
Synthesis of 4-Hydroxyquinolines as Potential Cytotoxic Agents †




Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
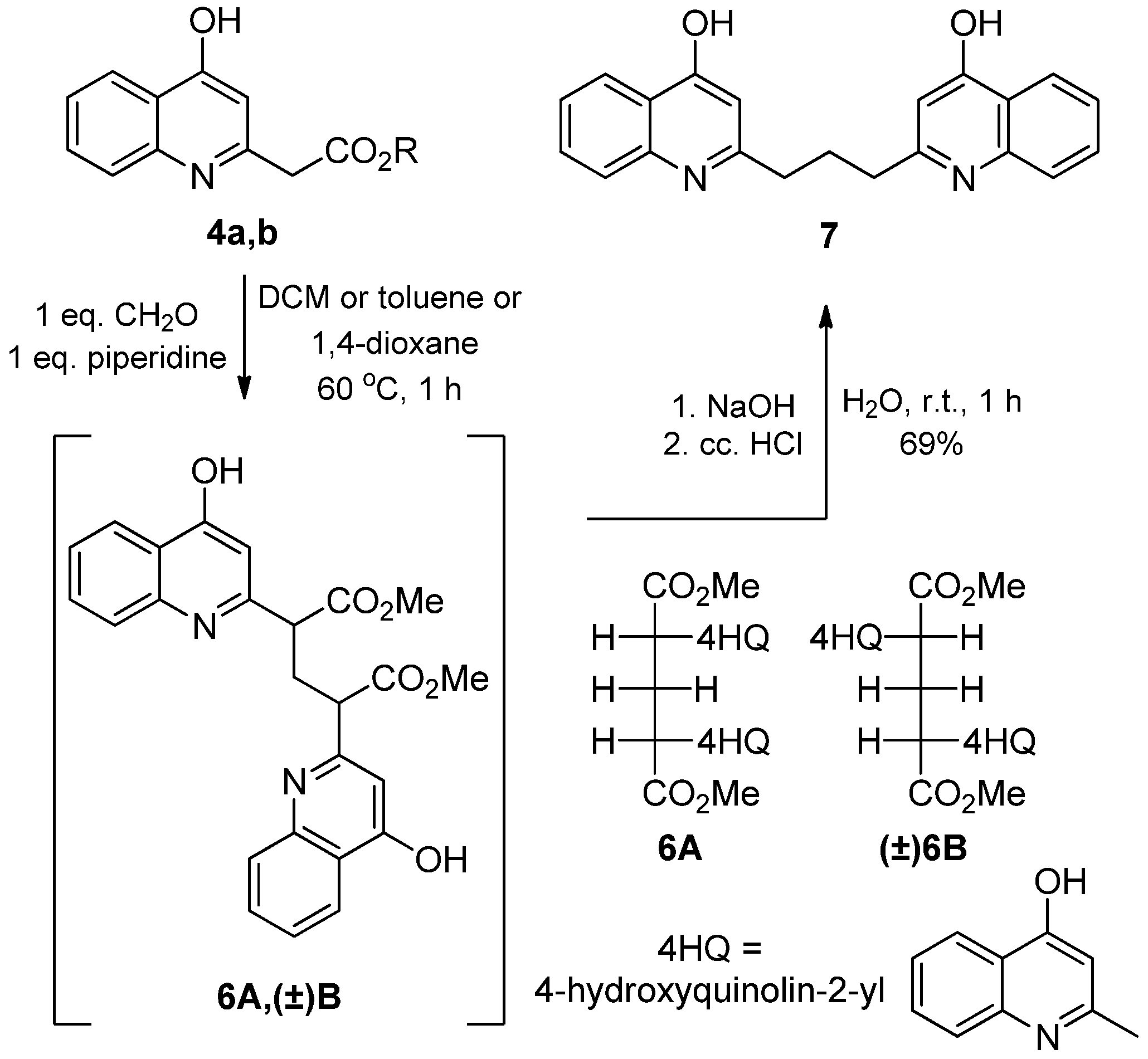
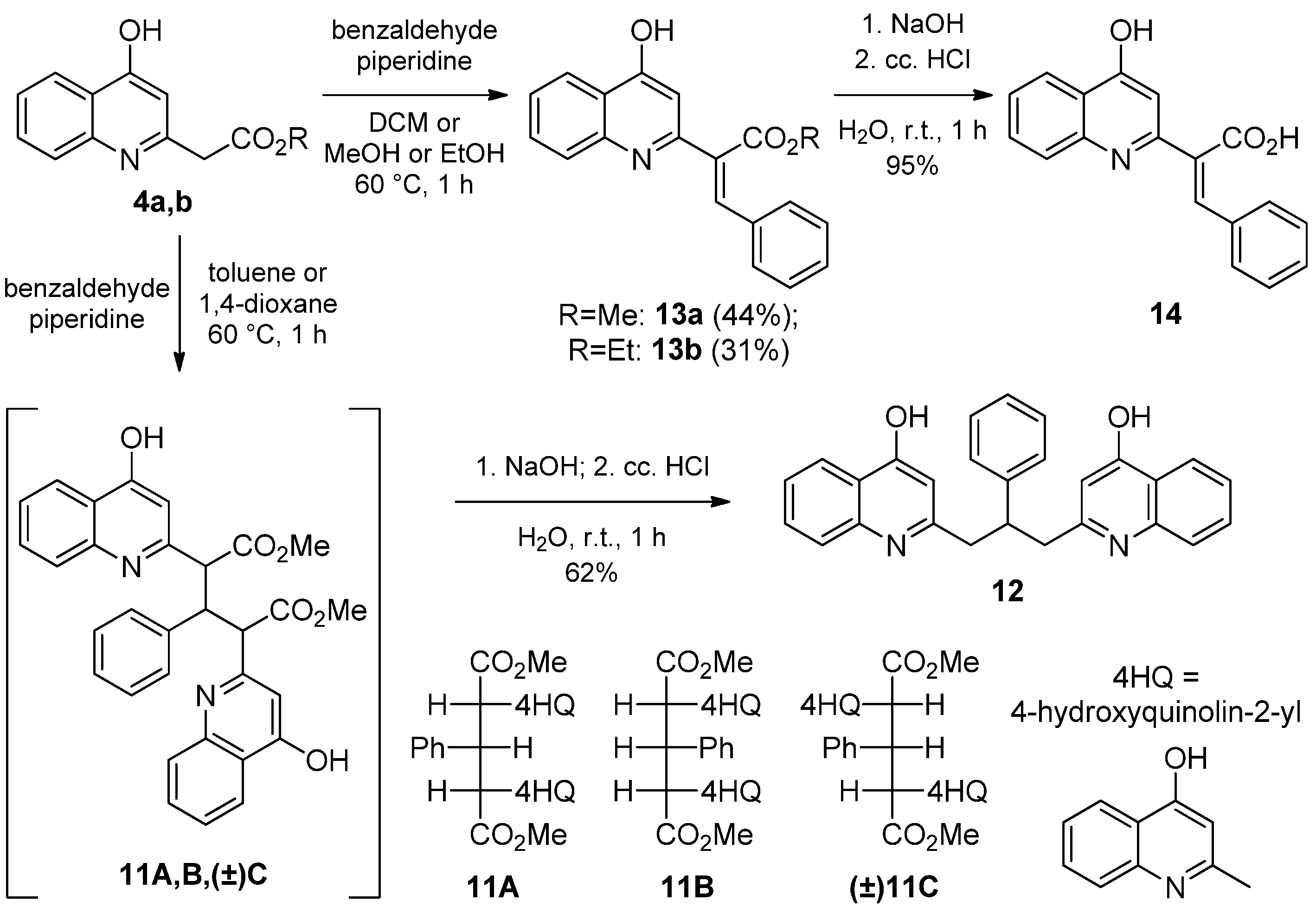
2.1.1. Syntheses of 4-Hydroxyquinolines
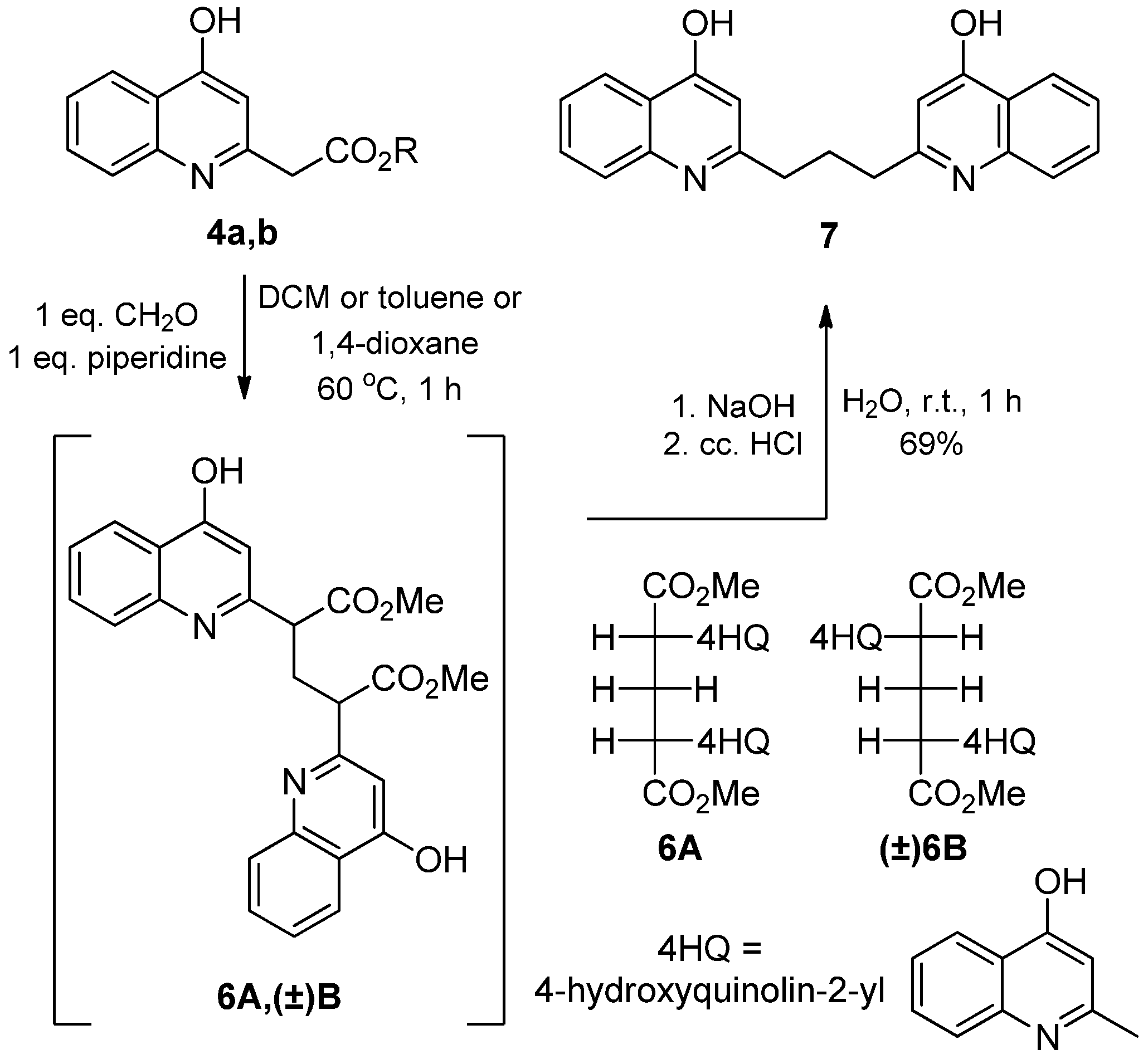
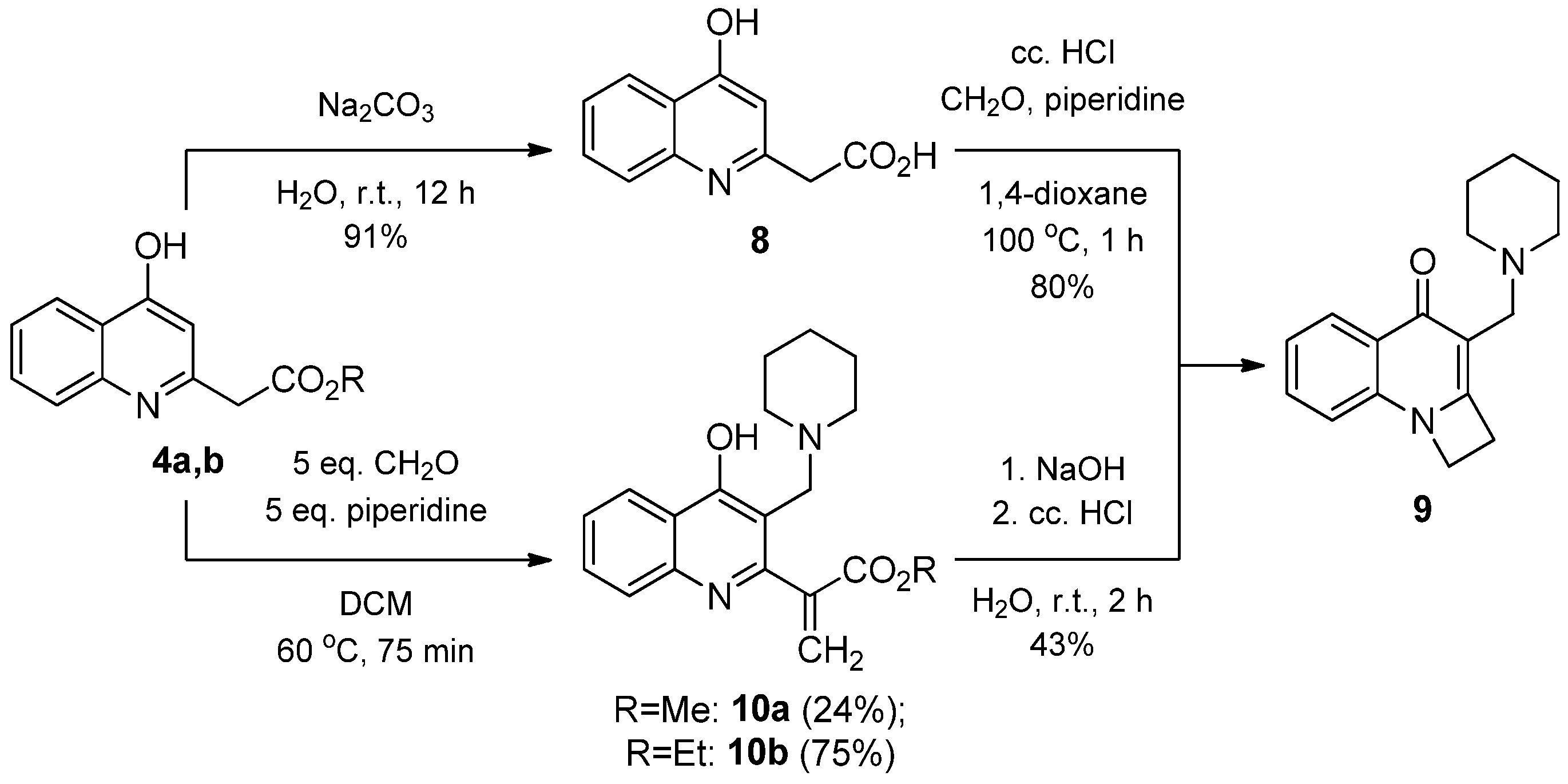
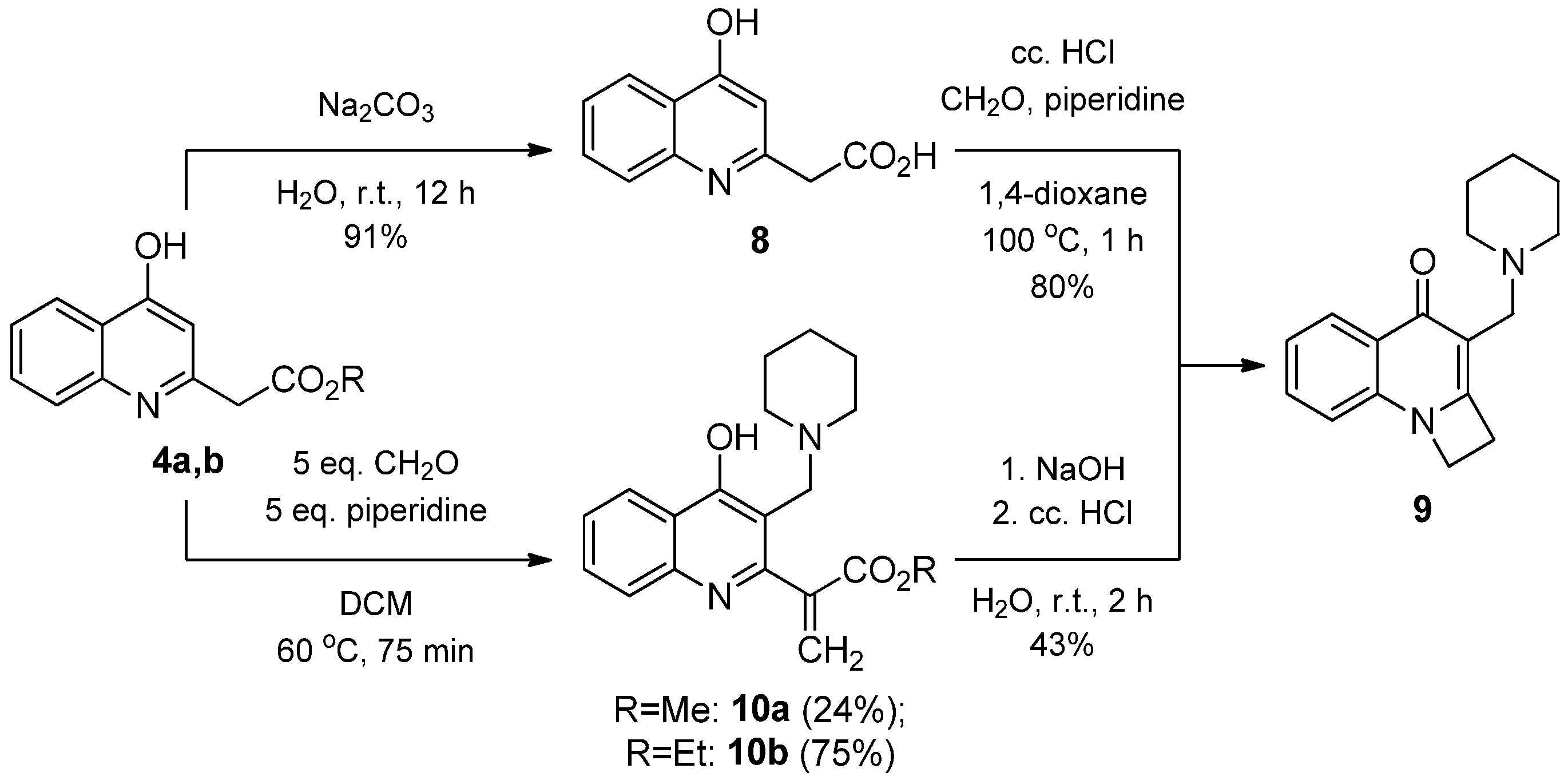
2.1.2. Reaction of 2-(4-Hydroxyquinolin-2-yl) Acetates with Paraformaldehyde and Piperidine
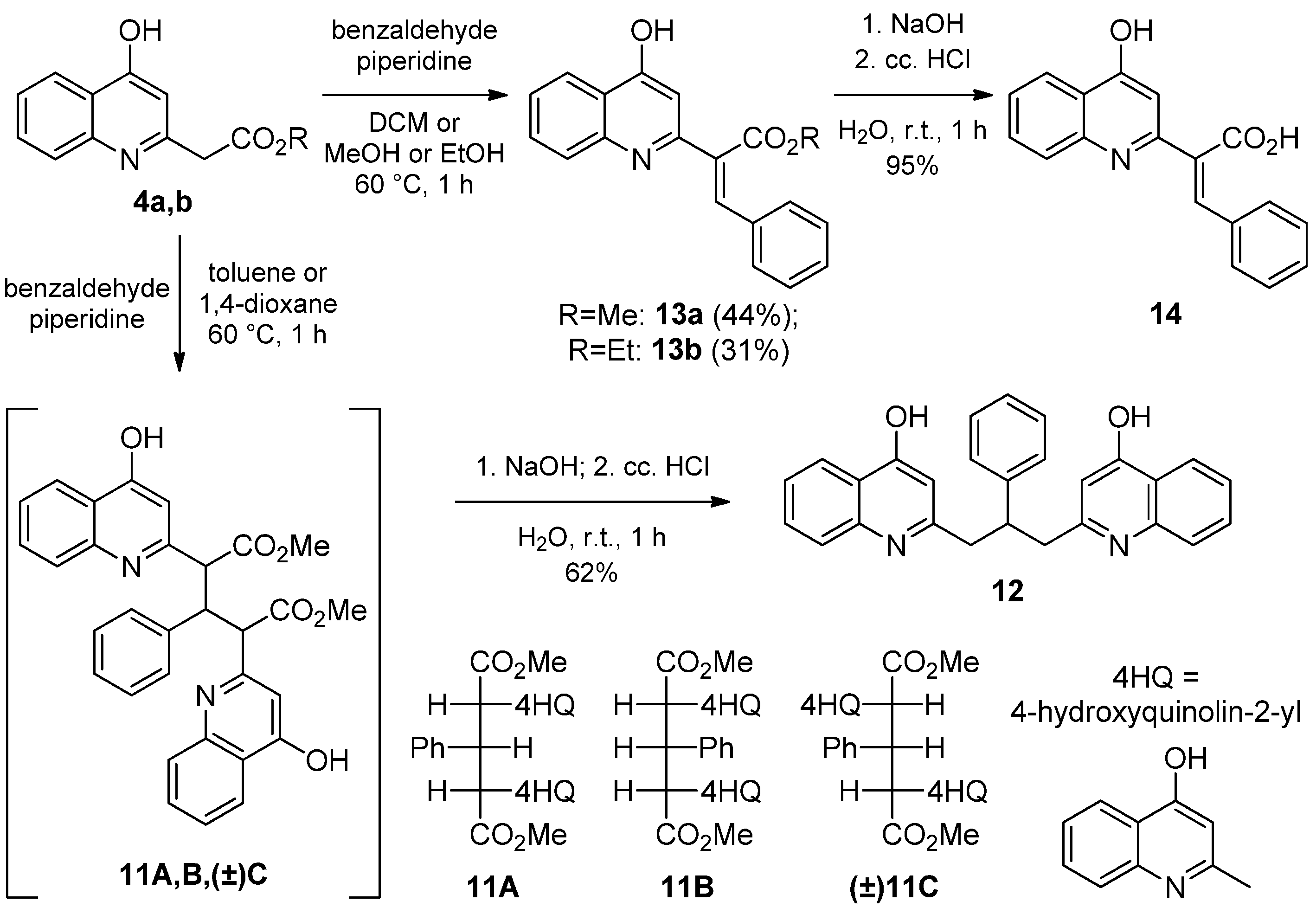
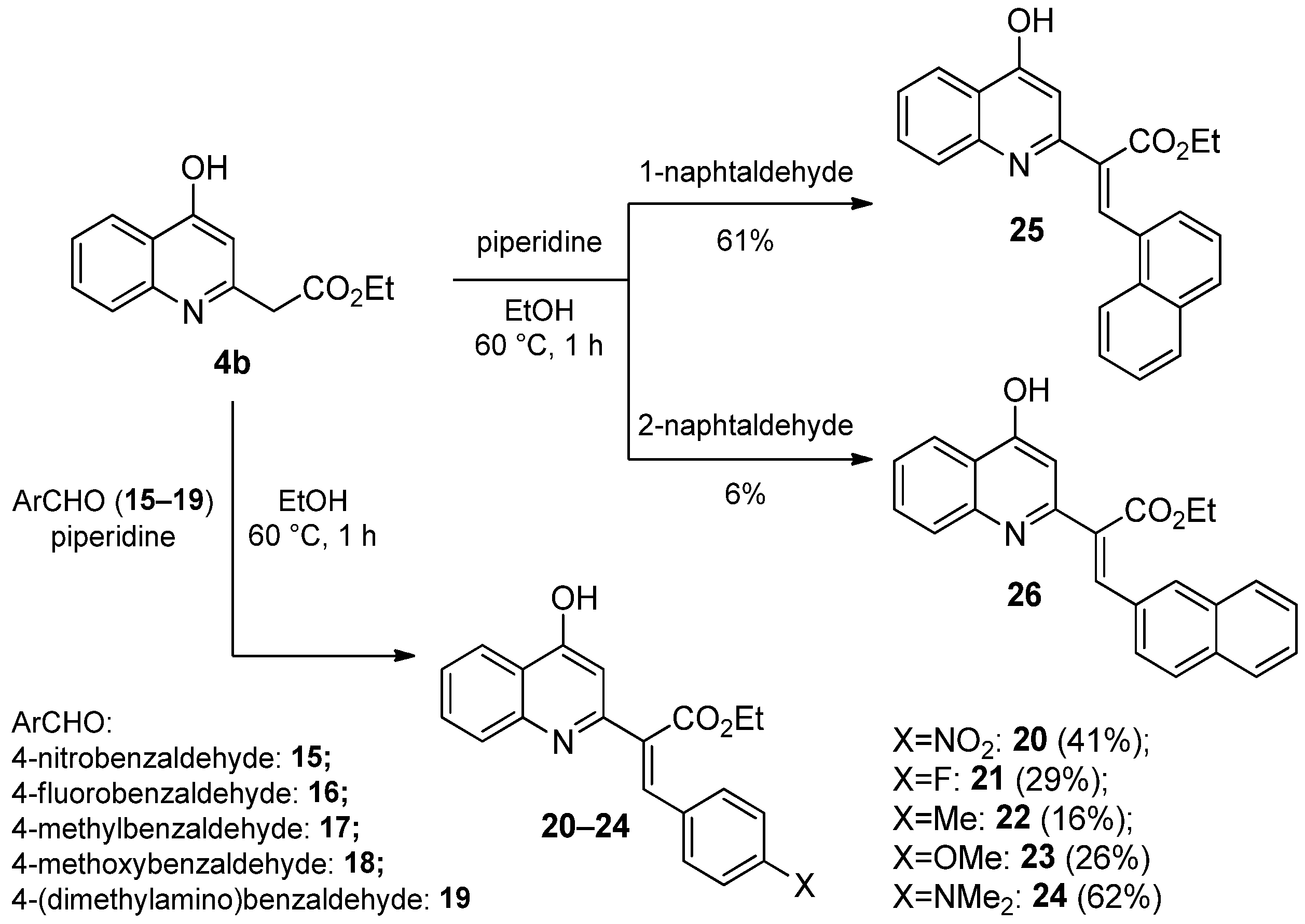
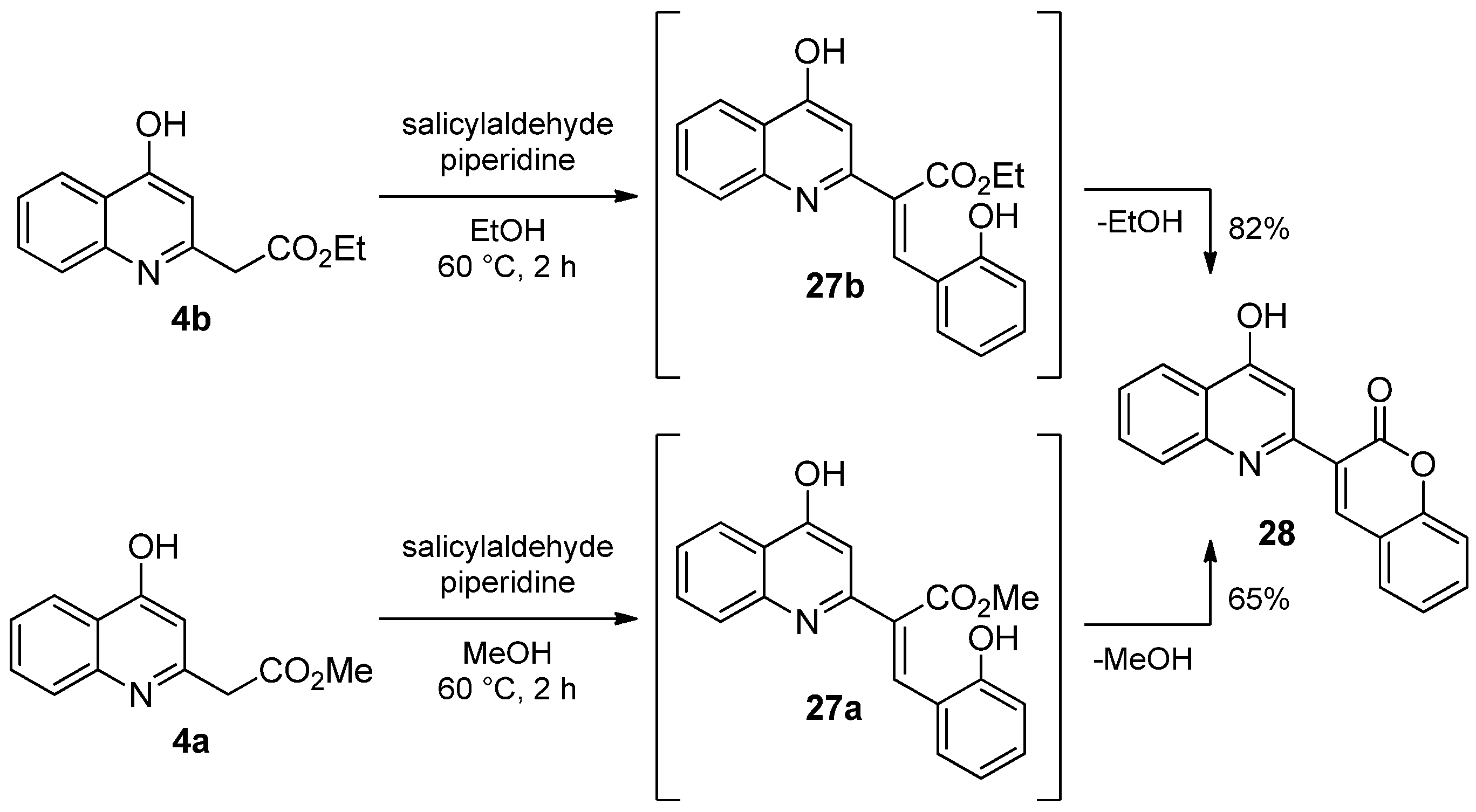
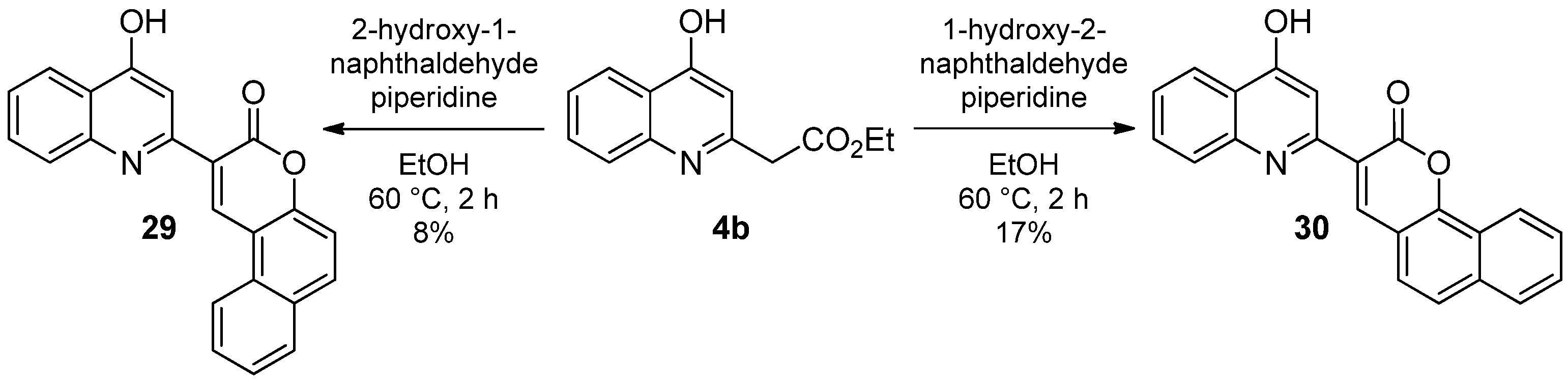
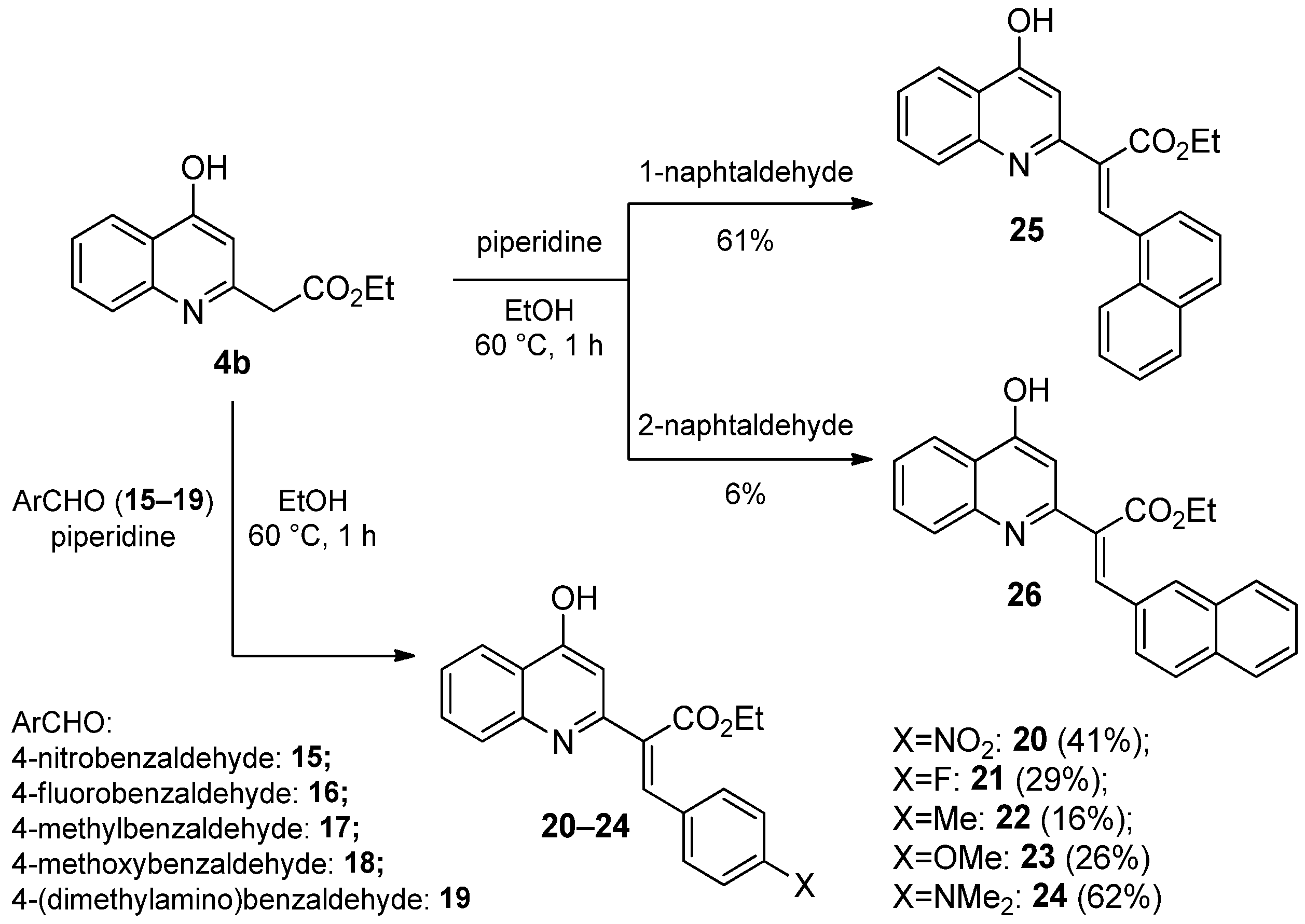
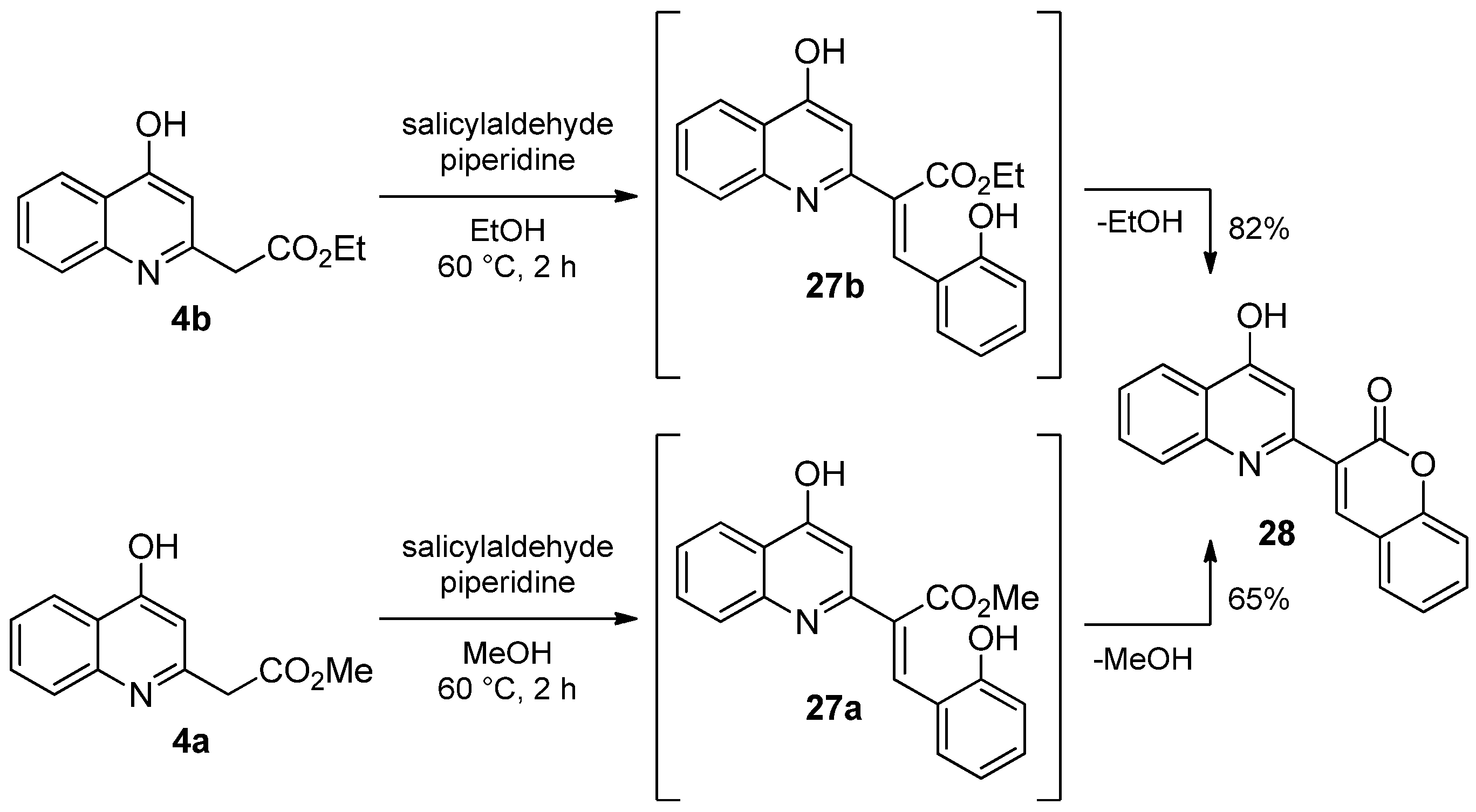
2.1.3. Reaction of 4-Hydroxyquinolines with Aromatic Aldehydes and Piperidine
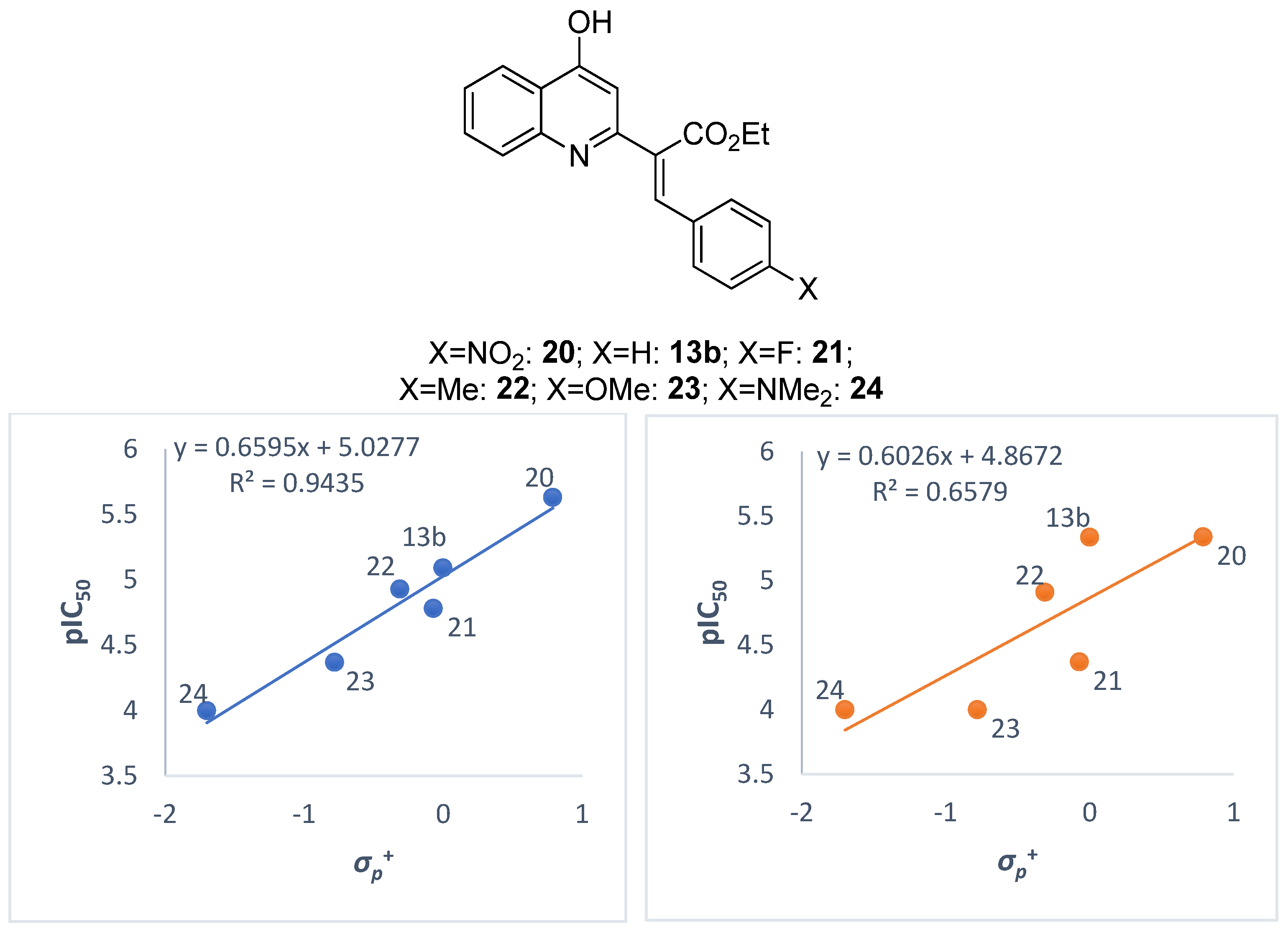
2.2. Biological Evaluations
3. Materials and Methods
3.1. Preparation Protocols for the Synthesis of the New Derivatives
3.1.1. General Procedure for the Synthesis of 4a,b and 5
3.1.2. General Procedure for the Synthesis of Bisquinoline Derivatives (7, 12)
3.1.3. Synthesis of 2-(4-Hydroxyquinolin-2-yl) Acetic Acid (8)
3.1.4. Synthesis of 3-(Piperidin-1-Ylmethyl)-1H-Azeto [1,2-a] Quinolin-4(2H)-One (9)
3.1.5. General Procedure for the Synthesis of Mannich Bases (10a,b)
3.1.6. General Procedure for the Synthesis of the Knoevenagel Derivatives 13a,b; 20–26; 28–30
3.1.7. Synthesis of (Z)-2-(4-hydroxyquinolin-2-yl)-3-phenylacrylic Acid (14)
3.2. Biological Assays
3.2.1. Cell Lines
3.2.2. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Large, M.S.; Smith, L.H. β-Adrenergic Blocking Agents. 24. Heterocyclic Substituted 1-(Aryloxy)-3-[[(amido)alkyl]amino]propan-2-ols. J. Med. Chem. 1982, 25, 1417–1422. [Google Scholar] [CrossRef]
