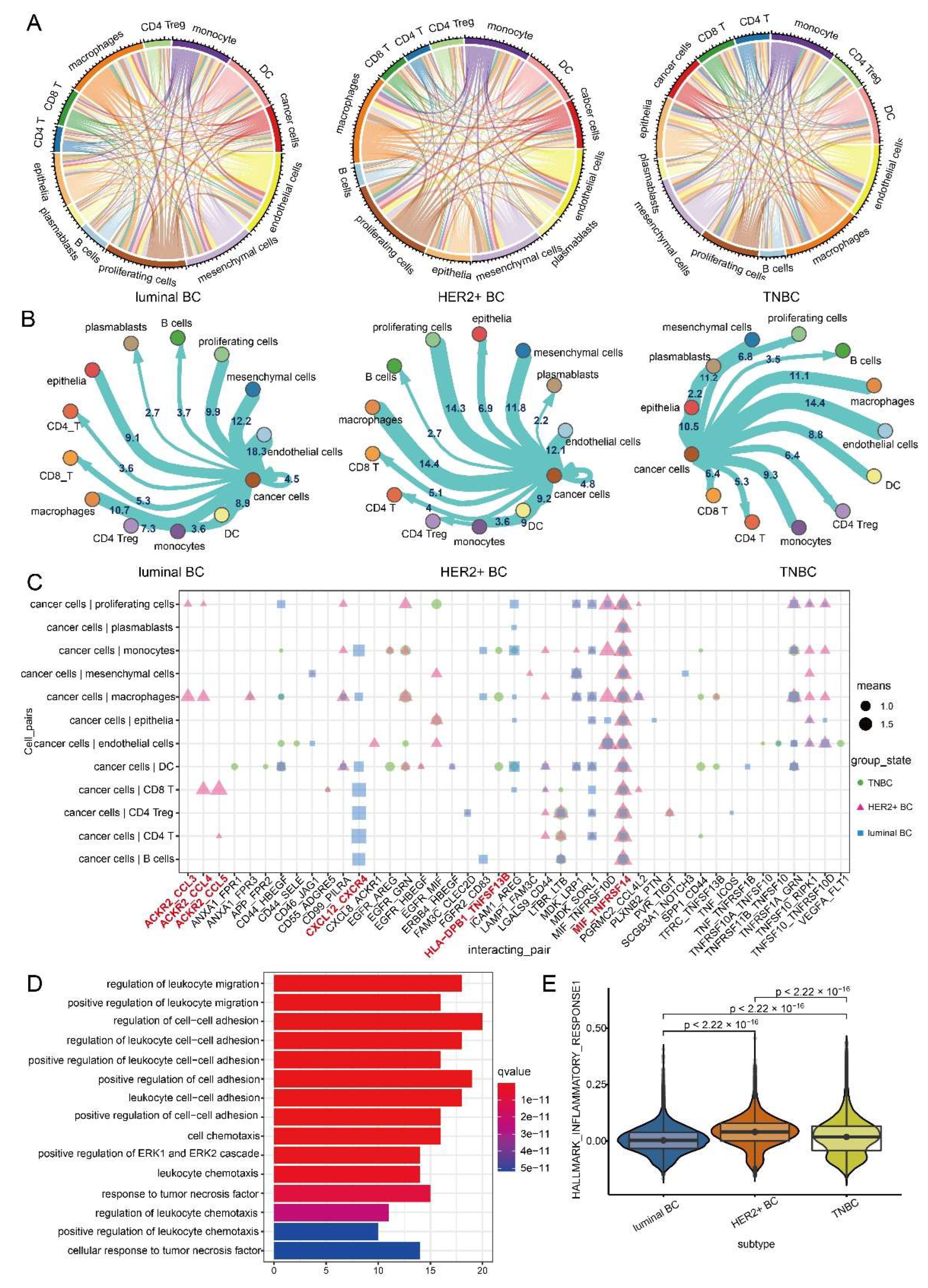
3.3. Intercellular Communication of Cancer Cells Leads to BC Inflammatory Microenvironment
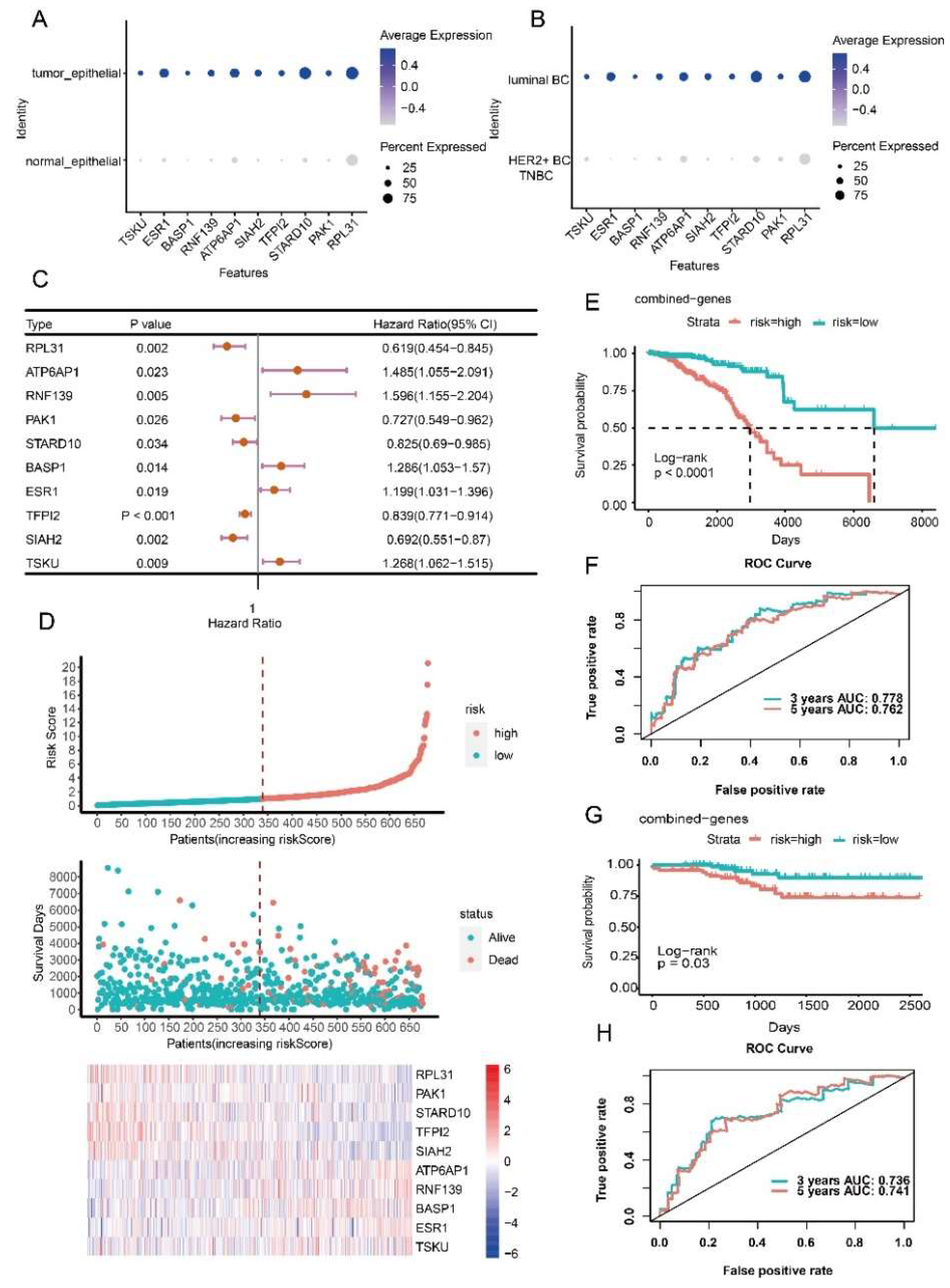
3.4. Prognostic Model Construction Using Cancer Cell-Specific Upregulated Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Januškevičienė, I.; Petrikaitė, V. Heterogeneity of breast cancer: The importance of interaction between different tumor cell populations. Life Sci. 2019, 239, 117009. [Google Scholar] [CrossRef] [PubMed]
- Franchet, C.; Duprez-Paumier, R.; Lacroix-Triki, M. Molecular taxonomy of luminal breast cancer in 2015. Bull. Cancer 2015, 102, S34–S46. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The trastuzumab era: Current and upcoming targeted HER2+ breast cancer therapies. Am. J. Cancer Res. 2020, 10, 1045–1067. [Google Scholar]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Alfarsi, L.; Johnston, S.; Liu, D.X.; Rakha, E.; Green, A.R. Current issues with luminal subtype classification in terms of prediction of benefit from endocrine therapy in early breast cancer. Histopathology 2018, 73, 545–558. [Google Scholar] [CrossRef]
- Veitch, Z.; Khan, O.F.; Tilley, D.; Ribnikar, D.; Kostaras, X.; King, K.; Tang, P.; Lupichuk, S. Real-World Outcomes of Adjuvant Chemotherapy for Node-Negative and Node-Positive HER2-Positive Breast Cancer. J. Natl. Compr. Canc. Netw. 2019, 17, 47–56. [Google Scholar] [CrossRef]
- Kumar, P.; Aggarwal, R. An overview of triple-negative breast cancer. Arch. Gynecol. Obstet. 2016, 293, 247–269. [Google Scholar] [CrossRef]
- Ren, C.; Han, C.; Fu, D.; Wang, D.; Chen, H.; Chen, Y.; Shen, M. Circulating tumor cells in breast cancer beyond the genotype of primary tumor for tailored therapy. Int. J. Cancer 2016, 138, 1586–1600. [Google Scholar] [CrossRef]
- Lane, A.N.; Tan, J.; Wang, Y.; Yan, J.; Higashi, R.M.; Fan, T.W. Probing the metabolic phenotype of breast cancer cells by multiple tracer stable isotope resolved metabolomics. Metab. Eng. 2017, 43, 125–136. [Google Scholar] [CrossRef]
