
Combined Treatment with Curcumin and Ferulic Acid Suppressed the Aβ-Induced Neurotoxicity More than Curcumin and Ferulic Acid Alone

 ,
, 
Abstract
:1. Introduction
2. Results
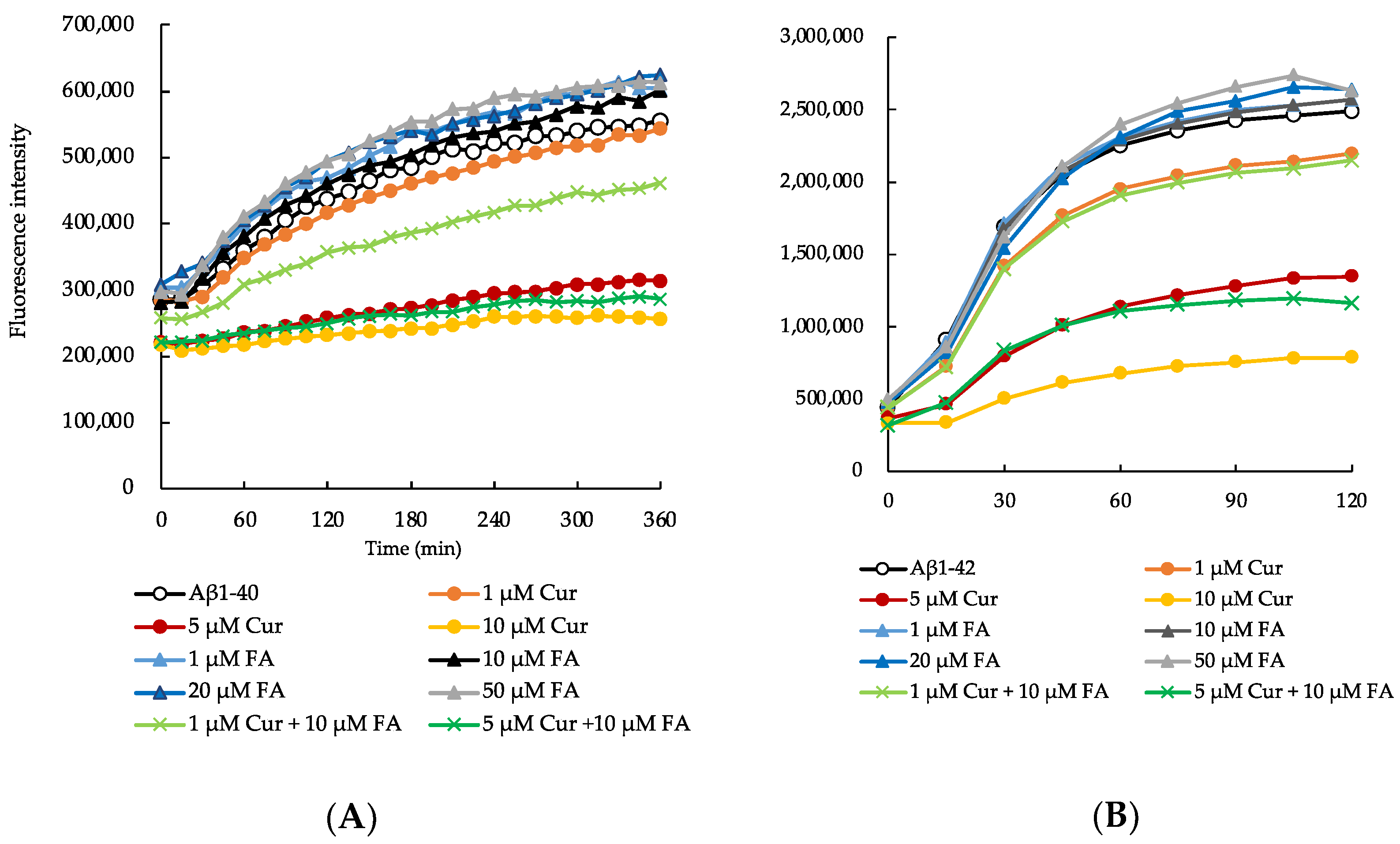
2.1. Effects of Cur, FA, and Their Combination on the Aggregation of Aβ1–40 and Aβ1–42
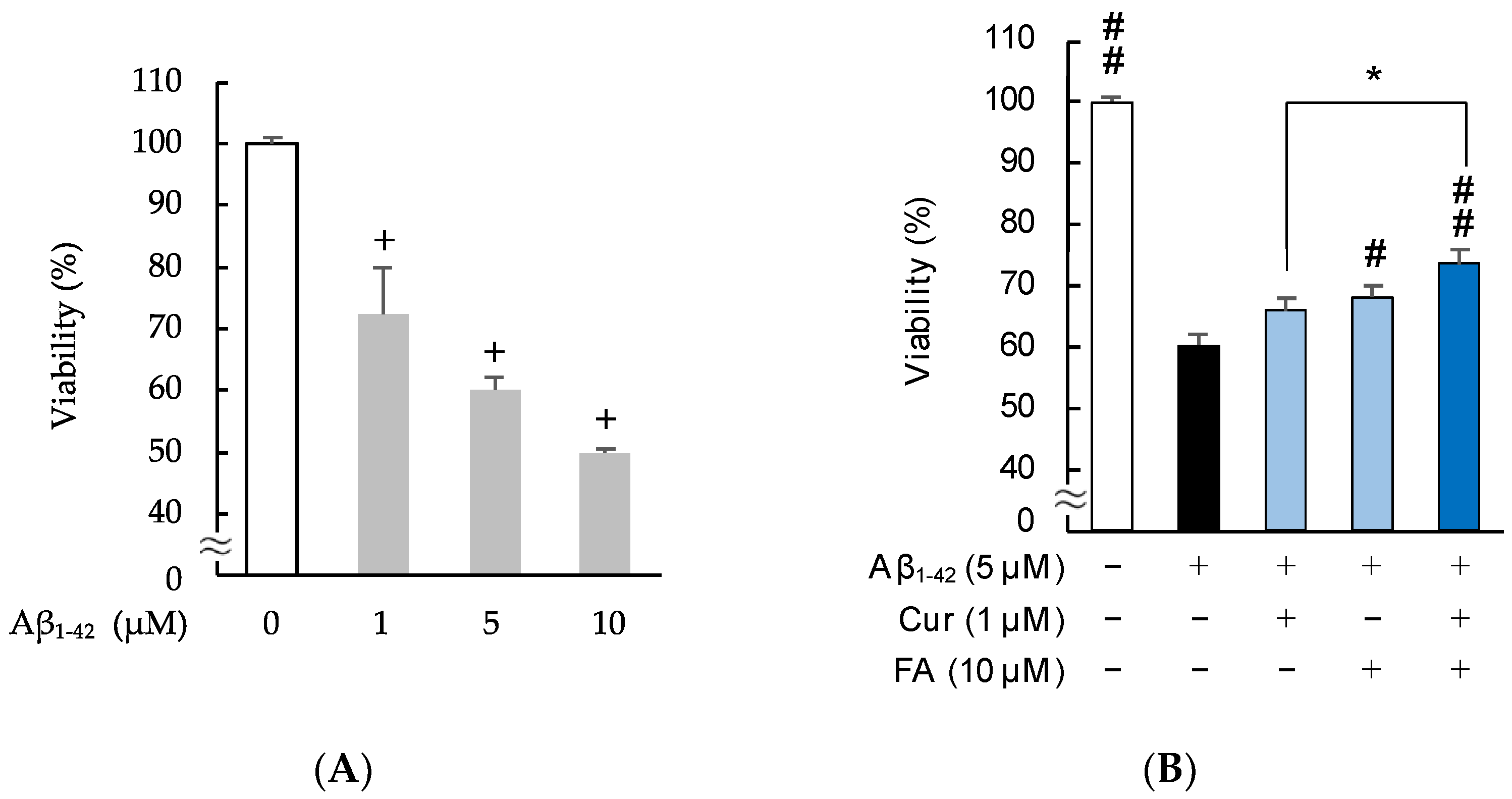
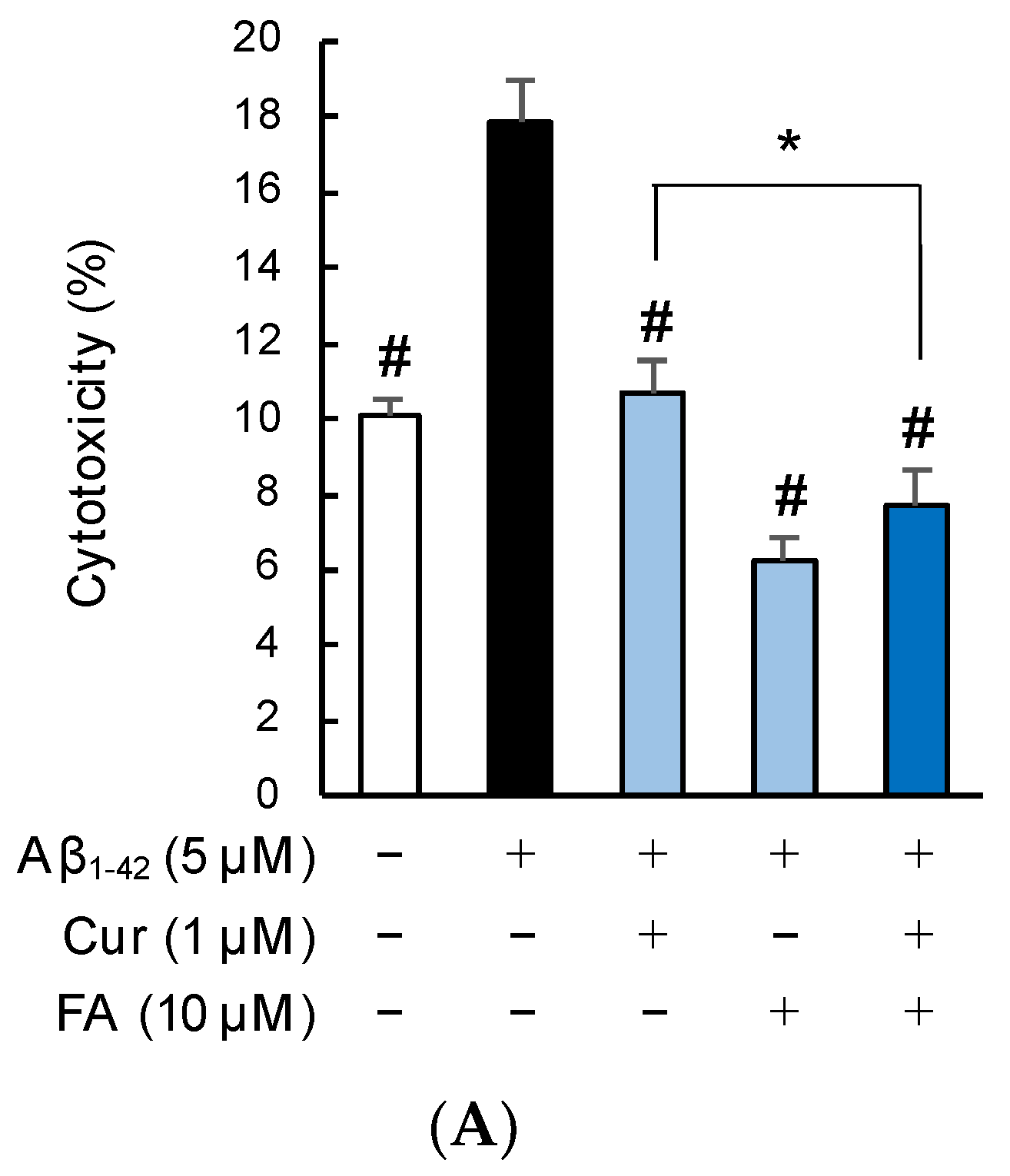
2.2. Effects of Cur, FA, and Their Combination on Viability and Neurotoxicity in Aβ1–42-Exposed Cells
2.2.1. Changes in Viability Assessed with MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide)
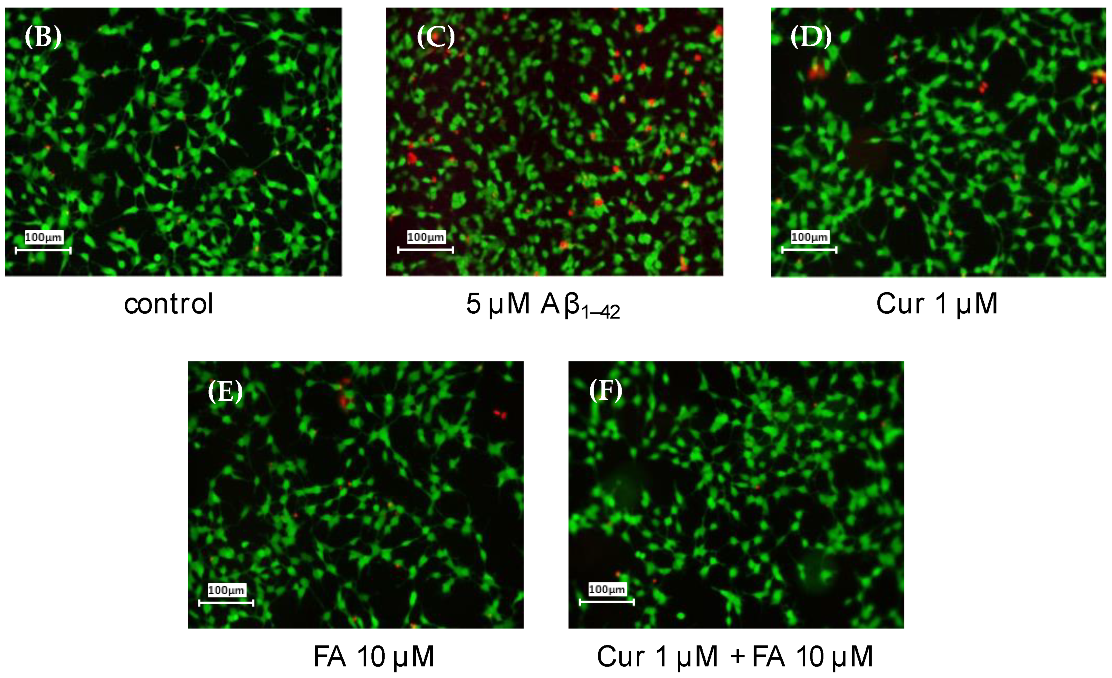
2.2.2. Staining with Calcein-AM/Ethidium Homodimer-1 (EthD-1)
2.3. Oxidative Stress
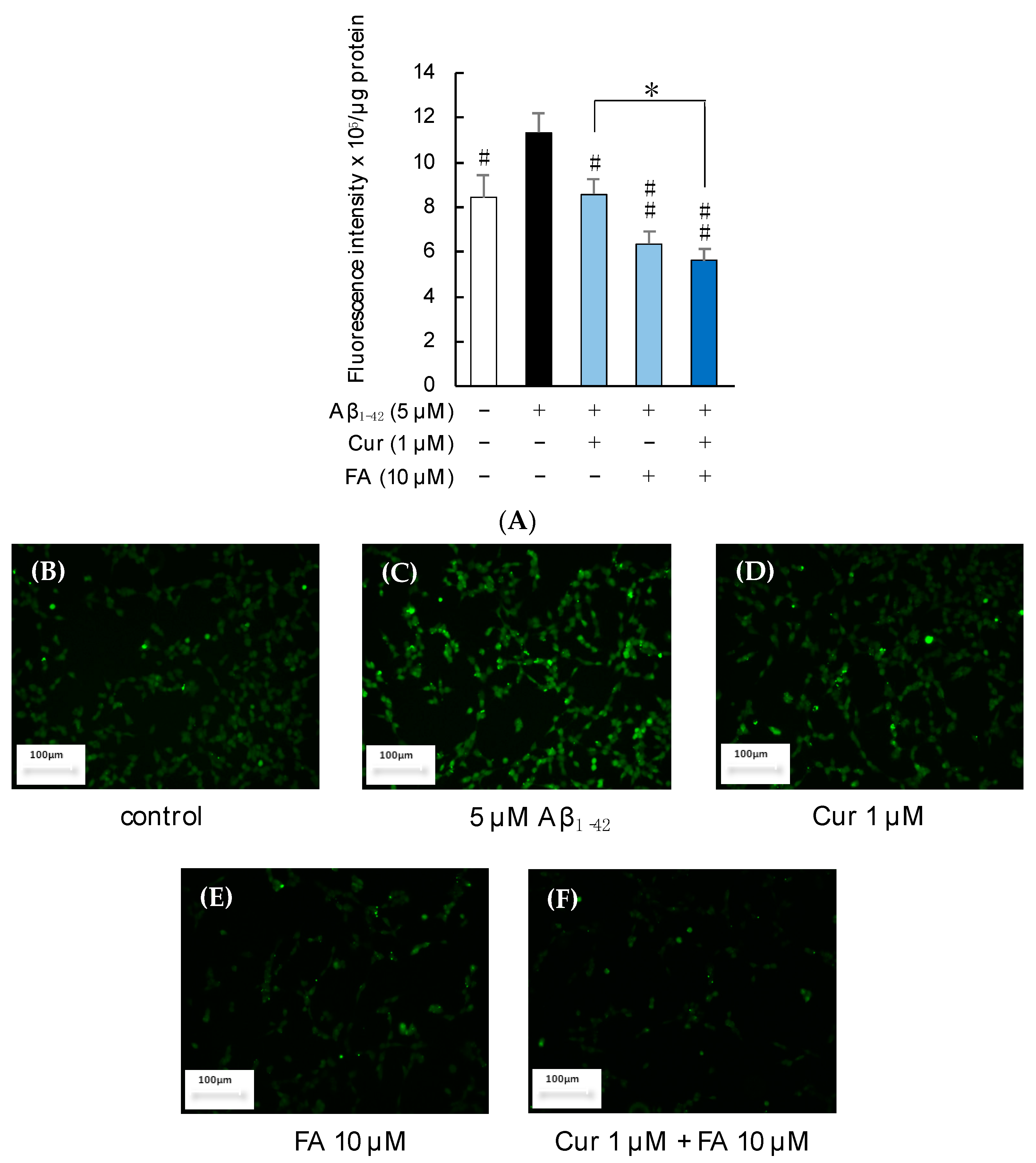
2.3.1. ROS Production
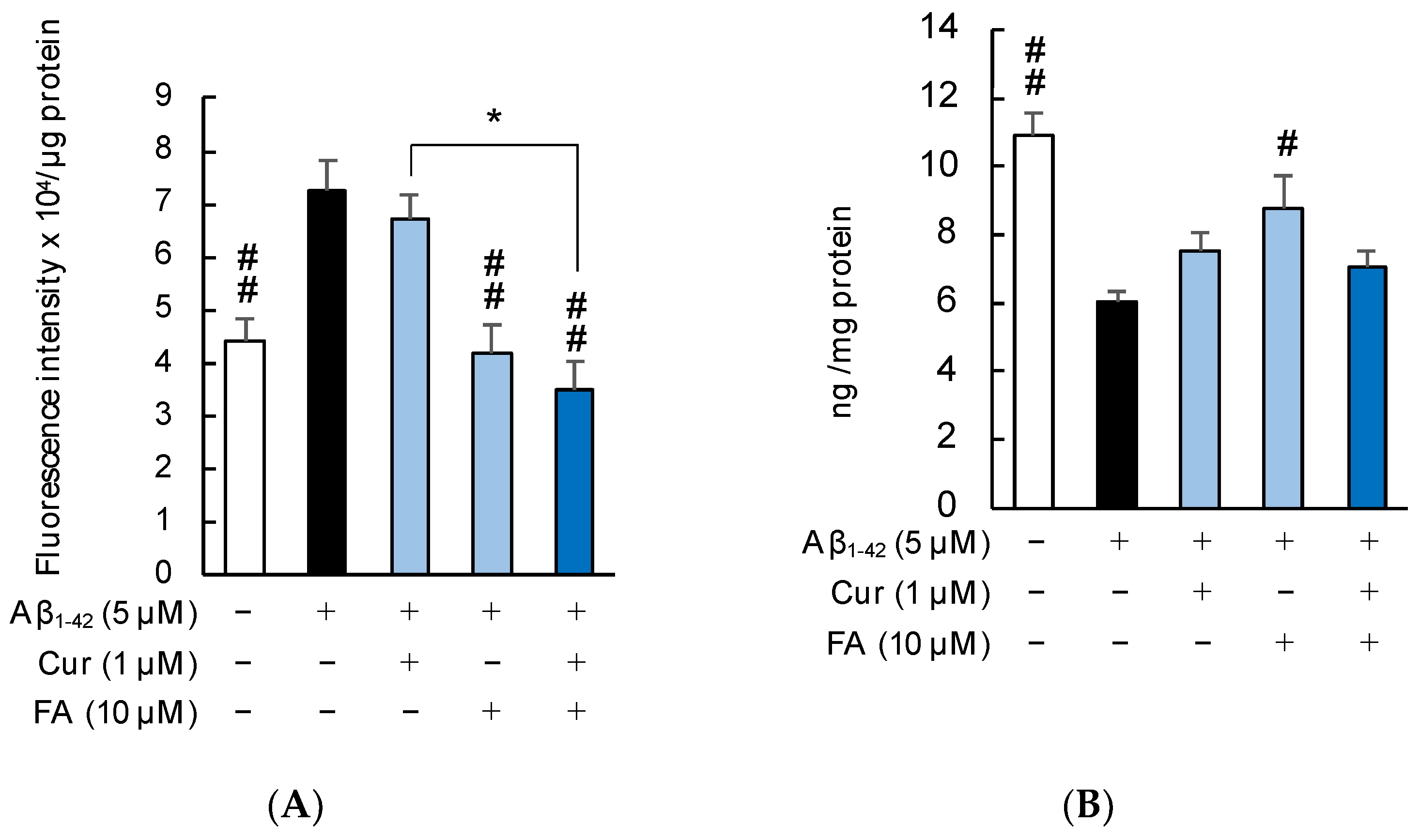
2.3.2. Mitochondrial ROS Production and Manganese Superoxide Dismutase (Mn-SOD) Levels
2.4. Effects of Cur or FA and Their Combination on Aβ1–42-Induced Disruption of Membrane Integrity
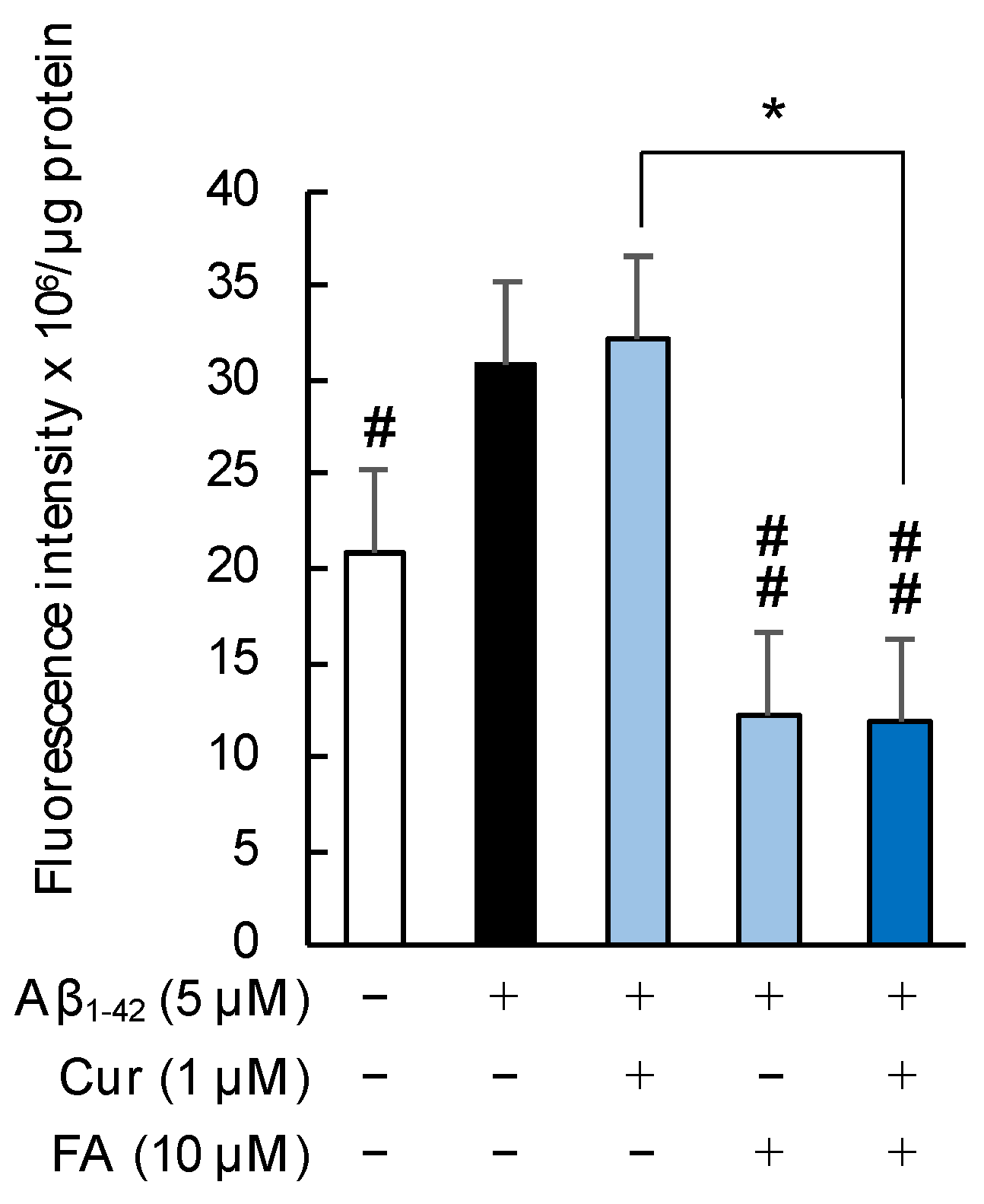
2.4.1. Fluidity of the Cell Membrane
2.4.2. Phospholipid Peroxidation in the Cell Membrane
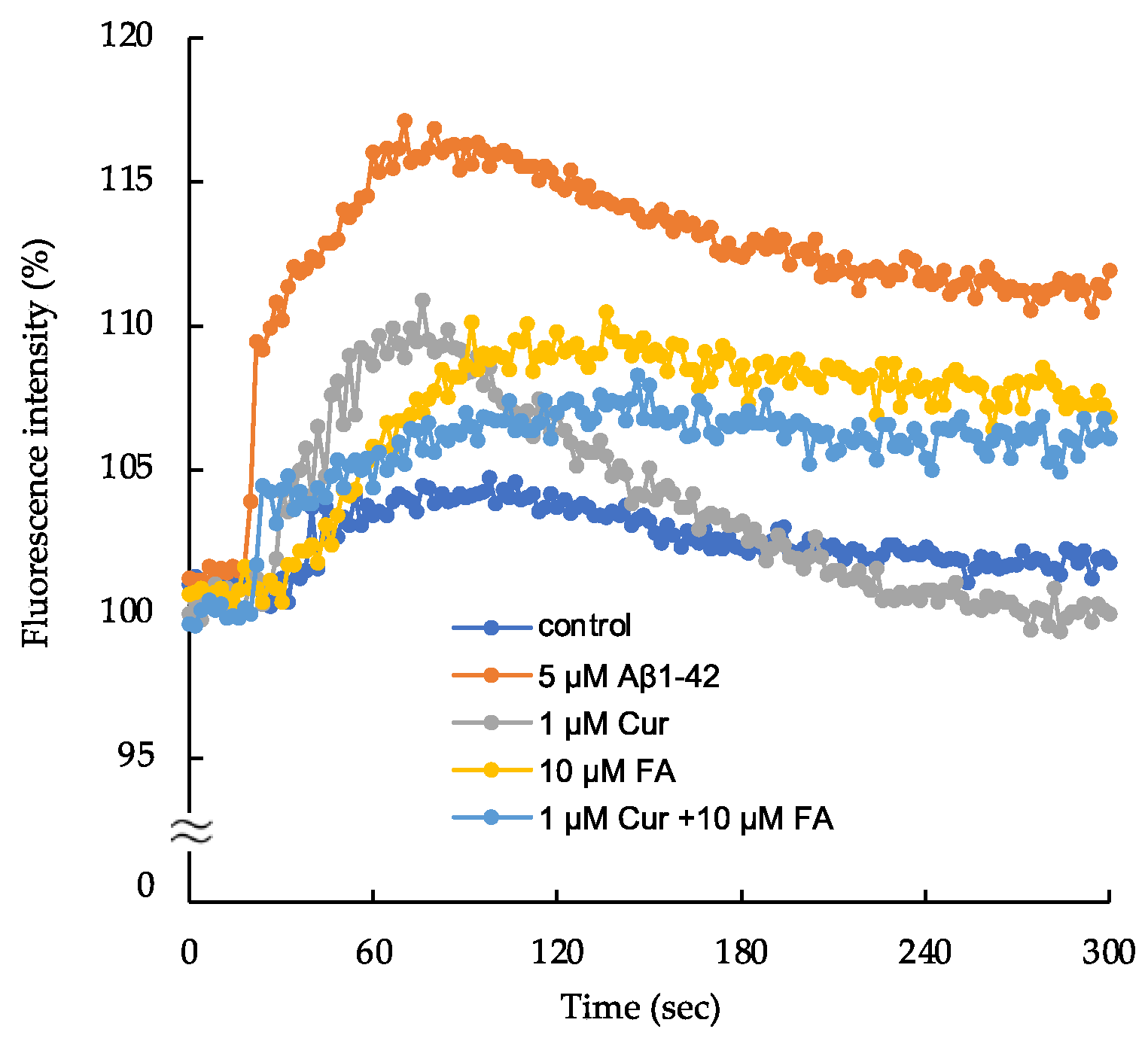
2.5. Changes in Intracellular Calcium ([Ca2+]i) following Treatment with Cur, FA, or a Combination of Both
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. The Aggregation Kinetics of Aβ1–40 and Aβ1–42
4.3. Cell Culture and Drug Treatment
4.4. Detection of Viability and Cytotoxicity in SH-SY5Y Cells
4.4.1. Cell Viability Assay
4.4.2. Calcein-AM and EthD-1 (Live/Dead) Cell Assay
4.5. Assay of Oxidative Stress
4.5.1. Reactive Oxygen Species (ROS) Detection
4.5.2. Mitochondrial ROS Assay
4.5.3. Detection of Manganese-Superoxide Dismutase (Mn-SOD)
4.6. Reaction to Cell Membrane
4.6.1. The Fluidity of Cell Membrane
4.6.2. Assay of Phospholipid Peroxidation in Cell Membranes
4.7. Detect of Changes in [Ca2+]i
