

Clustering Analysis, Structure Fingerprint Analysis, and Quantum Chemical Calculations of Compounds from Essential Oils of Sunflower (Helianthus annuus L.) Receptacles




Abstract
:1. Introduction
2. Results
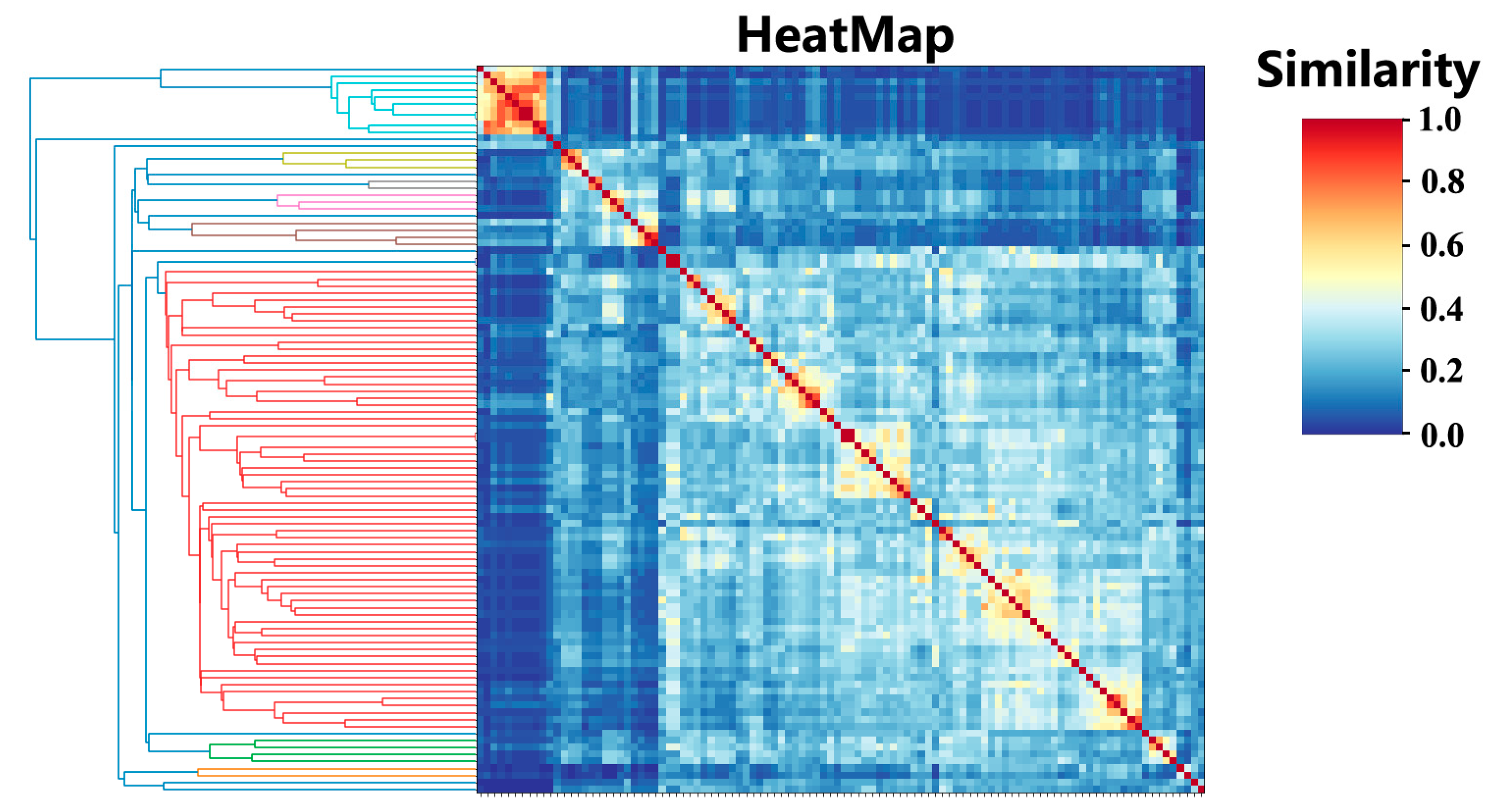
