
De Novo Transcriptome Analysis of R. nigrum cv. Aldoniai in Response to Blackcurrant Reversion Virus Infection




Abstract
:1. Introduction
2. Results
2.1. Quality Control of BioProject Data
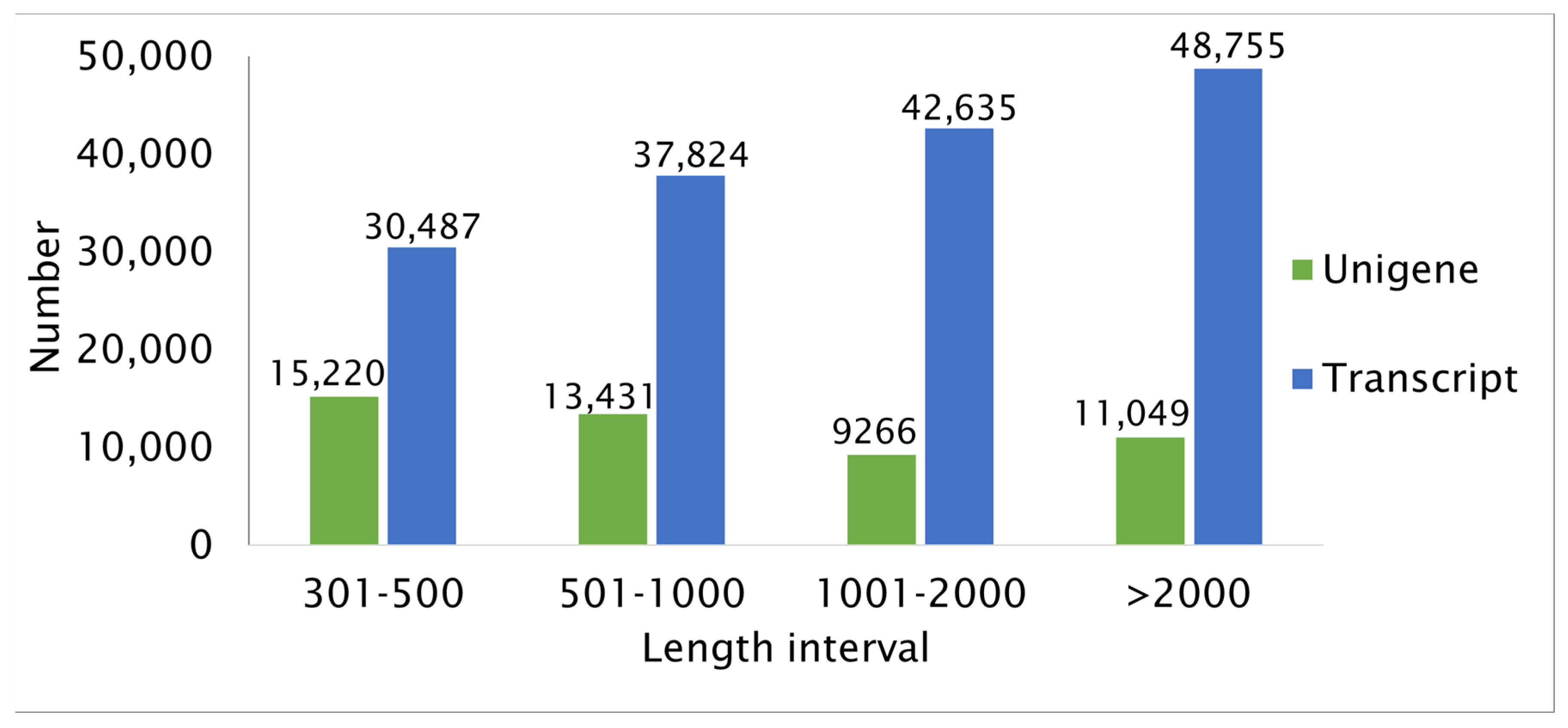
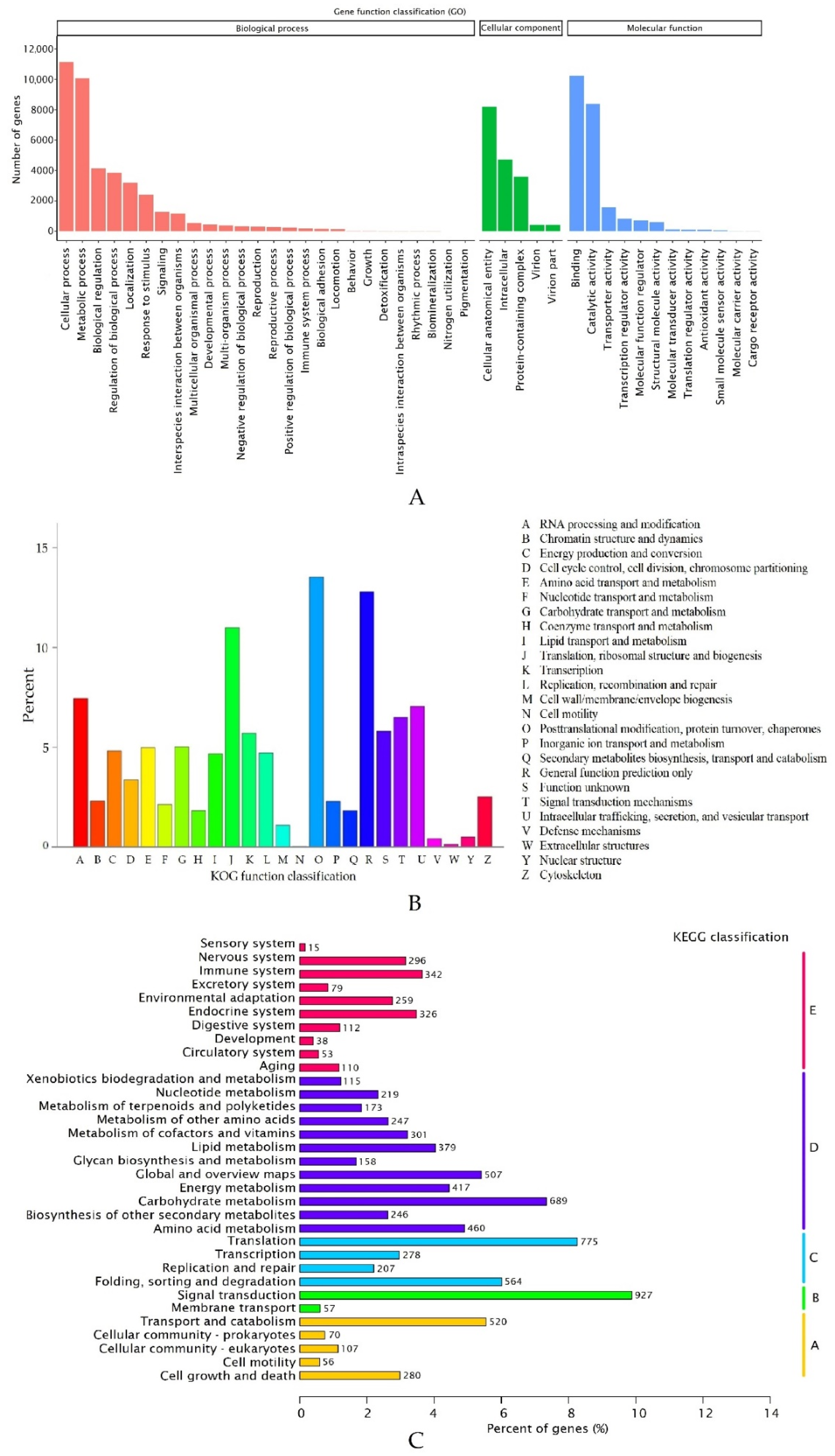
2.2. Annotation of Unigenes in R. nigrum Transcriptome
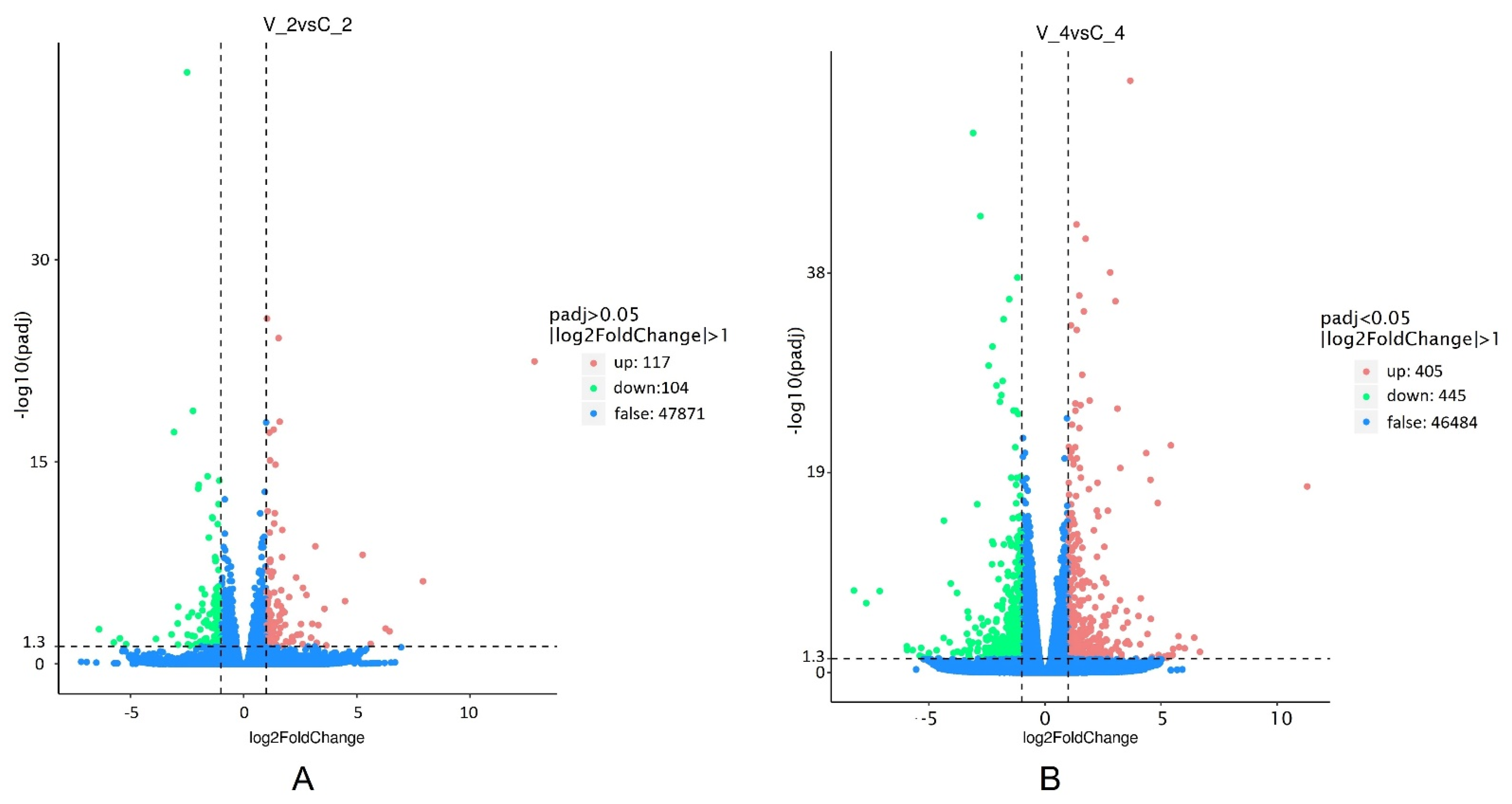
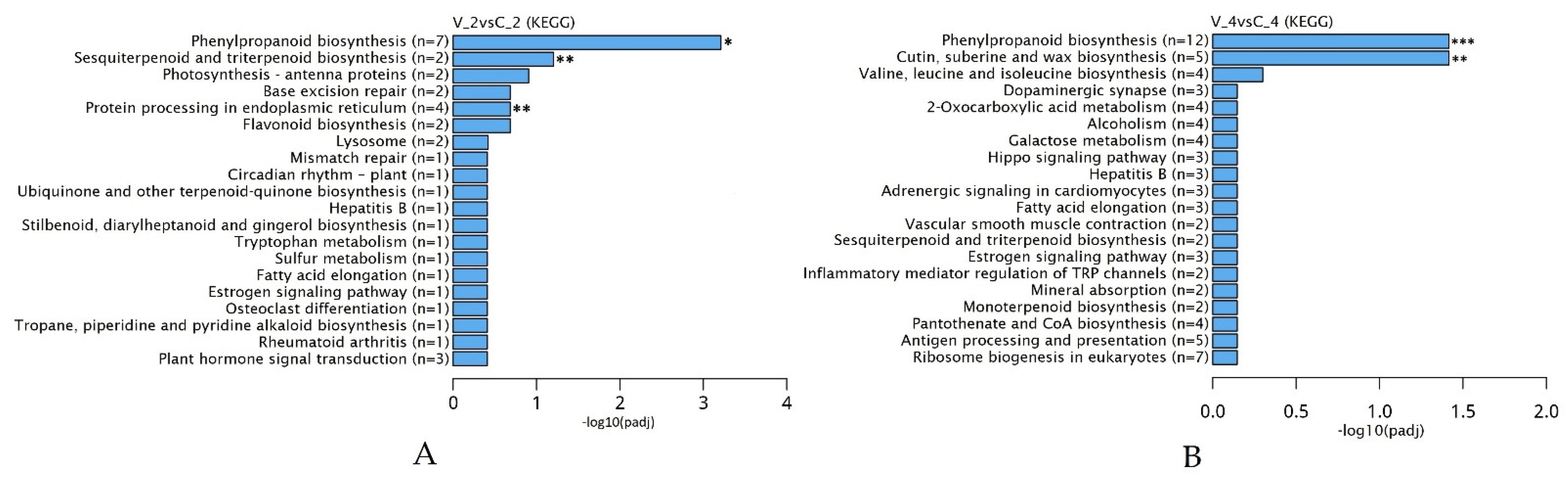
2.3. Differentially Expressed Genes (DEGs) to Virus Defense Response in R. nigrum cv. Aldoniai Microshoots
3. Discussion
3.1. Transcriptome of R. nigrum Microshoots
3.2. Virus Defense Response to BRV in R. nigrum
4. Materials and Methods
4.1. RNA Isolation, cDNA Library Preparation, and Sequencing
4.2. De Novo Transcriptome Analysis
4.3. Functional Annotation of Unigenes
4.4. Gene Functional Annotation
4.5. Gene Expression Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Šutic, D.D.; Ford, R.E.; Tošic, M.T. Virus diseases of small fruits. In Handbook of Plant Virus Diseases; Šutic, D.D., Ford, R.E., Tošic, M.T., Eds.; CRC Press: Boca Raton, FL, USA, 1999; pp. 433–475. [Google Scholar]
- Dolan, A.; MacFarlane, S.A.; McGavin, W.J.; Brennan, R.M.; McNicol, J.W. Blackcurrant reversion virus: Validation of an improved diagnostic test, accelerating testing in breeding and certification of blackcurrants. J. Berry Res. 2011, 1, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Adams, A.N.; Thresh, J.M. Reversion of black currant. In Virus Diseases of Small Fruits; Converse, R.H., Ed.; United States Department of Agriculture: Washington, DC, USA, 1987; pp. 133–136. [Google Scholar]