- Liu, X.; Peng, Y.; Wang, J. Integrative analysis of DNA methylation and gene expression profiles identified potential breast cancer-specific diagnostic markers. Biosci. Rep. 2020, 40, BSR20201053. [Google Scholar] [CrossRef]
- Wang, Y.; Mashock, M.; Tong, Z.; Mu, X.; Chen, H.; Zhou, X.; Zhang, H.; Zhao, G.; Liu, B.; Li, X. Changing Technologies of RNA Sequencing and Their Applications in Clinical Oncology. Front. Oncol. 2020, 10, 447. [Google Scholar] [CrossRef]
- Zheng, S.; Zou, Y.; Liang, J.Y.; Xiao, W.; Yang, A.; Meng, T.; Lu, S.; Luo, Z.; Xie, X. Identification and validation of a combined hypoxia and immune index for triple-negative breast cancer. Mol. Oncol. 2020, 14, 2814–2833. [Google Scholar] [CrossRef]
- Tang, J.; Luo, Y.; Wu, G. A glycolysis-related gene expression signature in predicting recurrence of breast cancer. Aging (Albany NY) 2020, 12, 24983–24994. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Olsen, T.K.; Baryawno, N. Introduction to Single-Cell RNA Sequencing. Curr. Protoc. Mol. Biol. 2018, 122, e57. [Google Scholar] [CrossRef]
- The Biology of Genomes. Single-cell sequencing tackles basic and biomedical questions. Science 2012, 336, 976–977. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed]
- AlJanahi, A.A.; Danielsen, M.; Dunbar, C.E. An Introduction to the Analysis of Single-Cell RNA-Sequencing Data. Mol. Ther. Methods Clin. Dev. 2018, 10, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.X.; Taramelli, R.; Pedrini, E.; Knijnenburg, T.; Huang, S. Extracting Intercellular Signaling Network of Cancer Tissues using Ligand-Receptor Expression Patterns from Whole-tumor and Single-cell Transcriptomes. Sci. Rep. 2017, 7, 8815. [Google Scholar] [CrossRef]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- AlMusawi, S.; Ahmed, M.; Nateri, A.S. Understanding cell-cell communication and signaling in the colorectal cancer microenvironment. Clin. Transl. Med. 2021, 11, e308. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020, 182, 1232–1251.e1222. [Google Scholar] [CrossRef]