- Baxter, A.D.; Owen, D.A.; Watson, R.J.; Hannah, D.; Batty, D. Preparation of Hydroxamic and Carboxylic Acid Derivatives for Treating Condition Associated with Matrix Metalloproteinase, ADAM or ADAM-TS Enzymes. WIPO (PCT) No. WO2000069827, 23 November 2000. [Google Scholar]
- Bisacchi, G.S. Origins of the Quinolone Class of Antibacterials: An Expanded “Discovery Story”. J. Med. Chem. 2015, 58, 4874–4882. [Google Scholar] [CrossRef]
- Giles, G.I.; Collins, C.A.; Stone, T.W.; Jacob, C. Electrochemical and in vitro evaluation of the redox-properties of kynurenine species. Biochem. Biophys. Res. Commun. 2003, 300, 719–724. [Google Scholar] [CrossRef]
- Mannich, C.; Krösche, W. About a condensation product of formaldehyde, ammonia and antipyrine. Archiv der Pharmazie 1912, 250, 647–667. [Google Scholar] [CrossRef]
- Betti, M. On the addition of benzyl amine to naphthol. Gazz. Chim. Ital. 1900, 30, 301–309. [Google Scholar]
- Betti, M. General condensation reaction between β-naphthol, aldehydes and amines. Gazz. Chim. Ital. 1900, 30, 310–316. [Google Scholar]
- Cardellicchio, C.; Capozzi, M.A.M.; Naso, F. The Betti base: The awakening of a sleeping beauty. Tetrahedron Asymmetry 2010, 21, 507–517. [Google Scholar] [CrossRef]
- Filho, J.F.A.; Lemos, B.C.; de Souza, A.S.; Pinheiro, S.; Greco, S.J. Multicomponent Mannich reactions: General aspects, methodologies and applications. Tetrahedron 2017, 73, 6977–7004. [Google Scholar] [CrossRef]
- Biersack, B.; Ahmed, K.; Padhye, S.; Schobert, R. Recent developments concerning the application of the Mannich reaction for drug design. Expert Opin. Drug. Discov. 2018, 13, 39–49. [Google Scholar] [CrossRef]