- Susi, P. Black currant reversion virus, a mite-transmitted nepovirus. Mol. Plant Pathol. 2004, 5, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Juškytė, A.D.; Mažeikienė, I.; Stanys, V. An effective method of Ribes spp. inoculation with blackcurrant reversion virus under in vitro conditions. Plants 2022, 11, 1635. [Google Scholar] [CrossRef] [PubMed]
- Seitsonen, J.J.T.; Susi, P.; Lemmetty, A.; Butcher, S.J. Structure of the mite-transmitted Blackcurrant reversion nepovirus using electron cryo-microscopy. Virology 2008, 378, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Latvala-Kilby, S.; Lehto, K. The complete nucleotide sequence of RNA2 of blackcurrant reversion nepovirus. Virus Res. 1999, 65, 87–92. [Google Scholar] [CrossRef]
- Pacot-Hiriart, C.; Latvala-Kilby, S.; Lehto, K. Nucleotide sequence of black currant reversion associated nepovirus RNA1. Virus Res. 2001, 79, 145–152. [Google Scholar] [CrossRef]
- Moročko-Bičevska, I.; Stalažs, A.; Lācis, G.; Laugale, V.; Baļķe, I.; Zuļģe, N.; Strautiņa, S. Cecidophyopsis mites and blackcurrant reversion virus on Ribes hosts: Current scientific progress and knowledge gaps. Ann. Appl. Biol. 2021, 180, 26–43. [Google Scholar] [CrossRef]
- Mažeikienė, I.; Stanys, V.; Juškytė, A.D.; Sasnauskas, A.; Šikšnianas, T. Juodojo serbento veislės ‘Aldoniai’ ir ‘Didikai’. Sodininkystė Daržininkystė 2017, 36, 3–14. [Google Scholar]
- Mazeikiene, I.; Juskyte, A.D.; Stanys, V. Application of marker-assisted selection for resistance to gall mite and Blackcurrant reversion virus in Ribes genus. Zemdirbyste 2019, 106, 359–366. [Google Scholar] [CrossRef]
- Ding, S.W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef]