- Li, F.; Wen, H.; Bukhari, I.; Liu, B.; Guo, C.; Ren, F.; Tang, Y.; Mi, Y.; Zheng, P. Relationship Between CNVs and Immune Cells Infiltration in Gastric Tumor Microenvironment. Front. Genet. 2022, 13, 869967. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Aibar, S.; González-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Yang, C. The Neuronal Transcription Factor Creb3l1 Potential Upregulates Ntrk2 in the Hypertensive Microenvironment to Promote Vascular Smooth Muscle Cell-Neuron Interaction and Prevent Neurons from Ferroptosis: A Bioinformatic Research of scRNA-seq Data. Dis. Markers 2022, 2022, 8339759. [Google Scholar] [CrossRef]
- Moons, K.G.; Altman, D.G.; Reitsma, J.B.; Ioannidis, J.P.; Macaskill, P.; Steyerberg, E.W.; Vickers, A.J.; Ransohoff, D.F.; Collins, G.S. Transparent Reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): Explanation and elaboration. Ann. Intern. Med. 2015, 162, W1–W73. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z. Increased Oxidative Stress as a Selective Anticancer Therapy. Oxid. Med. Cell Longev. 2015, 2015, 294303. [Google Scholar] [CrossRef] [Green Version]
- Moldogazieva, N.T.; Lutsenko, S.V.; Terentiev, A.A. Reactive Oxygen and Nitrogen Species-Induced Protein Modifications: Implication in Carcinogenesis and Anticancer Therapy. Cancer Res. 2018, 78, 6040–6047. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- DeSantis, C.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA Cancer J. Clin. 2014, 64, 52–62. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Ma, W.; Zhao, F.; Yu, X.; Guan, S.; Suo, H.; Tao, Z.; Qiu, Y.; Wu, Y.; Cao, Y.; Jin, F. Immune-related lncRNAs as predictors of survival in breast cancer: A prognostic signature. J. Transl. Med. 2020, 18, 442. [Google Scholar] [CrossRef]
- Lin, W.W.; Xu, L.T.; Chen, Y.S.; Go, K.; Sun, C.; Zhu, Y.J. Single-Cell Transcriptomics-Based Study of Transcriptional Regulatory Features in the Mouse Brain Vasculature. Biomed. Res. Int. 2021, 2021, 7643209. [Google Scholar] [CrossRef]
- Agarwal, N.; Theodorescu, D. The Role of Transcription Factor YY1 in the Biology of Cancer. Crit. Rev. Oncog. 2017, 22, 13–21. [Google Scholar] [CrossRef]
- Lai, Y.W.; Hsu, W.J.; Lee, W.Y.; Chen, C.H.; Tsai, Y.H.; Dai, J.Z.; Yang, C.C.; Lin, C.W. Prognostic Value of a Glycolytic Signature and Its Regulation by Y-Box-Binding Protein 1 in Triple-Negative Breast Cancer. Cells 2021, 10, 1890. [Google Scholar] [CrossRef]
