
Alpha-Synuclein: The Spark That Flames Dopaminergic Neurons, In Vitro and In Vivo Evidence


Abstract
:1. Introduction
2. Results
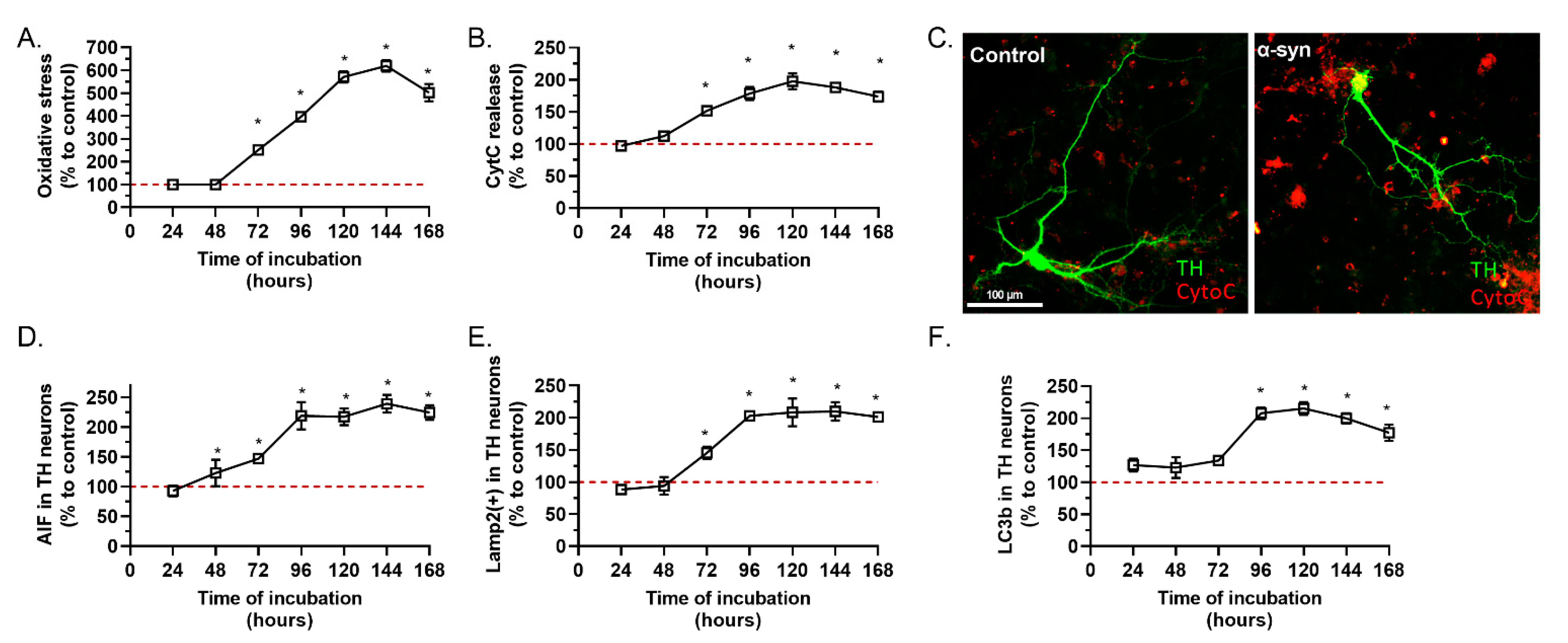
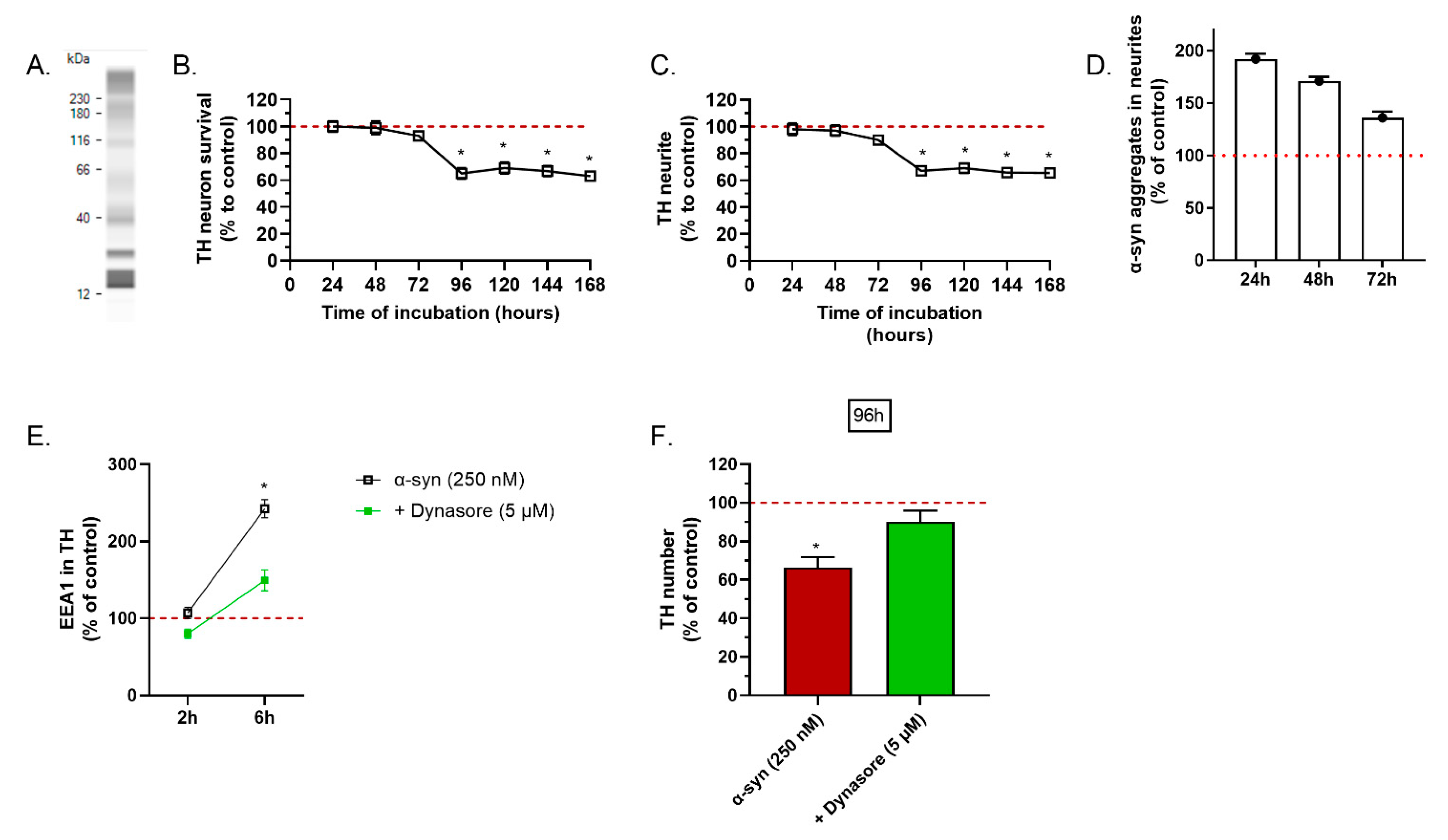
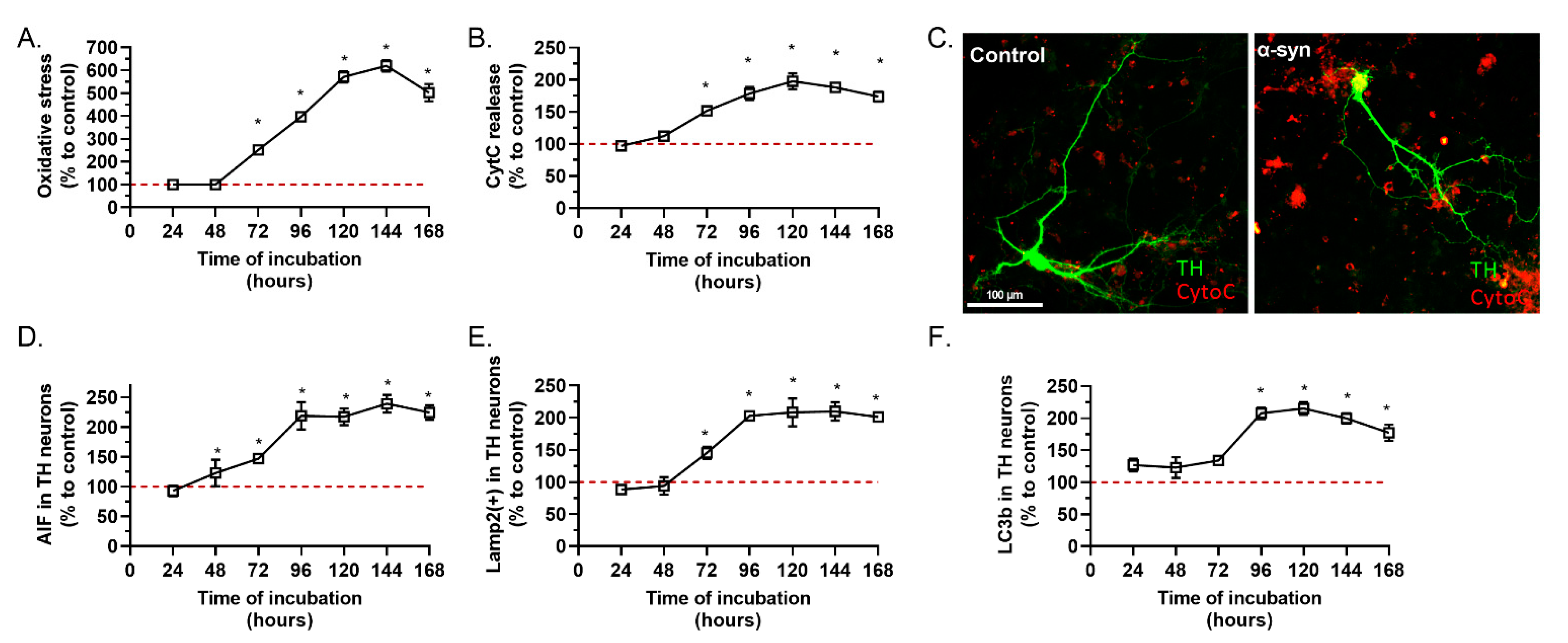
2.1. α-Synuclein Preparation Induced Toxicity on TH Positive Neurons
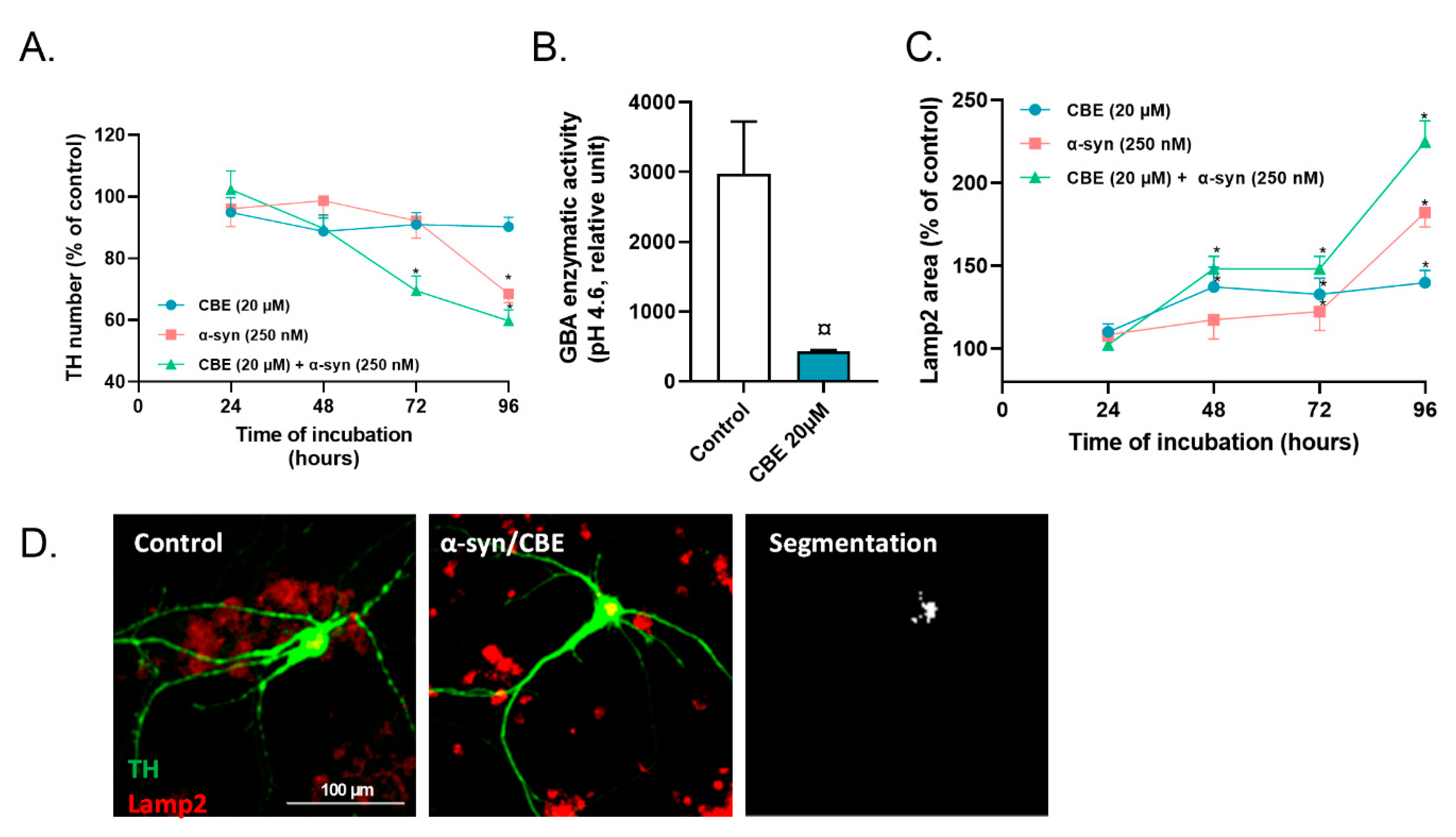
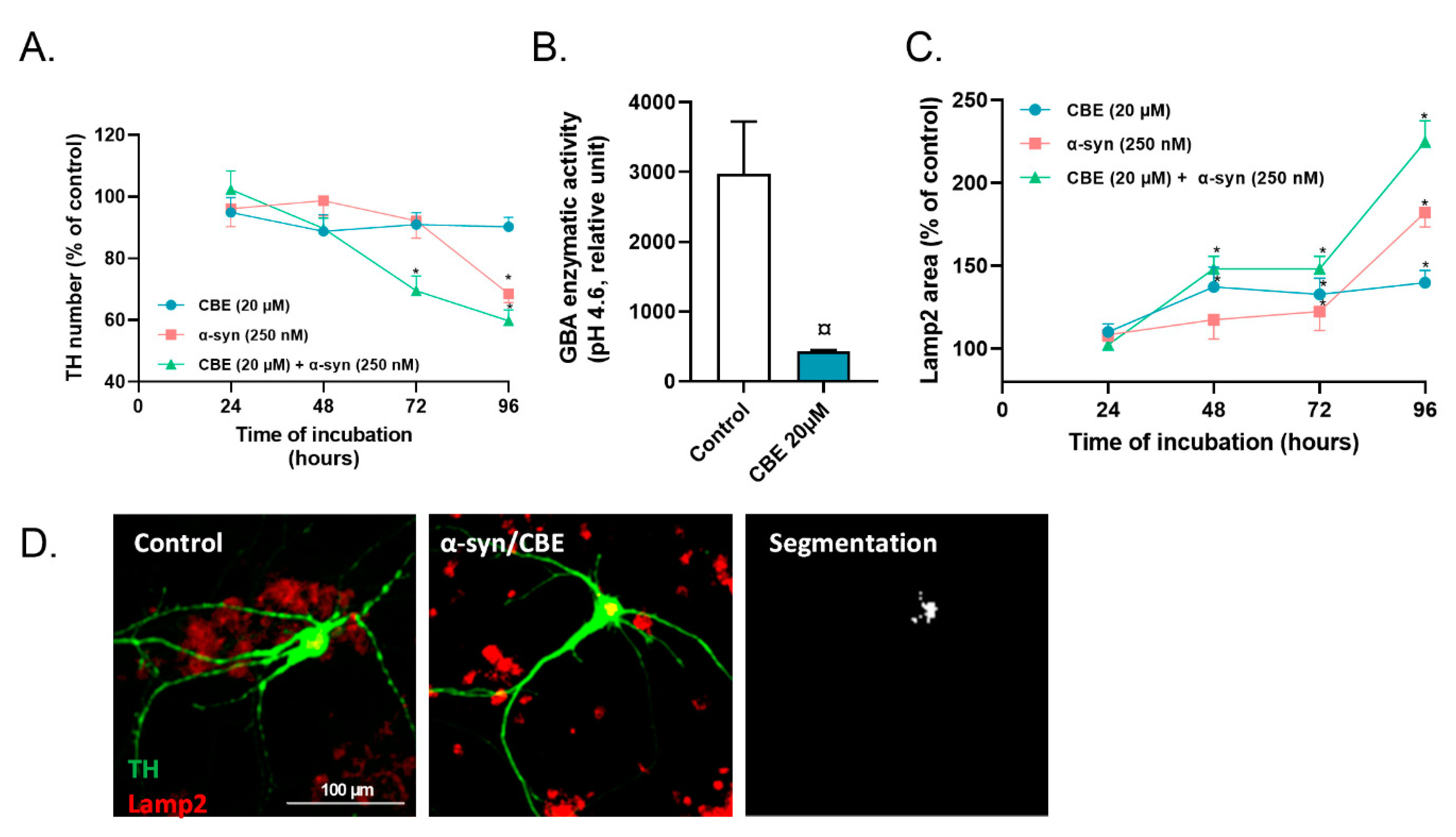
2.2. Reduced Activity of Lysosomal GBA Exacerbates the Toxicity of Alpha-Synuclein on Primary Dopaminergic Neurons
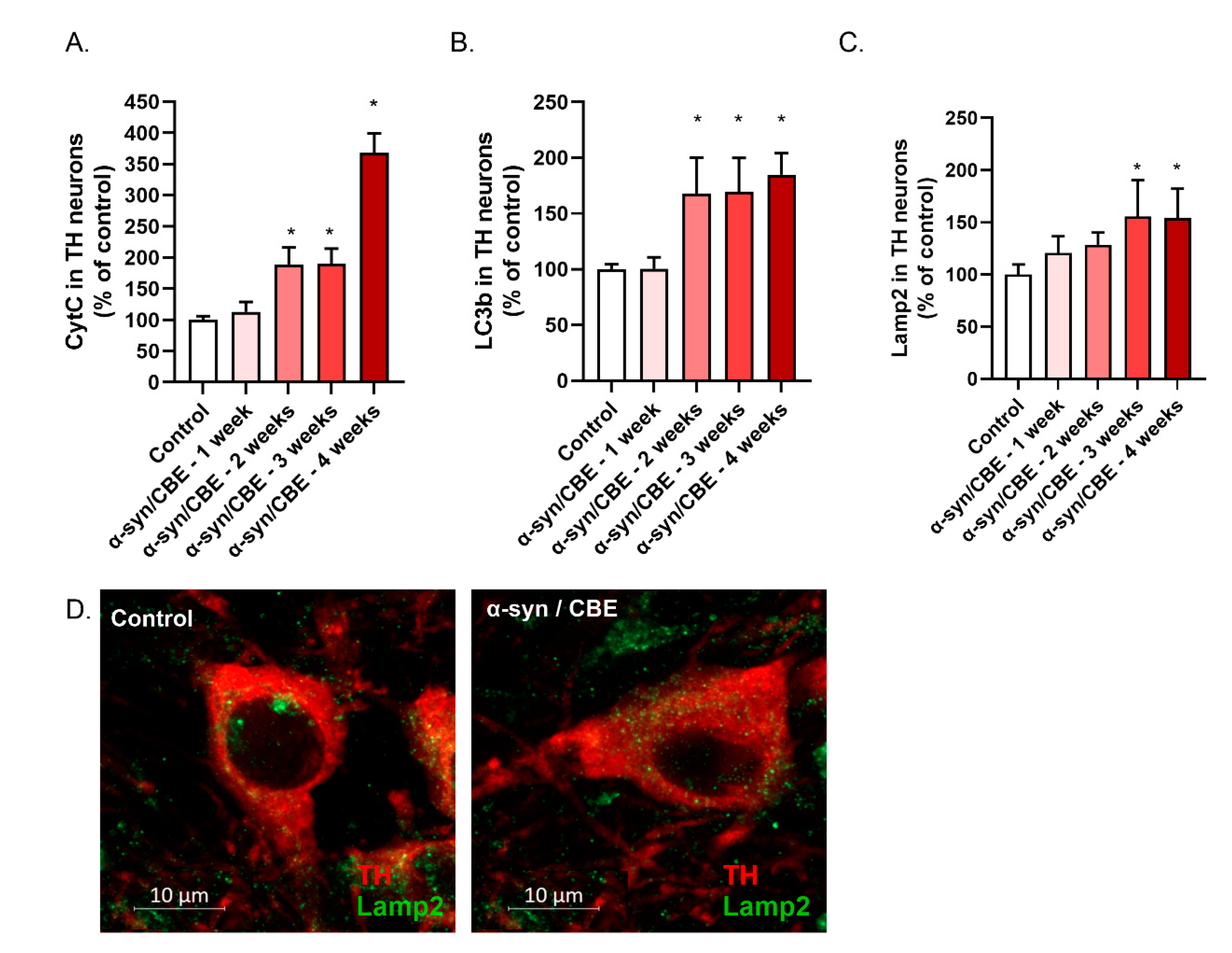
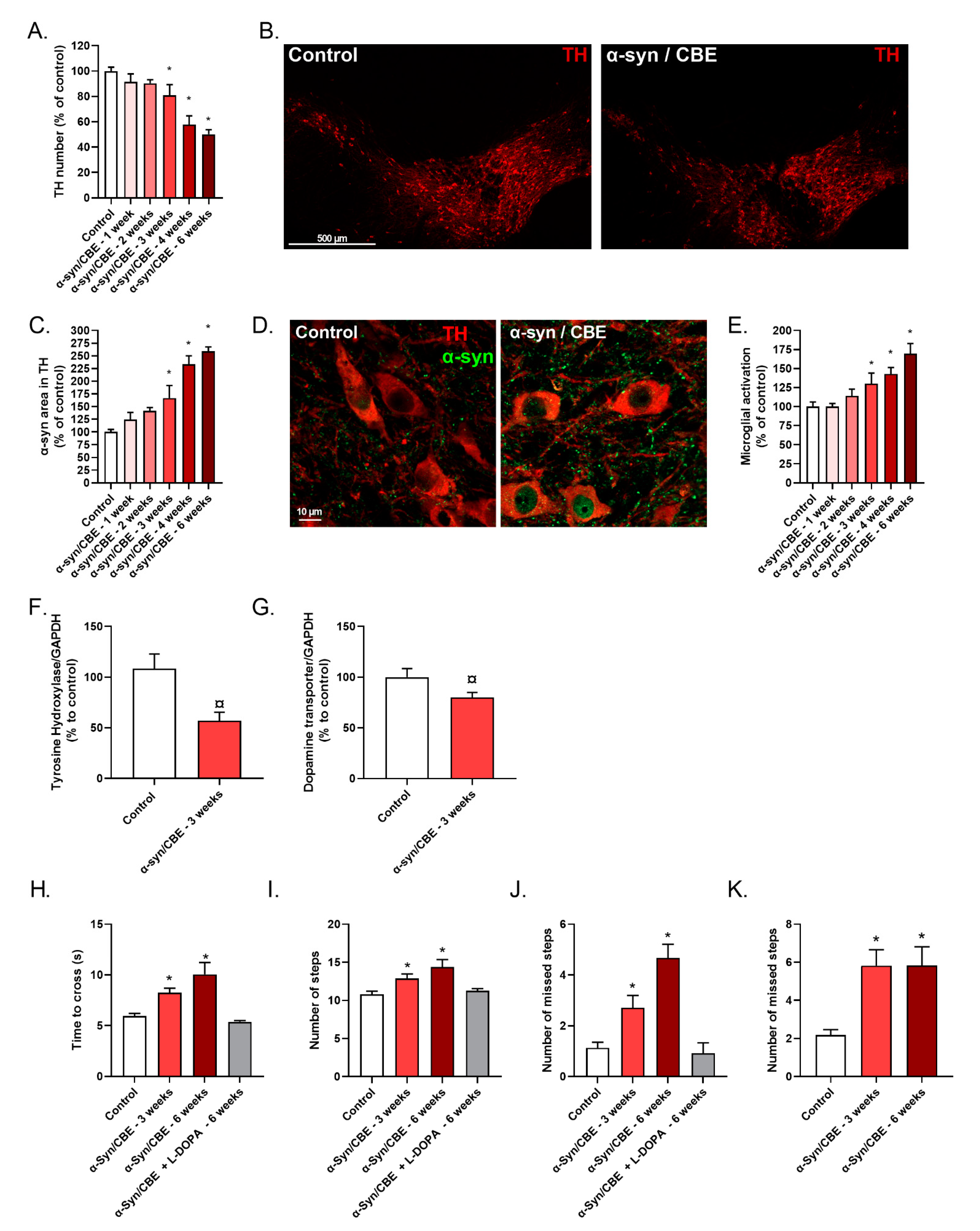
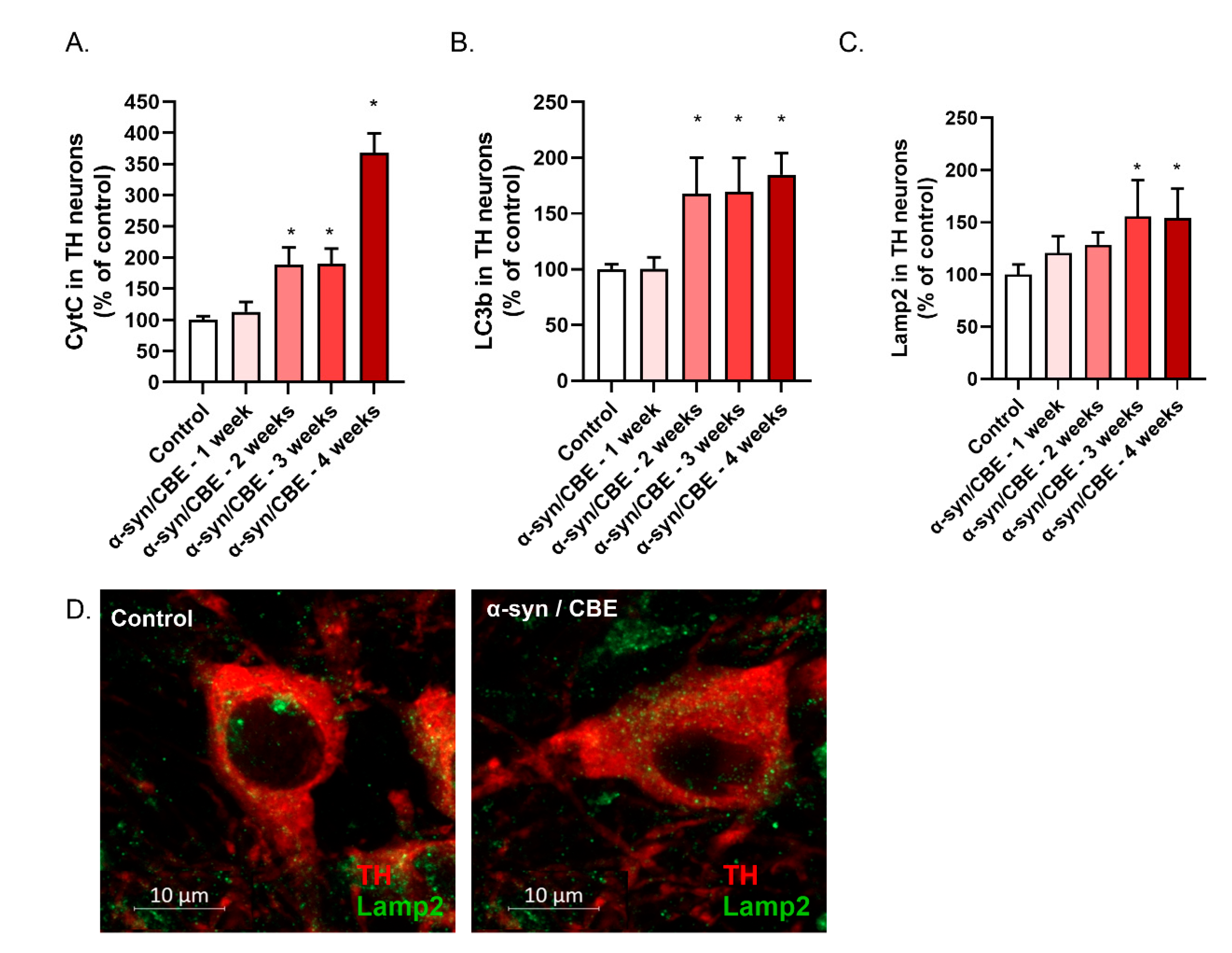
2.3. Intra-Nigral Injection of Alpha-Synuclein and Chronic Inhibition of GBA Induce Motor Dysfunction and Loss of Dopaminergic Neurons in Aged Mice
3. Discussion
4. Materials and Methods
4.1. Primary Culture of Mesencephalic Neurons
4.2. Treatment of Primary Mesencephalic Cultures
4.3. Immunostaining and Automatized Image Analysis
4.4. Mice; Stereotaxic Injection of Alpha-Synuclein and Chronic Inhibition of GBA
4.5. Histology and Automatized Image Analysis
4.6. Brain Protein Analysis
4.7. Evaluation of Motor Functions, Behavioral Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-Hydroxydopamine | 6-OHDA |
| α-synuclein | α-syn |
| Apoptotis Inducing Factor: Aif Antibody | Ab |
| Cellular Prion Protein | PrPc |
| Conduritol B Epoxide | CBE |
| Cytochrome C | CytC |
| Dopamine | DA |
| Dopamine Transporter | DAT |
| Endoplasmic Reticulum | ER |
| Glucocerebrosidase | GCase |
| Reactive Oxygen Species | ROS |
| Paraformaldehyde | PFA |
| Phosphate Buffer Saline | PBS |
| Pre-Formed Fibrils | PFF |
| Soluble Nsf Attachment Protein | SNAP |