- Brennan, R.; Jorgensen, L.; Hackett, C.; Woodhead, M.; Gordon, S.; Russell, J. The development of a genetic linkage map of blackcurrant (Ribes nigrum L.) and the identification of regions associated with key fruit quality and agronomic traits. Euphytica 2008, 161, 19–34. [Google Scholar] [CrossRef]
- Brennan, R.M.; Jorgensen, L.; Gordon, S.; Loades, K.; Hackett, C.; Russell, J. The development of a PCR-based marker linked to resistance to the blackcurrant gall mite (Cecidophyopsis ribis Acari: Eriophyidae). Theor. Appl. Genet. 2009, 118, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.M. Resistance to gall mite (Phytoptus ribis Nal.) in the Eucoreosma section of Ribes. Euphytica 1971, 20, 422–426. [Google Scholar] [CrossRef]
- Keep, E.; Knight, V.H.; Parker, J.H. Progress in the integration of characters in gall mite resistant black currants. J. Hortic. Sci. 1982, 57, 189–196. [Google Scholar] [CrossRef]
- Deng, S.; Ma, J.; Zhang, L.; Chen, F.; Sang, Z.; Jia, Z.; Ma, L. De novo transcriptome sequencing and gene expression profiling of Magnolia wufengensis in response to cold stress. BMC Plant Biol. 2019, 19, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thole, V.; Bassard, J.E.; Ramírez-González, R.; Trick, M.; Ghasemi Afshar, B.; Breitel, D.; Hill, L.; Foito, A.; Shepherd, L.; Freitag, S.; et al. RNA-seq, de novo transcriptome assembly and flavonoid gene analysis in 13 wild and cultivated berry fruit species with high content of phenolics. BMC Genom. 2019, 20, 995. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zhang, J.; Zhang, X.; He, S.; Xie, D.; Liu, Y.; Li, C.; Wang, Z.; Liu, Y. Development of SSR markers in Paeonia based on de novo transcriptomic assemblies. PLoS ONE 2020, 15, e0227794. [Google Scholar] [CrossRef] [Green Version]