- Roman, G. Anticancer Activity of Mannich Bases: A Review of Recent Literature. ChemMedChem 2022, 17, e202200258. [Google Scholar] [CrossRef]
- Bhilare, N.V.; Marulkar, V.S.; Shirote, P.J.; Dombe, S.A.; Pise, V.J.; Salve, P.L.; Biradar, S.M.; Yadav, V.D.; Jadhav, P.D.; Bodhe, A.A.; et al. Mannich Bases: Centrality in Cytotoxic Drug Design. Med. Chem. 2022, 18, 735–756. [Google Scholar] [CrossRef]
- Szatmári, I.; Fülöp, F. Syntheses, transformations and applications of aminonaphthol derivatives prepared via modified Mannich reactions. Tetrahedron 2013, 69, 1255–1278. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, T.N.; Kundu, A.K.; Chaudhuri, A.R. Search for New Antispasmodics. Part IV. J. Indian Chem. Soc. 1952, 29, 368–370. [Google Scholar]
- Zubkov, V.O.; Benzugly, P.O.; Taran, K.A.; Kamenetskaya, O.L.; Silayeva, L.F. Synthesis and antimicrobial activity of 3-aminomethyl substituted quinolones. Visnik Farmatsii 2003, 1, 1–4. [Google Scholar]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthesis of New C-3 Substituted Kynurenic Acid Derivatives. Molecules 2020, 25, 937. [Google Scholar] [CrossRef]
- Lőrinczi, B.; Csámpai, A.; Fülöp, F.; Szatmári, I. Synthetic- and DFT modelling studies on regioselective modified Mannich reactions of hydroxy-KYNA derivatives. RSC Adv. 2021, 11, 543–554. [Google Scholar] [CrossRef]
- Kaslow, C.E.; Nix, S.J. Substituted Quinolineacetic Acids. J. Org. Chem. 1951, 16, 895–898. [Google Scholar] [CrossRef]
- Munavalli, S.N.; Kulkarni, S.N.; Nargund, K.S. Use of ethyl acetonedicarboxylate in the Conrad–Limpach reaction. Preparation of substituted ethyl 4-hydroxyquinoline-2-acetates. J. Karnatak Univ. 1956, 1, 23–28. [Google Scholar]