| Soluble Nsf Attachment Protein Receptor | SNARE |
| Substantia Nigra Pars Compacta | SNpc |
| Tyrosine Hydroxylase | TH |
References
- Myhre, O.; Utkilen, H.; Duale, N.; Brunborg, G.; Hofer, T. Metal Dyshomeostasis and Inflammation in Alzheimer’s and Parkinson’s Diseases: Possible Impact of Environmental Exposures. Oxidative Med. Cell. Longev. 2013, 2013, 481827. [Google Scholar] [CrossRef]
- Obeso, I.; Casabona, E.; Rodríguez-Rojas, R.; Bringas, M.L.; Macías, R.; Pavón, N.; Obeso, J.A.; Jahanshahi, M. Unilateral subthalamotomy in Parkinson’s disease: Cognitive, psychiatric and neuroimaging changes. Cortex 2017, 94, 39–48. [Google Scholar] [CrossRef]
- Gonzalez-Latapi, P.; Bayram, E.; Litvan, I.; Marras, C. Cognitive Impairment in Parkinson’s Disease: Epidemiology, Clinical Profile, Protective and Risk Factors. Behav. Sci. 2021, 11, 74. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef]
- Alegre-Abarrategui, J.; Brimblecombe, K.R.; Roberts, R.F.; Velentza-Almpani, E.; Tilley, B.S.; Bengoa-Vergniory, N.; Proukakis, C. Selective vulnerability in α-synucleinopathies. Acta Neuropathol. 2019, 138, 681–704. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.; Scheller, R. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.R.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef]
- Carulla, N.; Caddy, G.L.; Hall, D.R.; Zurdo, J.; Gairí, M.; Feliz, M.; Giralt, E.; Robinson, C.; Dobson, C.M. Molecular recycling within amyloid fibrils. Nature 2005, 436, 554–558. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct Observation of the Interconversion of Normal and Toxic Forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Uversky, V.N. A Protein-Chameleon: Conformational Plasticity of α-synuclein, a Disordered Protein Involved in Neurodegenerative Disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Tarutani, A.; Arai, T.; Murayama, S.; Hisanaga, S.; Hasegawa, M. Potent prion-like behaviors of pathogenic α-synuclein and evaluation of inactivation methods. Acta Neuropathol. Commun. 2018, 6, 29. [Google Scholar] [CrossRef]
- Hoban, D.B.; Shrigley, S.; Mattsson, B.; Breger, L.S.; Jarl, U.; Cardoso, T.; Wahlestedt, J.N.; Luk, K.C.; Björklund, A.; Parmar, M. Impact of α-synuclein pathology on transplanted hESC-derived dopaminergic neurons in a humanized α-synuclein rat model of PD. Proc. Natl. Acad. Sci. USA 2020, 117, 15209–15220. [Google Scholar] [CrossRef]
- Vargas, J.Y.; Grudina, C.; Zurzolo, C. The prion-like spreading of α-synuclein: From in vitro to in vivo models of Parkinson’s disease. Ageing Res Rev. 2019, 50, 89–101. [Google Scholar] [CrossRef]
- Thom, T.; Schmitz, M.; Fischer, A.; Correia, A.; Correia, S.; Llorens, F.; Pique, A.; Möbius, W.; Domingues, R.; Zafar, S.; et al. Cellular Prion Protein Mediates α-Synuclein Uptake, Localization, and Toxicity In Vitro and In Vivo. Mov. Disord. 2022, 37, 39–51. [Google Scholar] [CrossRef]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Rammonhan, M.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca2+ Channel Agonists Protects Human Dopaminergic Neurons from α-synuclein Toxicity. J. Neurosci. 2019, 39, 5760–5772. [Google Scholar] [CrossRef]
- Bieri, G.; Gitler, A.D.; Brahic, M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 2018, 109, 219–225. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef]