- Simpson, M.G. Diversity and classification of flowering plants: Eudicots. In Plant Systematics, 3rd ed.; Simpson, M.G., Ed.; MA Academic Press: Burlington, NJ, USA, 2019; pp. 275–448. ISBN 978-0-12-812628-8. [Google Scholar]
- Jarret, D.A.; Morris, J.; Cullen, D.W.; Gordon, S.L.; Verrall, S.R.; Milne, L.; Hedley, P.E.; Allwood, J.W.; Brennan, R.M.; Hancock, R.D. A transcript and metabolite atlas of blackcurrant fruit development highlights hormonal regulation and reveals the role of key transcription factors. Front. Plant Sci. 2018, 9, 1235. [Google Scholar] [CrossRef]
- Starkevič, P.; Ražanskienė, A.; Starkevič, U.; Kazanavičiūtė, V.; Denkovskienė, E.; Bendokas, V.; Šikšnianas, T.; Rugienius, R.; Stanys, V.; Ražanskas, R. Isolation and analysis of anthocyanin pathway genes from Ribes genus reveals MYB gene with potent anthocyanin-inducing capabilities. Plants 2020, 9, 1078. [Google Scholar] [CrossRef]
- Mazeikiene, I.; Bendokas, V.; Stanys, V.; Siksnianas, T. Molecular markers linked to resistance to the gall mite in blackcurrant. Plant Breed. 2012, 131, 762–766. [Google Scholar] [CrossRef]
- Juškytė, A.D.; Mažeikienė, I.; Stanys, V. Putative genes of pathogenesis-related proteins and coronatine-insensitive protein 1 in Ribes spp. Plants 2022, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- One Thousand Plant Transcriptomes Initiative. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C. From player to pawn: Viral avirulence factors involved in plant immunity. Viruses 2021, 13, 688. [Google Scholar] [CrossRef]
- Matika, D.E.F.; Loake, G.J. Redox regulation in plant immune function. Antioxid. Redox Signal. 2014, 21, 1373–1388. [Google Scholar] [CrossRef] [PubMed]