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Ono, K.; Watanabe–Nakayama, T. Aggregation and structure of amyloid β–protein. Neurochem. Int. 2021, 151, 105208. [Google Scholar] [CrossRef]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini–Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef]
- Yamada, M.; Ono, K.; Hamaguchi, T.; Noguchi–Shinohara, M. Natural Phenolic Compounds as Therapeutic and Preventive Agents for Cerebral Amyloidosis. Adv. Exp. Med. Biol. 2015, 863, 79–94. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ono, K.; Murase, A.; Yamada, M. Phenolic compounds prevent Alzheimer’s pathology through different effects on the amyloid–beta aggregation pathway. Am. J. Pathol. 2009, 175, 2557–2565. [Google Scholar] [CrossRef]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti–amyloidogenic effects for Alzheimer’s beta–amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Brondino, N.; Re, S.; Boldrini, A.; Cuccomarino, A.; Lanati, N.; Barale, F.; Politi, P. Curcumin as a therapeutic agent in dementia: A mini systematic review of human studies. Sci. World J. 2014, 2014, 174282. [Google Scholar] [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Terao, J.; Karasawa, H.; Arai, H.; Nagao, A.; Suzuki, T.; Takama, K. Peroxyl Radical Scavenging Activity of Caffeic Acid and Its Related Phenolic Compounds in Solution. Biosci. Biotechnol. Biochem. 1993, 57, 1204–1205. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Basu, N.; Ghatak, N.; Gujral, P.K. Anti–inflammatory and irritant activities of curcumin analogues in rats. Agents Actions 1982, 12, 508–515. [Google Scholar] [CrossRef]
- Kawabata, K.; Yamamoto, T.; Hara, A.; Shimizu, M.; Yamada, Y.; Matsunaga, K.; Tanaka, T.; Mori, H. Modifying effects of ferulic acid on azoxymethane–induced colon carcinogenesis in F344 rats. Cancer Lett. 2000, 157, 15–21. [Google Scholar] [CrossRef]