- Angot, E.; Steiner, J.A.; Tomé, C.M.L.; Ekström, P.; Mattsson, B.; Björklund, A.; Brundin, P. Alpha-Synuclein Cell-to-Cell Transfer and Seeding in Grafted Dopaminergic Neurons In Vivo. PLoS ONE 2012, 7, e39465. [Google Scholar] [CrossRef]
- Visanji, N.P.; Orsi, A.; Johnston, T.H.; Howson, P.A.; Dixon, K.; Callizot, N.; Brotchie, J.; Rees, D.D. PYM50028, a novel, orally active, nonpeptide neurotrophic factor inducer, prevents and reverses neuronal damage induced by MPP+ in mesencephalic neurons and by MPTP in a mouse model of Parkinson’s disease. FASEB J. 2008, 22, 2488–2497. [Google Scholar] [CrossRef]
- Callizot, N.; Combes, M.; Henriques, A.; Poindron, P. Necrosis, apoptosis, necroptosis, three modes of action of dopaminergic neuron neurotoxins. PLoS ONE 2019, 14, e0215277. [Google Scholar] [CrossRef]
- Villar-Piqué, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J. Neurochem. 2016, 139, 240–255. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, Prefibrillar α-synuclein Oligomers Promote Complex I-dependent, Ca2+-induced Mitochondrial Dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef]
- Sepúlveda, D.; Cisternas-Olmedo, M.; Arcos, J.; Nassif, M.; Vidal, R.L. Contribution of Autophagy-Lysosomal Pathway in the Exosomal Secretion of Alpha-Synuclein and Its Impact in the Progression of Parkinson’s Disease. Front. Mol. Neurosci. 2022, 15, 805087. [Google Scholar] [CrossRef]
- Brockmann, K. GBA-Associated Synucleinopathies: Prime Candidates for Alpha-Synuclein Targeting Compounds. Front. Cell Dev. Biol. 2020, 8, 562522. [Google Scholar] [CrossRef]
- Wallings, R.L.; Humble, S.W.; Ward, M.E.; Wade-Martins, R. Lysosomal Dysfunction at the Centre of Parkinson’s Disease and Frontotemporal Dementia/Amyotrophic Lateral Sclerosis. Trends Neurosci. 2019, 42, 899–912. [Google Scholar] [CrossRef]