- Shilabin, A.G.; Dzhekieva, L.; Misra, P.; Jayaram, B.; Pratt, R.F. 4-Quinolones as Noncovalent Inhibitors of High Molecular Mass Penicillin-Binding Proteins. ACS Med. Chem. Lett. 2012, 9, 592–595. [Google Scholar] [CrossRef]
- Rahn, T.; Appel, B.; Baumann, W.; Jiao, H.; Börner, A.; Fischer, C.; Langer, P. Synthesis of chromones and 4-hydroxyquinolines based on uncatalyzed condensations of 1-methoxy-1,3-bis(trimethylsilyloxy)-1,3-butadiene with 2-alkoxy- and 2-nitrobenzoyl chlorides and related reactions. Org. Biomol. Chem. 2009, 7, 1931–1938. [Google Scholar] [CrossRef]
- Mee, J.D. 1-Phenylpiperidine-2,4,6-Trione. J. Org. Chem. 1975, 40, 2135–2136. [Google Scholar] [CrossRef]
- Nadaraj, V.; Selvi, S.T. Synthesis of quinoline derivatives by acid catalyzed microwave-assisted approach. Org. Chem. Indian J. 2010, 6, 108–111. [Google Scholar]
- Tatemitsu, H.; Ogura, F.; Nakagawa, Y.; Nakagawa, M.; Naemura, K.; Nakazaki, M. Optical Activity and Absolute Configuration of 2,3:6,7-Dibenzobicyclo [3.3.1]nona-2,6-diene Derivatives. Bull. Chem. Soc. Jpn. 1975, 48, 2473–2483. [Google Scholar] [CrossRef]
- Valdéz-Camacho, J.R.; Rivera-Ramírez, J.D.; Escalante, J. Studies and Mechanism of Olefination Reaction in Aryl-Enolates with Paraformaldehyde. Int. J. Org. Chem. 2019, 9, 10–22. [Google Scholar] [CrossRef]
- Ayad, F.; Tilley, L.; Deady, L.W. Synthesis, Antimalarial Activity and Inhibition of Haem Detoxification of Novel Bisquinolines. Bioorg. Med. Chem. Lett. 2001, 11, 2075–2077. [Google Scholar] [CrossRef]