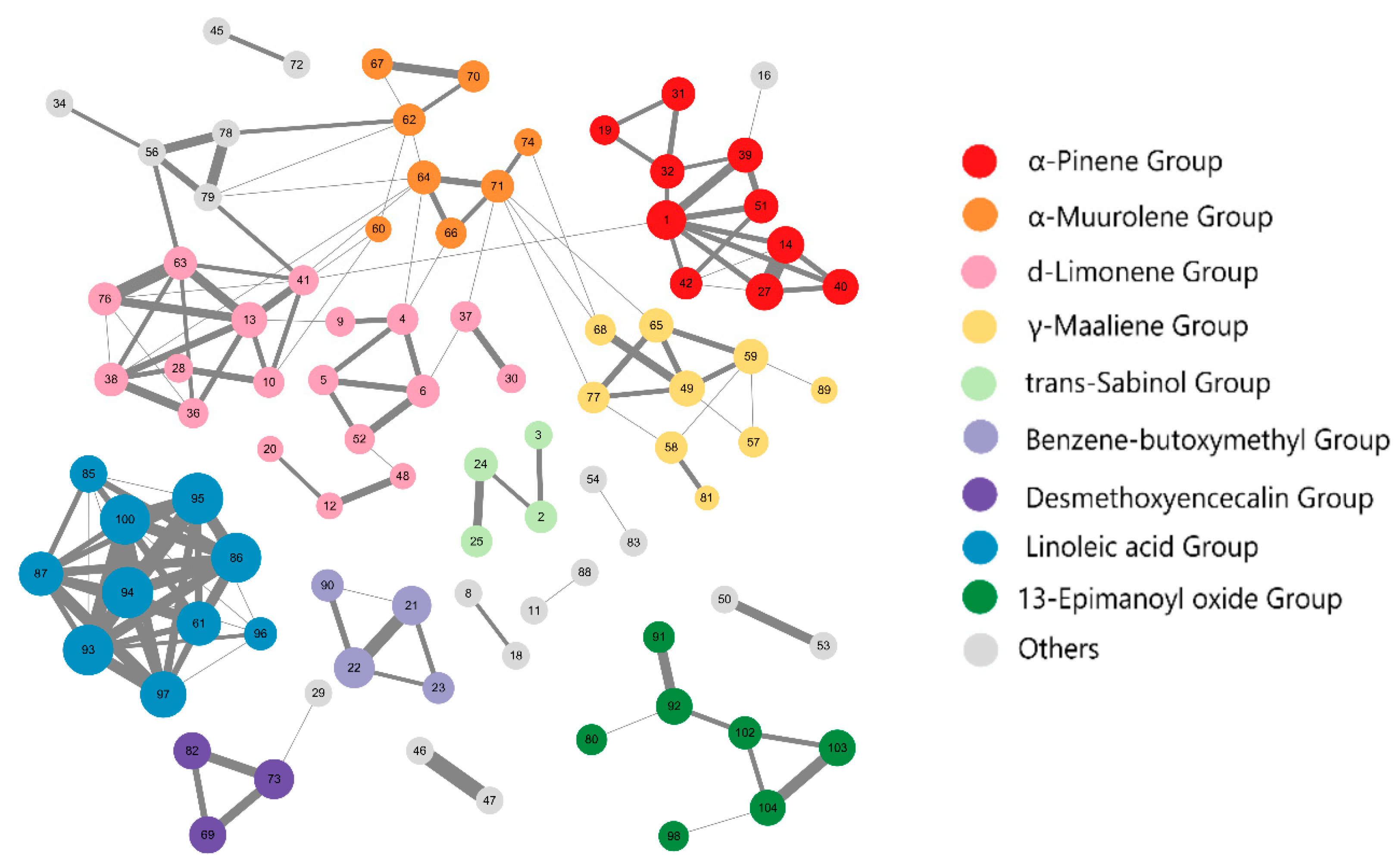
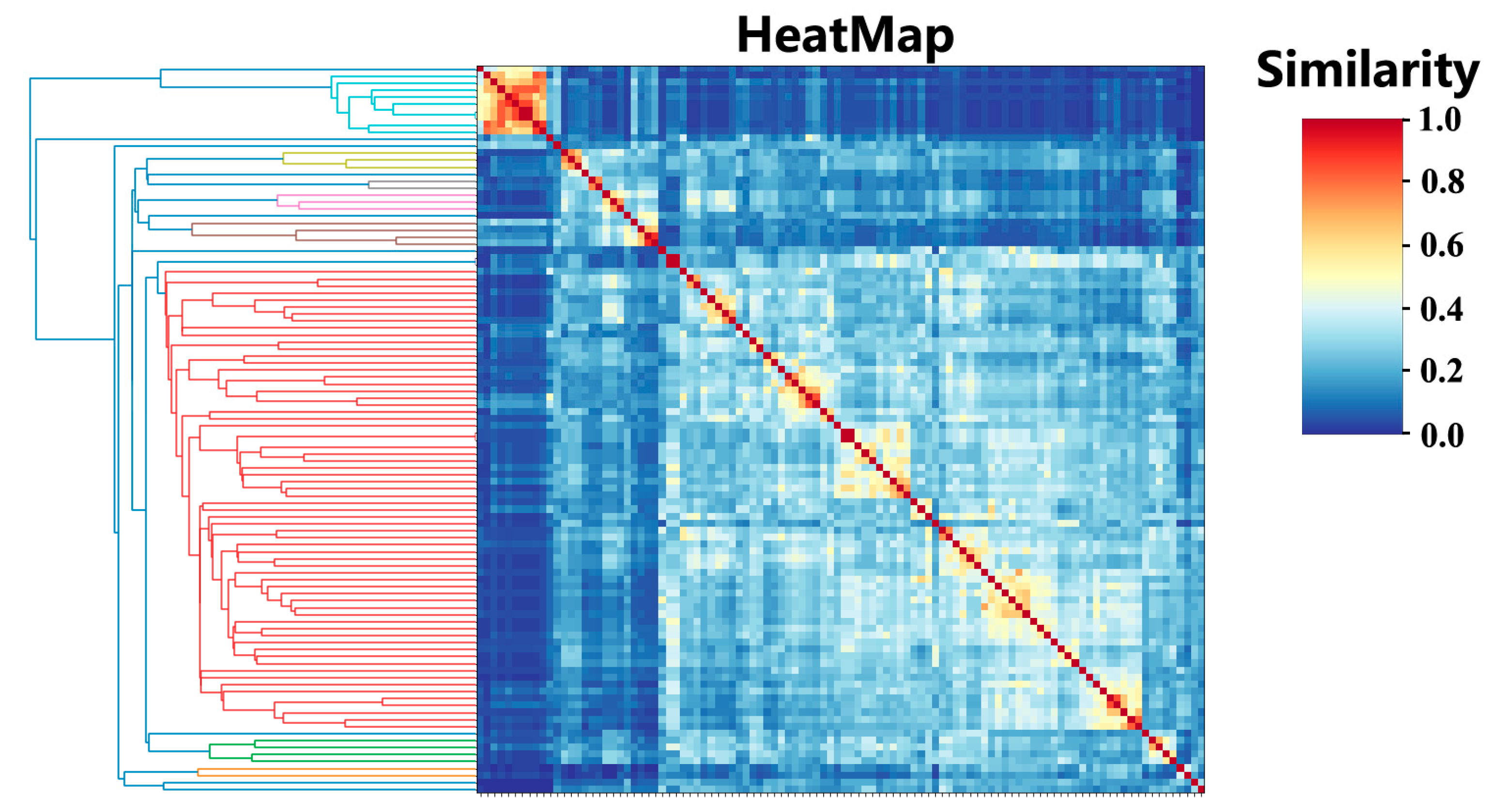