- Ghatak, S.; Trudler, D.; Dolatabadi, N.; Ambasudhan, R. Parkinson’s disease: What the model systems have taught us so far. J. Genet. 2018, 97, 729–751. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of -Glucocerebrosidase Reduces Pathological -Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.A.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013, 136, 1128–1138. [Google Scholar] [CrossRef]
- Henderson, M.X.; Cornblath, E.J.; Darwich, A.; Zhang, B.; Brown, H.; Gathagan, R.J.; Sandler, R.; Bassett, D.S.; Trojanowski, J.Q.; Lee, V.M.Y. Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 2019, 22, 1248–1257. [Google Scholar] [CrossRef]
- Chu, Y.; Muller, S.; Tavares, A.; Barret, O.; Alagille, D.; Seibyl, J.; Tamagnan, G.; Marek, K.; Luk, K.; Trojanowski, J.Q.; et al. Intrastriatal alpha-synuclein fibrils in monkeys: Spreading, imaging and neuropathological changes. Brain 2019, 142, 3565–3579. [Google Scholar] [CrossRef]
- Fakhree, M.A.A.; Konings, I.B.M.; Kole, J.; Cambi, A.; Blum, C.; Claessens, M.M.A.E. The Localization of Alpha-synuclein in the Endocytic Pathway. Neuroscience 2021, 457, 186–195. [Google Scholar] [CrossRef]
- Pozo Devoto, V.M.; Falzone, T.L. Mitochondrial dynamics in Parkinson’s disease: A role for α-synuclein? Dis. Model. Mech. 2017, 10, 1075–1087. [Google Scholar] [CrossRef]
- Helley, M.P.; Pinnell, J.; Sportelli, C.; Tieu, K. Mitochondria: A Common Target for Genetic Mutations and Environmental Toxicants in Parkinson’s Disease. Front. Genet. 2017, 8, 177. [Google Scholar] [CrossRef]
- Decressac, M.; Björklund, A. TFEB: Pathogenic role and therapeutic target in Parkinson disease. Autophagy 2013, 9, 1244–1246. [Google Scholar] [CrossRef]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef]