- Obora, Y.; Anno, Y.; Okamoto, R.; Matsu-ura, T.; Ishii, Y. Iridium-Catalyzed Reactions of ω-Arylalkanols to α,ω-Diarylalkanes. Angew. Chem. Int. Ed. 2011, 50, 8618–8622. [Google Scholar] [CrossRef]
- Chen, J.; Deady, L.W. Synthesis of Some Quinolizine Derivatives. Tetrahedron 1998, 54, 2785–2794. [Google Scholar] [CrossRef]
- Musiol, R. Styrylquinoline—A Versatile Scaffold in Medicinal Chemistry. Med. Chem. 2020, 16, 141–154. [Google Scholar] [CrossRef]
- Bahner, C.T. Effect of compounds related to 4-(p-dimethylaminostyryl) quinolone methiodide on lymphoma 8. Cancer Res. 1955, 15, 588–592. [Google Scholar]
- Bahner, C.T.; Pace, E.S.; Prevost, R. Quaternary Salts of Styryl Pyridines and Quinolines. J. Am. Chem. Soc. 1951, 73, 3407–3408. [Google Scholar] [CrossRef]
- Mrozek-Wilczkiewicz, A.; Kuczak, M.; Malarz, K.; Cieślik, W.; Spaczyńska, E.; Musiol, R. The synthesis and anticancer activity of 2-styrylquinoline derivatives. A P53 independent mechanism of action. Eur. J. Med. Chem. 2019, 177, 338–349. [Google Scholar] [CrossRef]
- Podeszwa, B.; Niedbala, H.; Polanski, J.; Musiol, R.; Tabak, D.; Finster, J.; Serafin, K.; Milczarek, M.; Wietrzyk, J.; Boryczka, S.; et al. Investigating the antiproliferative activity of quinoline-5,8-diones and styrylquinolinecarboxylic acids on tumor cell lines. Bioorg. Med. Chem. Lett. 2007, 17, 6138–6141. [Google Scholar] [CrossRef]
- Shablykina, O.V.; Khilya, O.V.; Ishchenko, V.V.; Khilya, V.P. 3-(2-Quinolyl)- and 3-(5-carbethoxyfuryl-2)coumarins. Chem. Nat. Compd. 2005, 41, 529–532. [Google Scholar] [CrossRef]