- Shepherd, J.H.; Uray, I.P.; Mazumdar, A.; Tsimelzon, A.; Savage, M.; Hilsenbeck, S.G.; Brown, P.H. The SOX11 transcription factor is a critical regulator of basal-like breast cancer growth, invasion, and basal-like gene expression. Oncotarget 2016, 7, 13106–13121. [Google Scholar] [CrossRef]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.G.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, J.; Wu, J.; Xu, B.; Wang, Z.; Giamas, G.; Stebbing, J.; Yu, Z. A Pan-Cancer Analysis of SMARCA4 Alterations in Human Cancers. Front. Immunol. 2021, 12, 762598. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer—Overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ruan, S.; Ming, J.; Dong, F. Nuclear expression of XBP1s is correlated with breast cancer survival: A retrospective analysis based on tissue microarray. Onco Targets Ther. 2017, 10, 5927–5934. [Google Scholar] [CrossRef]
- Yan, M.; Xu, H.; Waddell, N.; Shield-Artin, K.; Haviv, I.; McKay, M.J.; Fox, S.B. Enhanced RAD21 cohesin expression confers poor prognosis in BRCA2 and BRCAX, but not BRCA1 familial breast cancers. Breast Cancer Res. 2012, 14, R69. [Google Scholar] [CrossRef]
- Xiong, S.; Dong, L.; Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. J. Hematol. Oncol. 2021, 14, 173. [Google Scholar] [CrossRef]
- Lodge, K.M.; Cowburn, A.S.; Li, W.; Condliffe, A.M. The Impact of Hypoxia on Neutrophil Degranulation and Consequences for the Host. Int. J. Mol. Sci. 2020, 21, 1183. [Google Scholar] [CrossRef]
- Choi, E.K.; Kim, W.K.; Sul, O.J.; Park, Y.K.; Kim, E.S.; Suh, J.H.; Yu, R.; Choi, H.S. TNFRSF14 deficiency protects against ovariectomy-induced adipose tissue inflammation. J. Endocrinol. 2014, 220, 25–33. [Google Scholar] [CrossRef]