- Henderson, M.X.; Sedor, S.; McGeary, I.; Cornblath, E.J.; Peng, C.; Riddle, D.M.; Li, H.L.; Zhang, B.; Brown, H.J.; Olufemi, M.F.; et al. Glucocerebrosidase Activity Modulates Neuronal Susceptibility to Pathological α-synuclein Insult. Neuron 2020, 105, 822–836.e7. [Google Scholar] [CrossRef]
- Tayebi, N.; Parisiadou, L.; Berhe, B.; Gonzalez, A.N.; Serra-Vinardell, J.; Tamargo, R.J.; Maniwang, E.; Sorrentino, Z.; Fujiwara, H.; Grey, R.J.; et al. Glucocerebrosidase haploinsufficiency in A53T α-synuclein mice impacts disease onset and course. Mol. Genet. Metab. 2017, 122, 198–208. [Google Scholar] [CrossRef]
- Polinski, N.K.; Martinez, T.N.; Gorodinsky, A.; Gareus, R.; Sasner, M.; Herberth, M.; Switzer, R.; Ahmad, S.O.; Cosden, M.; Kandebo, M.; et al. Decreased glucocerebrosidase activity and substrate accumulation of glycosphingolipids in a novel GBA1 D409V knock-in mouse model. PLoS ONE 2021, 16, e0252325. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef]
- Valdinocci, D.; Kovarova, J.; Neuzil, J.; Pountney, D.L. Alpha-Synuclein Aggregates Associated with Mitochondria in Tunnelling Nanotubes. Neurotox. Res. 2021, 39, 429–443. [Google Scholar] [CrossRef]
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef]
- Guo, Y.J.; Xiong, H.; Chen, K.; Zou, J.J.; Lei, P. Brain regions susceptible to alpha-synuclein spreading. Mol. Psychiatry 2022, 27, 758–770. [Google Scholar] [CrossRef]
- George, S.; Rey, N.L.; Tyson, T.; Esquibel, C.; Meyerdirk, L.; Schulz, E.; Pierce, S.; Burmeister, A.R.; Madaj, Z.; Steiner, J.A.; et al. Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 2019, 14, 34. [Google Scholar] [CrossRef]