- Sankaran, M.; Chandraprakash, K.; Uvarani, C.; Vennila, K.N.; Velmurugan, D.; Mohan, P.S. A Convenient Method of Facilitating Aryl–Aryl Bond-Formation Reactions in the Synthesis of Biquinoline- and Quinoline-Bearing Chromene Derivatives. Synlett 2012, 23, 2858–2864. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, X.-R.; Sun, R.; Xu, Y.-J.; Ge, J.-F. The fluorescent biomarkers for lipid droplets with quinolone-coumarin unit. Org. Biomol. Chem. 2018, 16, 7619–7625. [Google Scholar] [CrossRef]
- Hande, P.E.; Mishra, M.; Ali, F.; Kapoor, S.; Datta, A.; Gharpure, S.J. Design and Expeditious Synthesis of Quinoline-Pyrene-Based Ratiometric Fluorescent Probes for Targeting Lysosomal pH. ChemBioChem 2020, 21, 1492–1498. [Google Scholar] [CrossRef]
- Gajdács, M.; Spengler, G.; Sanmartín, C.; Marć, M.A.; Handzlik, J.; Domínguez-Álvarez, E. Selenoesters and selenoanhydrides as novel multidrug resistance reversing agents: A confirmation study in a colon cancer MDR cell line. Bioorg. Med. Chem. Lett. 2017, 27, 797–802. [Google Scholar] [CrossRef] [Green Version]
- Hansch, C.; Leo, A.; Taft, R.W. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Ackroyd, J.; Scheinmann, F. A New Route to 1H-1,5-Benzodiazepinones. J. Chem. Soc. Chem. Commun. 1981, 339–340. [Google Scholar] [CrossRef]
- Balkenhohl, F.; Bohnenpoll, M.; Winterfeldt, E. Cyclopentenone Derivatives, XIV. Stereoselective Formation of Prostacyclin Intermediates. Chemische Berichte 1989, 122, 797–800. [Google Scholar] [CrossRef]
- Troin, Y.; Sinibaldi, M.E.; Gramain, J.C.; Rubiralta, M.; Diez, A. Preparation of Spiroindolines as Potential Intermediates in Aspidosperma Alkaloid Synthesis. Tetrahedron Lett. 1991, 32, 6129–6132. [Google Scholar] [CrossRef]