- Neto–Neves, E.M.; da Silva Maia Bezerra Filho, C.; Dejani, N.N.; de Sousa, D.P. Ferulic Acid and Cardiovascular Health: Therapeutic and Preventive Potential. Mini. Rev. Med. Chem. 2021, 21, 1625–1637. [Google Scholar] [CrossRef]
- Chaikijurajai, T.; Tang, W.H.W. Myeloperoxidase: A potential therapeutic target for coronary artery disease. Expert Opin. Ther. Targets 2020, 24, 695–705. [Google Scholar] [CrossRef]
- Chmielowski, R.A.; Abdelhamid, D.S.; Faig, J.J.; Petersen, L.K.; Gardner, C.R.; Uhrich, K.E.; Joseph, L.B.; Moghe, P.V. Athero–inflammatory nanotherapeutics: Ferulic acid–based poly(anhydride–ester) nanoparticles attenuate foam cell formation by regulating macrophage lipogenesis and reactive oxygen species generation. Acta Biomater. 2017, 57, 85–94. [Google Scholar] [CrossRef]
- Yan, J.J.; Jung, J.S.; Kim, T.K.; Hasan, A.; Hong, C.W.; Nam, J.S.; Song, D.K. Protective effects of ferulic acid in amyloid precursor protein plus presenilin–1 transgenic mouse model of Alzheimer disease. Biol. Pharm. Bull. 2013, 36, 140–143. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Hirohata, M.; Yamada, M. Ferulic acid destabilizes preformed beta–amyloid fibrils in vitro. Biochem. Biophys. Res. Commun. 2005, 336, 444–449. [Google Scholar] [CrossRef]
- Mori, T.; Koyama, N.; Guillot–Sestier, M.V.; Tan, J.; Town, T. Ferulic acid is a nutraceutical β–secretase modulator that improves behavioral impairment and alzheimer–like pathology in transgenic mice. PLoS ONE 2013, 8, e55774. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dong, C. Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 2009, 16, 386–394. [Google Scholar] [CrossRef]
- Shozawa, H.; Oguchi, T.; Tsuji, M.; Yano, S.; Kiuchi, Y.; Ono, K. Supratherapeutic concentrations of cilostazol inhibits β–amyloid oligomerization in vitro. Neurosci. Lett. 2018, 677, 19–25. [Google Scholar] [CrossRef]
- Ono, K.; Tsuji, M. Protofibrils of Amyloid–β are Important Targets of a Disease–Modifying Approach for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 952. [Google Scholar] [CrossRef]