- Alazem, M.; Lin, N.S. Roles of plant hormones in the regulation of host-virus interactions. Mol. Plant Pathol. 2015, 16, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Kamle, M.; Borah, R.; Bora, H.; Jaiswal, A.K.; Singh, R.K.; Kumar, P. Systemic acquired resistance (SAR) and induced systemic resistance (ISR): Role and mechanism of action against phytopathogens. In Fungal Biotechnology and Bioengineering; Hesham, A.L., Upadhyay, R., Sharma, G., Manoharachary, C., Gupta, V., Eds.; Springer: Cham, Switzerland, 2020; pp. 457–470. ISBN 978-3-030-41870-0. [Google Scholar] [CrossRef]
- Thakur, M.; Sohal, B.S. Role of elicitors in inducing resistance in plants against pathogen infection: A review. Int. Sch. Res. Not. 2013, 2013, 762412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, T. Phenylpropanoid biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kong, L.; Zhi, P.; Chang, C. Update on cuticular wax biosynthesis and its roles in plant disease resistance. Int. J. Mol. Sci. 2020, 21, 5514. [Google Scholar] [CrossRef]
- Raffaele, S.; Leger, A.; Roby, D. Very long chain fatty acid and lipid signaling in the response of plants to pathogens. Plant Signal. Behav. 2009, 4, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. Available online: http://genomebiology.com/2014/15/8/410 (accessed on 5 July 2022). [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Library ID | Raw Reads | Raw Bases, G | Clean Reads | Clean Bases, G | Error Rate, % | Q20 | GC_pct, % |
|---|---|---|---|---|---|---|---|
| C_2_1 | 28,086,156 | 8.4 | 27,637,344 | 8.3 | 0.03 | 97.45 | 43.53 |
| C_2_2 | 24,977,381 | 7.5 | 24,594,501 | 7.4 | 0.03 | 97.51 | 43.32 |
| C_2_3 | 24,061,978 | 7.2 | 23,627,353 | 7.1 | 0.03 | 97.55 | 43.36 |
| V_2_1 | 27,826,308 | 8.3 | 27,343,527 | 8.2 | 0.03 | 97.52 | 43.57 |