- Charvát, T.; Potáček, M.; Humpa, O.; Marek, J. Isomerization of N-Aryl Substituted Diethyl 3-Aminoglutaconates. Part I. J. Mol. Struct. 1996, 377, 271–276. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Time (min) | Temperature (°C) | Yield of 4a/4b (%) | Yield of 5 (%) |
|---|---|---|---|---|
| 4a | 20 | 240 | 28 | 0 |
| 20 | 245 | 31 | 0 | |
| 20 | 250 | 27 | 0 | |
| 20 | 245 | 0 | 53 1 | |
| 4b | 20 | 220 | 7 | 0.5 |
| 20 | 230 | 16 | 0.6 | |
| 20 | 235 | 28 | 0.3 | |
| 20 | 240 | 35 | 0.1 | |
| 20 | 245 | 41 | 0 | |
| 20 | 250 | 35 | 0 | |
| 15 | 245 | 37 | 0 | |
| 30 | 245 | 36 | 0 |
| Compound | Colo 205 IC50 (µM) | Colo 320 IC50 (µM) | MRC-5 IC50 (µM) | |||
|---|---|---|---|---|---|---|
| Mean | SD (+/−) | Mean | SD (+/−) | Mean | SD (+/−) | |
| 4a | >100 | − | 92.78 | 1.68 | >100 | − |
| 4b | >100 | − | >100 | − | >100 | − |
| 5 | >100 | − | >100 | − | >100 | − |
| 7 | >100 | − | >100 | − | >100 | − |
| 8 | >100 | − | >100 | − | >100 | − |
| 9 | >100 | − | >100 | − | >100 | − |
| 10a | >100 | − | >100 | − | >100 | − |
| 10b | >100 | − | >100 | − | >100 | − |
| 12 | >100 | − | >100 | − | >100 | − |
| 13a | 11.86 | 1.07 | 8.19 | 1.35 | 28.56 | 1.11 |
| 13b | 8.10 | 0.11 | 4.58 | 0.18 | 18.94 | 1.83 |
| 14 | >100 | − | >100 | − | >100 | − |
| 20 | 2.34 | 0.24 | 4.61 | 0.24 | 9.89 | 0.00 |
| 21 | 16.54 | 1.97 | 42.65 | 2.26 | 21.94 | 0.82 |
| 22 | 11.79 | 0.27 | 12.29 | 0.55 | 30.64 | 0.64 |
| 23 | 42.76 | 2.35 | >100 | − | 65.18 | 1.13 |
| 24 | >100 | − | >100 | − | >100 | − |
| 25 | >100 | − | 32.40 | 3.37 | 36.54 | 1.01 |
| 26 | 12.63 | 0.49 | 11.00 | 0.37 | 17.58 | 0.27 |
| 28 | >100 | − | 14.08 | 0.35 | 6.52 | 0.46 |
| 29 | >100 | − | 9.86 | 0.90 | >100 | − |
| 30 | >100 | − | >100 | − | >100 | − |
| DOXO | 2.30 | 0.12 | 3.61 | 0.34 | >10 | − |
| Compound | SI (Selectivity Index) | |