- Chang, C.C.; Edwald, E.; Veatch, S.; Steel, D.G.; Gafni, A. Interactions of amyloid–β peptides on lipid bilayer studied by single molecule imaging and tracking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1616–1624. [Google Scholar] [CrossRef]
- Meker, S.; Chin, H.; Sut, T.N.; Cho, N.J. Amyloid–β Peptide Triggers Membrane Remodeling in Supported Lipid Bilayers Depending on Their Hydrophobic Thickness. Langmuir 2018, 34, 9548–9560. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Dolai, S.; Shi, W.; Corbo, C.; Sun, C.; Averick, S.; Obeysekera, D.; Farid, M.; Alonso, A.; Banerjee, P.; Raja, K. “Clicke” sugar–curcumin conjugate: Modulator of amyloid–β and tau peptide aggregation at ultralow concentrations. ACS Chem. Neurosci. 2011, 2, 694–699. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.; Nishijo, H.; et al. Phenolic compounds prevent amyloid β–protein oligomerization and synaptic dysfunction by site–specific binding. J. Biol. Chem. 2012, 287, 14631–14643. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Kotani, S.; Mano, Y.; Ueno, A.; Ito, Y.; Kitaba, T.; Takata, T.; Fujii, N. Ferulic Acid Suppresses Amyloid β Production in the Human Lens Epithelial Cell Stimulated with Hydrogen Peroxide. Biomed. Res. Int. 2017, 2017, 5343010. [Google Scholar] [CrossRef] [PubMed]
- Ono, K. Alzheimer’s disease as oligomeropathy. Neurochem. Int. 2018, 119, 57–70. [Google Scholar] [CrossRef]
- Masuda, Y.; Fukuchi, M.; Yatagawa, T.; Tada, M.; Takeda, K.; Irie, K.; Akagi, K.; Monobe, Y.; Imazawa, T.; Takegoshi, K. Solid–state NMR analysis of interaction sites of curcumin and 42–residue amyloid β–protein fibrils. Bioorg. Med. Chem. 2011, 19, 5967–5974. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti–amyloidogenic and fibril–destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Ngo, S.T.; Truong, D.T.; Tam, N.M.; Nguyen, M.T. EGCG inhibits the oligomerization of amyloid beta (16–22) hexamer: Theoretical studies. J. Mol. Graph. Model. 2017, 76, 1–10. [Google Scholar] [CrossRef]