- Lin, W.C.; Lu, C.H.; Chiu, P.Y.; Yang, S.Y. Plasma Total α-synuclein and Neurofilament Light Chain: Clinical Validation for Discriminating Parkinson’s Disease from Normal Control. Dement. Geriatr. Cogn. Disord. 2020, 49, 401–409. [Google Scholar] [CrossRef]
- Bouscary, A.; Quessada, C.; Mosbach, A.; Callizot, N.; Spedding, M.; Loeffler, J.P.; Henriques, A. Ambroxol Hydrochloride Improves Motor Functions and Extends Survival in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2019, 10, 883. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Characteristic | Fixation | Dilution | Provider | Reference |
|---|---|---|---|---|---|
| Tyrosine hydroxylase (TH) | Mouse monoclonal | PFA | 1/2000 | Sigma | T1299-.2ML |
| alpha-synuclein (α-syn) | Rabbit polyclonal | PFA | 1/200 | Cell signaling | 2642S |
| Early endosome antigen 1 (EEA1) | Mouse monoclonal | PFA | 1/100 | Abcam | Ab70521 |
| Lysosome-associated membrane protein 2A (Lamp2) | Rabbit polyclonal | PFA | 1/200 | Abcam | Ab203224 |
| Microtubule-associated protein light chain 3 beta (LC3B) | Rabbit polyclonal | PFA | 1/100 | Cell signaling | 3868S |
| Reactive oxygen species (ROS) | On live cells | 1/2000 | Fisher | 12648335 | |
| Allograft inflammatory factor-1 (AIF) | Mouse monoclonal | PFA | 1/100 | Santa Cruz biotechnology | SC-13116 |
| Cytochrome C (CytC) | Rabbit polyclonal | PFA | 1/100 | Abcam | ab90529 |
| Caspase 3 | Rabbit polyclonal | PFA | 1/100 | cell signaling | 9662 |
| Antibody | Characteristic | Fixation | Dilution | Provider | Reference |
|---|---|---|---|---|---|
| Tyrosine hydroxylase (TH) | Polyclonal, chicken | PFA | 1/1000 | Abcam | Ab76442 |