- Choi, E.K.; Park, H.J.; Sul, O.J.; Rajasekaran, M.; Yu, R.; Choi, H.S. Carbon monoxide reverses adipose tissue inflammation and insulin resistance upon loss of ovarian function. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E621–E630. [Google Scholar] [CrossRef]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and chemokine receptors during COVID-19 infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Lu, B.; Cao, C.; Li, H.; Yang, D.; Huang, L.; Ding, T.; Wu, M.; Lu, G. Plasma TNFSF13B and TNFSF14 Function as Inflammatory Indicators of Severe Adenovirus Pneumonia in Pediatric Patients. Front. Immunol. 2020, 11, 614781. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef]
- Zappavigna, S.; Cossu, A.M.; Grimaldi, A.; Bocchetti, M.; Ferraro, G.A.; Nicoletti, G.F.; Filosa, R.; Caraglia, M. Anti-Inflammatory Drugs as Anticancer Agents. Int. J. Mol. Sci. 2020, 21, 2605. [Google Scholar] [CrossRef]
- Greenland, K.J.; Jantke, I.; Jenatschke, S.; Bracken, K.E.; Vinson, C.; Gellersen, B. The human NAD+-dependent 15-hydroxyprostaglandin dehydrogenase gene promoter is controlled by Ets and activating protein-1 transcription factors and progesterone. Endocrinology 2000, 141, 581–597. [Google Scholar] [CrossRef]
- Bray, K.; Gillette, M.; Young, J.; Loughran, E.; Hwang, M.; Sears, J.C.; Vargo-Gogola, T. Cdc42 overexpression induces hyperbranching in the developing mammary gland by enhancing cell migration. Breast Cancer Res. 2013, 15, R91. [Google Scholar] [CrossRef]
- He, Y.; Luo, Y.; Zhang, D.; Wang, X.; Zhang, P.; Li, H.; Ejaz, S.; Liang, S. PGK1-mediated cancer progression and drug resistance. Am. J. Cancer Res. 2019, 9, 2280–2302. [Google Scholar]
- Zheng, X.; Weigert, A.; Reu, S.; Guenther, S.; Mansouri, S.; Bassaly, B.; Gattenlöhner, S.; Grimminger, F.; Pullamsetti, S.; Seeger, W.; et al. Spatial Density and Distribution of Tumor-Associated Macrophages Predict Survival in Non-Small Cell Lung Carcinoma. Cancer Res. 2020, 80, 4414–4425. [Google Scholar] [CrossRef]
- Chao, Y.; Ou, Q.; Shang, J. Expression and prognostic value of SULT1A2 in bladder cancer. Exp. Ther. Med. 2021, 22, 779. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Zhang, S. Prognostic and immunological value of ATP6AP1 in breast cancer: Implications for SARS-CoV-2. Aging 2021, 13, 16904–16921. [Google Scholar] [CrossRef]