- Przygońska, K.; Pacewicz, M.; Sadowska, W.; Poznański, J.; Bal, W.; Dadlez, M. His6, His13, and His14 residues in Aβ 1–40 peptide significantly and specifically affect oligomeric equilibria. Sci. Rep. 2019, 9, 9449. [Google Scholar] [CrossRef]
- Nedumpully–Govindan, P.; Kakinen, A.; Pilkington, E.H.; Davis, T.P.; Chun Ke, P.; Ding, F. Stabilizing Off–pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition. Sci. Rep. 2016, 6, 19463. [Google Scholar] [CrossRef]
- Ishii, T.; Mori, T.; Tanaka, T.; Mizuno, D.; Yamaji, R.; Kumazawa, S.; Nakayama, T.; Akagawa, M. Covalent modification of proteins by green tea polyphenol (–)–epigallocatechin–3–gallate through autoxidation. Free Radic. Biol. Med. 2008, 45, 1384–1394. [Google Scholar] [CrossRef]
- Sato, M.; Murakami, K.; Uno, M.; Nakagawa, Y.; Katayama, S.; Akagi, K.; Masuda, Y.; Takegoshi, K.; Irie, K. Site–specific inhibitory mechanism for amyloid β42 aggregation by catechol–type flavonoids targeting the Lys residues. J. Biol. Chem. 2013, 288, 23212–23224. [Google Scholar] [CrossRef] [Green Version]
- Perry, G.; Nunomura, A.; Hirai, K.; Takeda, A.; Aliev, G.; Smith, M.A. Oxidative damage in Alzheimer’s disease: The metabolic dimension. Int. J. Dev. Neurosci. 2000, 18, 417–421. [Google Scholar] [CrossRef]
- Lovell, M.A.; Markesbery, W.R. Ratio of 8–hydroxyguanine in intact DNA to free 8–hydroxyguanine is increased in Alzheimer disease ventricular cerebrospinal fluid. Arch. Neurol. 2001, 58, 392–396. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe–Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β(1–42) oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef]
- Watanabe–Nakayama, T.; Ono, K.; Itami, M.; Takahashi, R.; Teplow, D.B.; Yamada, M. High–speed atomic force microscopy reveals structural dynamics of amyloid β1–42 aggregates. Proc. Natl. Acad. Sci. USA 2016, 113, 5835–5840. [Google Scholar] [CrossRef]