| V_2_2 | 24,844,092 | 7.5 | 24,476,571 | 7.3 | 0.03 | 97.28 | 43.49 |
| V_2_3 | 26,612,831 | 8.0 | 26,276,865 | 7.9 | 0.03 | 97.41 | 43.47 |
| C_4_1 | 23,144,738 | 6.9 | 22,864,209 | 6.9 | 0.03 | 97.50 | 43.60 |
| C_4_2 | 21,330,139 | 6.4 | 21,028,990 | 6.3 | 0.03 | 97.18 | 43.27 |
| C_4_3 | 30,235,731 | 9.1 | 29,812,238 | 8.9 | 0.03 | 97.25 | 43.36 |
| V_4_1 | 23,810,909 | 7.1 | 23,507,940 | 7.1 | 0.03 | 97.21 | 42.93 |
| V_4_2 | 28,857,614 | 8.7 | 28,387,016 | 8.5 | 0.03 | 97.58 | 42.61 |
| V_4_3 | 25,987,937 | 7.8 | 25,614,722 | 7.7 | 0.03 | 97.02 | 42.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mažeikienė, I.; Juškytė, A.D.; Bendokas, V.; Stanys, V. De Novo Transcriptome Analysis of R. nigrum cv. Aldoniai in Response to Blackcurrant Reversion Virus Infection. Int. J. Mol. Sci. 2022, 23, 9560. https://doi.org/10.3390/ijms23179560
Mažeikienė I, Juškytė AD, Bendokas V, Stanys V. De Novo Transcriptome Analysis of R. nigrum cv. Aldoniai in Response to Blackcurrant Reversion Virus Infection. International Journal of Molecular Sciences. 2022; 23(17):9560. https://doi.org/10.3390/ijms23179560
Chicago/Turabian StyleMažeikienė, Ingrida, Ana Dovilė Juškytė, Vidmantas Bendokas, and Vidmantas Stanys. 2022. "De Novo Transcriptome Analysis of R. nigrum cv. Aldoniai in Response to Blackcurrant Reversion Virus Infection" International Journal of Molecular Sciences 23, no. 17: 9560. https://doi.org/10.3390/ijms23179560
APA StyleMažeikienė, I., Juškytė, A. D., Bendokas, V., & Stanys, V. (2022). De Novo Transcriptome Analysis of R. nigrum cv. Aldoniai in Response to Blackcurrant Reversion Virus Infection. International Journal of Molecular Sciences, 23(17), 9560. https://doi.org/10.3390/ijms23179560