| Alpha-synuclein (α-syn) | Polyclonal, rabbit | PFA | 1/200 | Cell Signaling | 2642S |
| Ionized calcium-binding adaptator molecule 1 (IBA1) | Polyclonal, rabbit | PFA | 1/500 | Novus | NBP2-19019 |
| Cytochrome C (CytC) | Polyclonal, rabbit | PFA | 1/100 | Abcam | Ab90529 |
| Microtubule-associated protein light chain 3 beta (LC3b) | Monoclonal, rabbit | PFA | 1/200 | Cell Signaling | 3868S |
| Lysosome-associated membrane protein 2A (Lamp2A) | Polyclonal, rabbit | PFA | 1/100 | Fisher scientific | 10332473 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henriques, A.; Rouvière, L.; Giorla, E.; Farrugia, C.; El Waly, B.; Poindron, P.; Callizot, N. Alpha-Synuclein: The Spark That Flames Dopaminergic Neurons, In Vitro and In Vivo Evidence. Int. J. Mol. Sci. 2022, 23, 9864. https://doi.org/10.3390/ijms23179864
Henriques A, Rouvière L, Giorla E, Farrugia C, El Waly B, Poindron P, Callizot N. Alpha-Synuclein: The Spark That Flames Dopaminergic Neurons, In Vitro and In Vivo Evidence. International Journal of Molecular Sciences. 2022; 23(17):9864. https://doi.org/10.3390/ijms23179864
Chicago/Turabian StyleHenriques, Alexandre, Laura Rouvière, Elodie Giorla, Clémence Farrugia, Bilal El Waly, Philippe Poindron, and Noëlle Callizot. 2022. "Alpha-Synuclein: The Spark That Flames Dopaminergic Neurons, In Vitro and In Vivo Evidence" International Journal of Molecular Sciences 23, no. 17: 9864. https://doi.org/10.3390/ijms23179864
APA StyleHenriques, A., Rouvière, L., Giorla, E., Farrugia, C., El Waly, B., Poindron, P., & Callizot, N. (2022). Alpha-Synuclein: The Spark That Flames Dopaminergic Neurons, In Vitro and In Vivo Evidence. International Journal of Molecular Sciences, 23(17), 9864. https://doi.org/10.3390/ijms23179864