- Kimura, A.M.; Tsuji, M.; Yasumoto, T.; Mori, Y.; Oguchi, T.; Tsuji, Y.; Umino, M.; Umino, A.; Nishikawa, T.; Nakamura, S.; et al. Myricetin prevents high molecular weight Aβ(1–42) oligomer–induced neurotoxicity through antioxidant effects in cell membranes and mitochondria. Free Radic. Biol. Med. 2021, 171, 232–244. [Google Scholar] [CrossRef]
- Sultana, R.; Ravagna, A.; Mohmmad–Abdul, H.; Calabrese, V.; Butterfield, D.A. Ferulic acid ethyl ester protects neurons against amyloid beta– peptide(1–42)–induced oxidative stress and neurotoxicity: Relationship to antioxidant activity. J. Neurochem. 2005, 92, 749–758. [Google Scholar] [CrossRef]
- Picone, P.; Bondi, M.L.; Montana, G.; Bruno, A.; Pitarresi, G.; Giammona, G.; Di Carlo, M. Ferulic acid inhibits oxidative stress and cell death induced by Ab oligomers: Improved delivery by solid lipid nanoparticles. Free Radic. Res. 2009, 43, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; De, R.; Mukhopadhyay, A.K. Curcumin as a potential therapeutic candidate for Helicobacter pylori associated diseases. World J. Gastroenterol. 2016, 22, 2736–2748. [Google Scholar] [CrossRef]
- Casley, C.S.; Canevari, L.; Land, J.M.; Clark, J.B.; Sharpe, M.A. Beta–amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J. Neurochem. 2002, 80, 91–100. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein–membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef] [PubMed]
- Peters, I.; Igbavboa, U.; Schütt, T.; Haidari, S.; Hartig, U.; Rosello, X.; Böttner, S.; Copanaki, E.; Deller, T.; Kögel, D.; et al. The interaction of beta–amyloid protein with cellular membranes stimulates its own production. Biochim. Biophys. Acta 2009, 1788, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, G.Y.; Eckert, G.P.; Lee, J.C. Cellular membrane fluidity in amyloid precursor protein processing. Mol. Neurobiol. 2014, 50, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.J.; Cai, J.X. Aniracetam restores the effects of amyloid–beta protein or ageing on membrane fluidity and intracellular calcium concentration in mice synaptosomes. J. Neural Transm. 2007, 114, 1407–1411. [Google Scholar] [CrossRef]