- Xu, Y.; Luo, H.; Hu, Q.; Zhu, H. Identification of Potential Driver Genes Based on Multi-Genomic Data in Cervical Cancer. Front. Genet. 2021, 12, 598304. [Google Scholar] [CrossRef]
- Jaikumarr Ram, A.; Girija As, S.; Jayaseelan, V.P.; Arumugam, P. Overexpression of BASP1 Indicates a Poor Prognosis in Head and Neck Squamous Cell Carcinoma. Asian Pac. J. Cancer Prev. 2020, 21, 3435–3439. [Google Scholar] [CrossRef]
- Carausu, M.; Bidard, F.C.; Callens, C.; Melaabi, S.; Jeannot, E.; Pierga, J.Y.; Cabel, L. ESR1 mutations: A new biomarker in breast cancer. Expert Rev. Mol. Diagn. 2019, 19, 599–611. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, D.; Fu, J.; Zhao, L.; Li, D.; Sun, H.; Liu, X.; Xu, J.; Tian, T.; Zhang, L.; et al. Tsukushi is a novel prognostic biomarker and correlates with tumor-infiltrating B cells in non-small cell lung cancer. Aging (Albany NY) 2021, 13, 4428–4451. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 66. [Google Scholar] [CrossRef]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Qin, S.; Yi, Y.; Gao, H.; Liu, X.; Ma, F.; Guan, M. Delving into the Heterogeneity of Different Breast Cancer Subtypes and the Prognostic Models Utilizing scRNA-Seq and Bulk RNA-Seq. Int. J. Mol. Sci. 2022, 23, 9936. https://doi.org/10.3390/ijms23179936
Xu J, Qin S, Yi Y, Gao H, Liu X, Ma F, Guan M. Delving into the Heterogeneity of Different Breast Cancer Subtypes and the Prognostic Models Utilizing scRNA-Seq and Bulk RNA-Seq. International Journal of Molecular Sciences. 2022; 23(17):9936. https://doi.org/10.3390/ijms23179936
Chicago/Turabian StyleXu, Jieyun, Shijie Qin, Yunmeng Yi, Hanyu Gao, Xiaoqi Liu, Fei Ma, and Miao Guan. 2022. "Delving into the Heterogeneity of Different Breast Cancer Subtypes and the Prognostic Models Utilizing scRNA-Seq and Bulk RNA-Seq" International Journal of Molecular Sciences 23, no. 17: 9936. https://doi.org/10.3390/ijms23179936
APA StyleXu, J., Qin, S., Yi, Y., Gao, H., Liu, X., Ma, F., & Guan, M. (2022). Delving into the Heterogeneity of Different Breast Cancer Subtypes and the Prognostic Models Utilizing scRNA-Seq and Bulk RNA-Seq. International Journal of Molecular Sciences, 23(17), 9936. https://doi.org/10.3390/ijms23179936