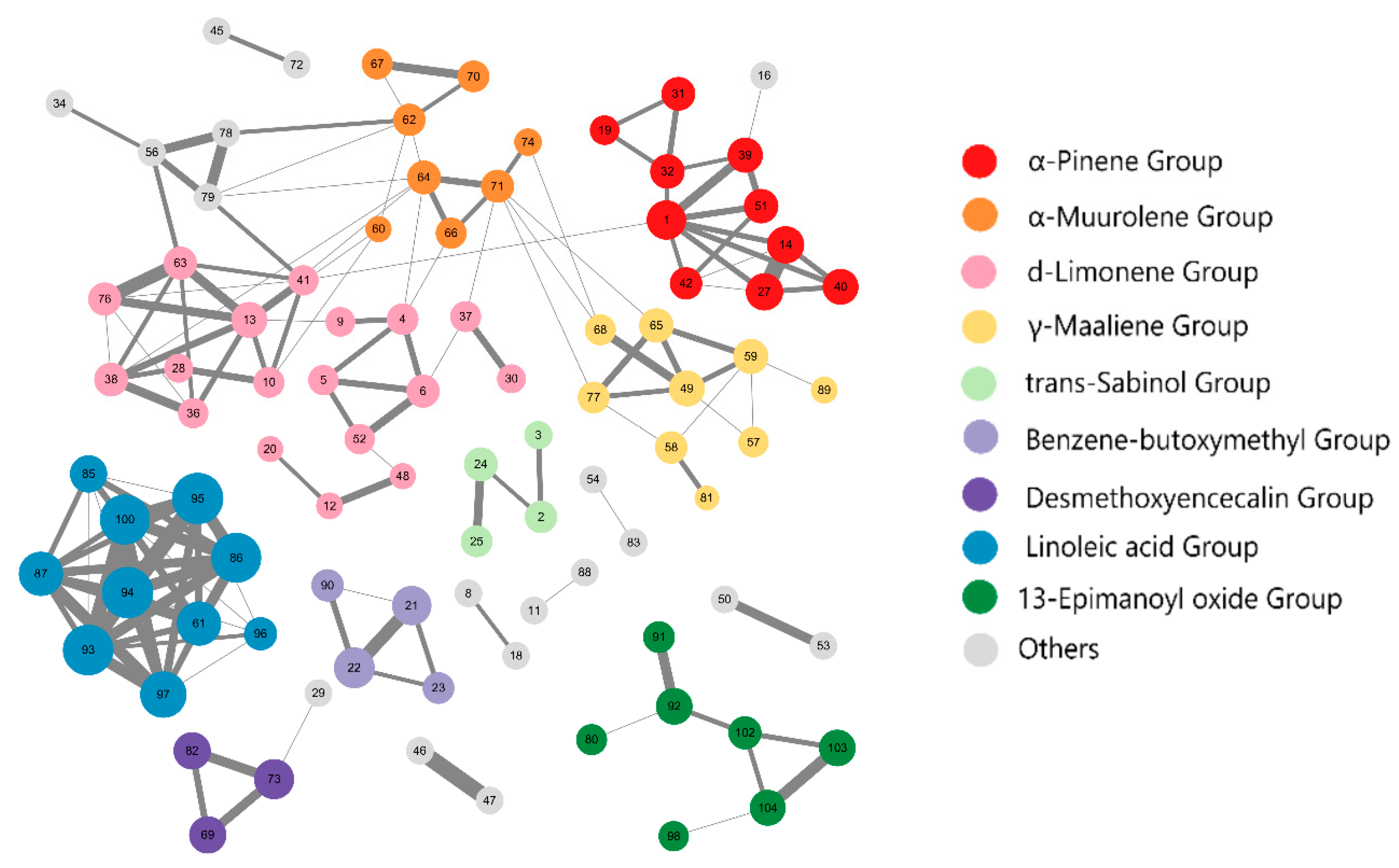
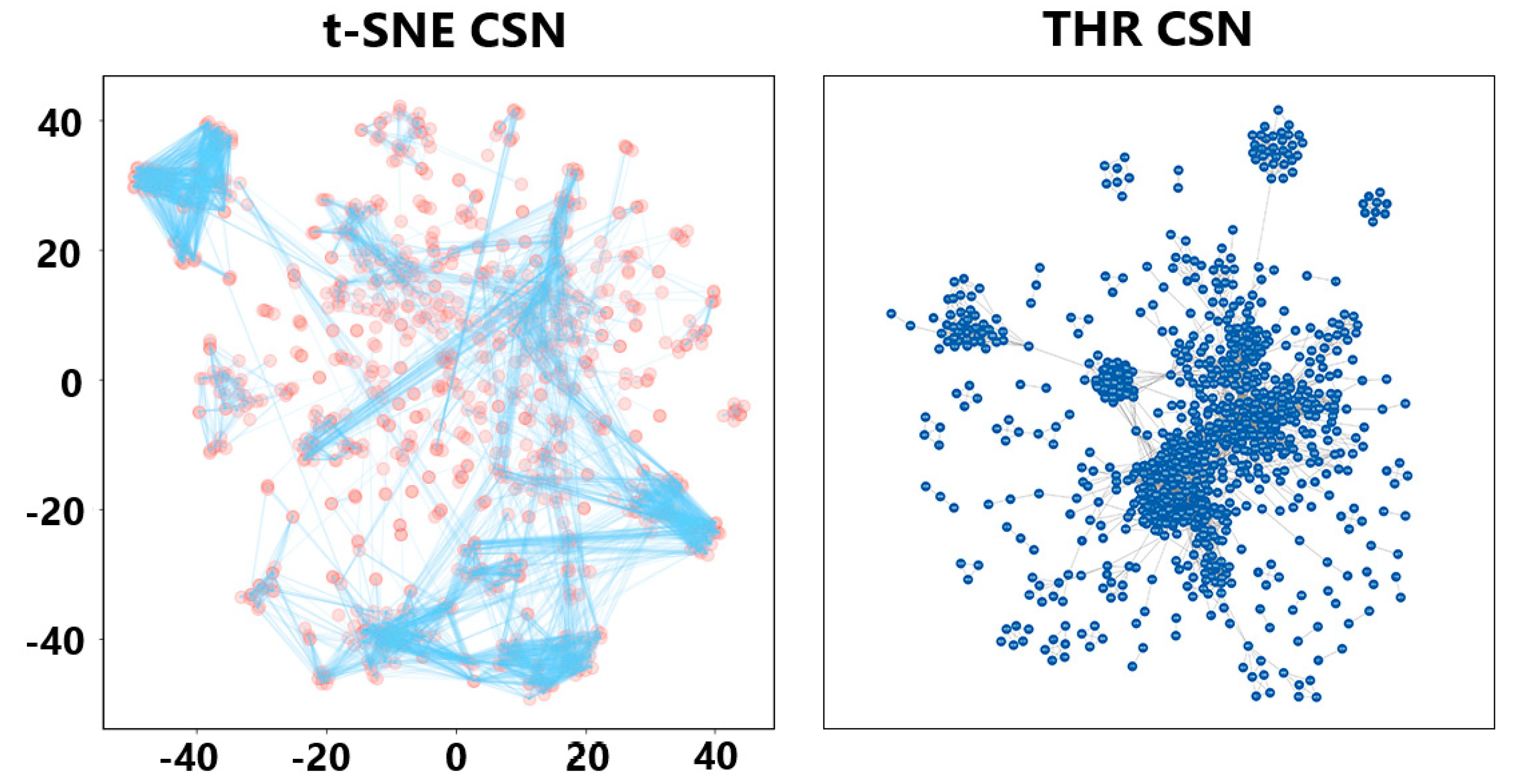