- Piotr, D.; Broncel, M.; Podsędek, A.; Koter–Michalak, M. Hypolipidemic and antioxidant effects of hydroxycinnamic acids, quercetin, and cyanidin 3–glucoside in hypercholesterolemic erythrocytes (in vitro study). Eur. J. Nutr. 2012, 51, 435–443. [Google Scholar] [CrossRef]
- Leite, N.B.; Martins, D.B.; Fazani, V.E.; Vieira, M.R.; Dos Santos Cabrera, M.P. Cholesterol modulates curcumin partitioning and membrane effects. Biochim. Biophys. Acta Biomembr. 2018, 1860, 2320–2328. [Google Scholar] [CrossRef]
- Srimal, R.C.; Dhawan, B.N. Pharmacology of diferuloyl methane (curcumin), a non–steroidal anti–inflammatory agent. J. Pharm. Pharmacol. 1973, 25, 447–452. [Google Scholar] [CrossRef]
- Paiva, L.B.; Goldbeck, R.; Santos, W.D.; Squina, F.M. Ferulic acid and derivatives: Molecules with potential application in the pharmaceutical field. Braz. J. Pharm. Sci. 2013, 49, 395–411. [Google Scholar] [CrossRef]
- Vareed, S.K.; Kakarala, M.; Ruffin, M.T.; Crowell, J.A.; Normolle, D.P.; Djuric, Z.; Brenner, D.E. Pharmacokinetics of curcumin conjugate metabolites in healthy human subjects. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1411–1417. [Google Scholar] [CrossRef]
- Yang, C.; Tian, Y.; Zhang, Z.; Xu, F.; Chen, Y. High–performance liquid chromatography–electrospray ionization mass spectrometry determination of sodium ferulate in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 945–950. [Google Scholar] [CrossRef]
- Nagata, M.; Tsuji, M.; Oguchi, T.; Momma, Y.; Nohara, T.; Ohashi, H.; Ito, N.; Yamamoto, K.; Udaka, Y.; Sasaki, A.; et al. Protective Effects of the Alga Fucoidan Against Amyloid–β–Induced Neurotoxicity in SH–SY5Y Cells. BPB Rep. 2021, 4, 206–213. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activities | Measurement Item | Cur | FA | Cur + FA |