|---|---|---|
| MRC-5/Colo 205 | MRC-5/Colo 320 | |
| 4a | - | - |
| 4b | - | - |
| 5 | - | - |
| 7 | - | - |
| 8 | - | - |
| 9 | - | - |
| 10a | - | - |
| 10b | - | - |
| 12 | - | - |
| 13a | 2.41 | 3.49 |
| 13b | 2.34 | 4.14 |
| 14 | - | - |
| 20 | 4.23 | 2.15 |
| 21 | 1.33 | 0.51 |
| 22 | 2.60 | 2.49 |
| 23 | 1.52 | - |
| 24 | - | - |
| 25 | - | 1.13 |
| 26 | 1.39 | 1.60 |
| 28 | - | 0.46 |
| 29 | - | >6 |
| 30 | - | - |
| Compound | X | σp+ | Colo 205 pIC50 | Colo 320 pIC50 |
|---|---|---|---|---|
| 20 | NO2 | 0.79 | 5.63 | 5.34 |
| 13b | H | 0.00 | 5.09 | 5.34 |
| 21 | F | –0.07 | 4.78 | 4.37 |
| 22 | Me | –0.31 | 4.93 | 4.91 |
| 23 | OMe | –0.78 | 4.37 | <4.00 |
| 24 | NMe2 | –1.70 | <4.00 | <4.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csuvik, O.; Szemerédi, N.; Spengler, G.; Szatmári, I. Synthesis of 4-Hydroxyquinolines as Potential Cytotoxic Agents. Int. J. Mol. Sci. 2022, 23, 9688. https://doi.org/10.3390/ijms23179688
Csuvik O, Szemerédi N, Spengler G, Szatmári I. Synthesis of 4-Hydroxyquinolines as Potential Cytotoxic Agents. International Journal of Molecular Sciences. 2022; 23(17):9688. https://doi.org/10.3390/ijms23179688
Chicago/Turabian StyleCsuvik, Oszkár, Nikoletta Szemerédi, Gabriella Spengler, and István Szatmári. 2022. "Synthesis of 4-Hydroxyquinolines as Potential Cytotoxic Agents" International Journal of Molecular Sciences 23, no. 17: 9688. https://doi.org/10.3390/ijms23179688
APA StyleCsuvik, O., Szemerédi, N., Spengler, G., & Szatmári, I. (2022). Synthesis of 4-Hydroxyquinolines as Potential Cytotoxic Agents. International Journal of Molecular Sciences, 23(17), 9688. https://doi.org/10.3390/ijms23179688