|---|---|---|---|---|
| Suppression of Aβ aggregation | Aβ1–40, Aβ1–42 | +++ | ± | +++ |
| Antioxidant effects | ROS, mitochondrial ROS | + | ++ | +++ |
| Cell membrane effects | Fluidity, DPPP | + | +++ | +++ |
| Ca2+ homeostasis | [Ca2+]i | +++ | + | ++ |
| Neuroprotective effects | EthD, MTT | ++ | ++ | +++ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohashi, H.; Tsuji, M.; Oguchi, T.; Momma, Y.; Nohara, T.; Ito, N.; Yamamoto, K.; Nagata, M.; Kimura, A.M.; Kiuchi, Y.; et al. Combined Treatment with Curcumin and Ferulic Acid Suppressed the Aβ-Induced Neurotoxicity More than Curcumin and Ferulic Acid Alone. Int. J. Mol. Sci. 2022, 23, 9685. https://doi.org/10.3390/ijms23179685
Ohashi H, Tsuji M, Oguchi T, Momma Y, Nohara T, Ito N, Yamamoto K, Nagata M, Kimura AM, Kiuchi Y, et al. Combined Treatment with Curcumin and Ferulic Acid Suppressed the Aβ-Induced Neurotoxicity More than Curcumin and Ferulic Acid Alone. International Journal of Molecular Sciences. 2022; 23(17):9685. https://doi.org/10.3390/ijms23179685
Chicago/Turabian StyleOhashi, Hideaki, Mayumi Tsuji, Tatsunori Oguchi, Yutaro Momma, Tetsuhito Nohara, Naohito Ito, Ken Yamamoto, Miki Nagata, Atsushi Michael Kimura, Yuji Kiuchi, and et al. 2022. "Combined Treatment with Curcumin and Ferulic Acid Suppressed the Aβ-Induced Neurotoxicity More than Curcumin and Ferulic Acid Alone" International Journal of Molecular Sciences 23, no. 17: 9685. https://doi.org/10.3390/ijms23179685
APA StyleOhashi, H., Tsuji, M., Oguchi, T., Momma, Y., Nohara, T., Ito, N., Yamamoto, K., Nagata, M., Kimura, A. M., Kiuchi, Y., & Ono, K. (2022). Combined Treatment with Curcumin and Ferulic Acid Suppressed the Aβ-Induced Neurotoxicity More than Curcumin and Ferulic Acid Alone. International Journal of Molecular Sciences, 23(17), 9685. https://doi.org/10.3390/ijms23179685