2.1. Cluster Analysis
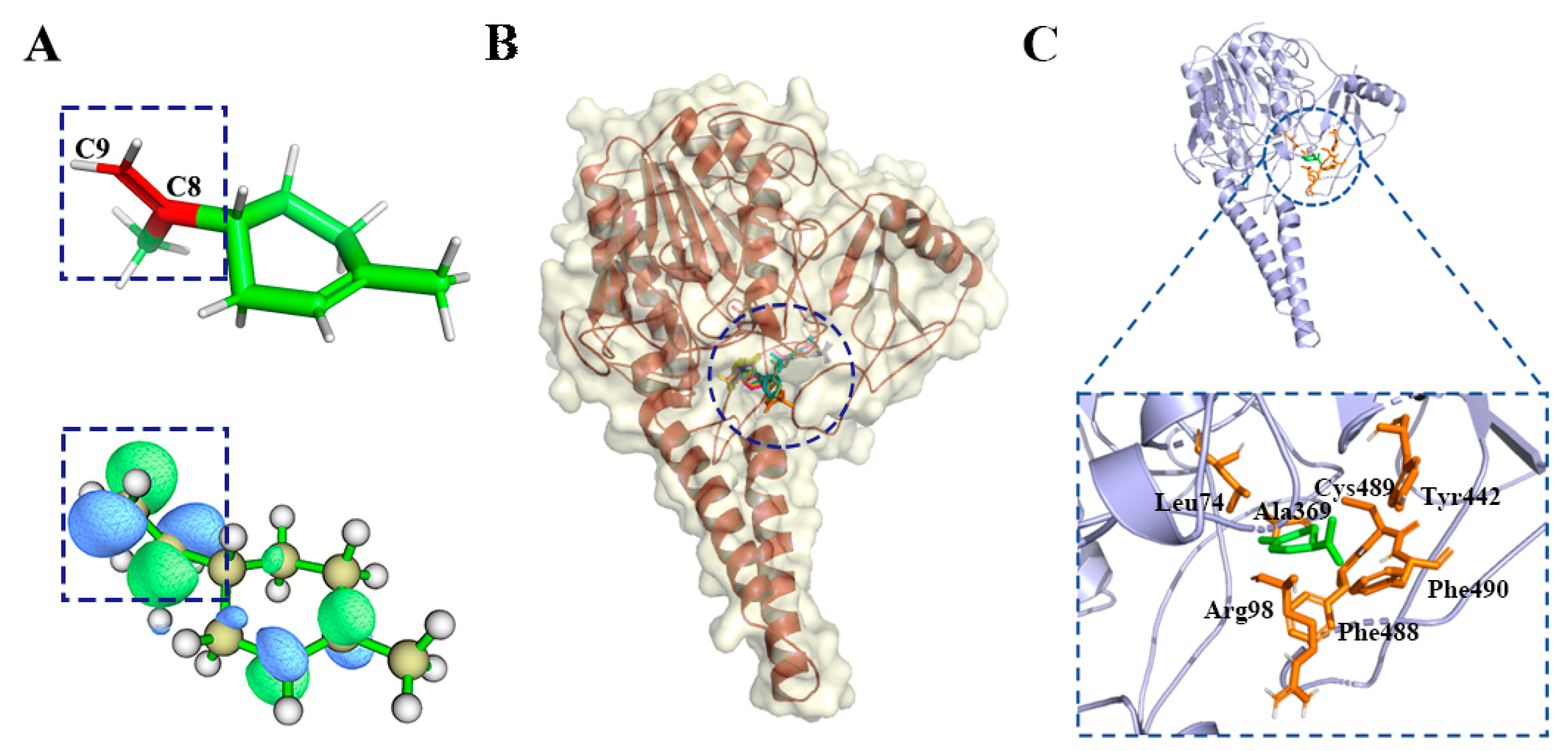
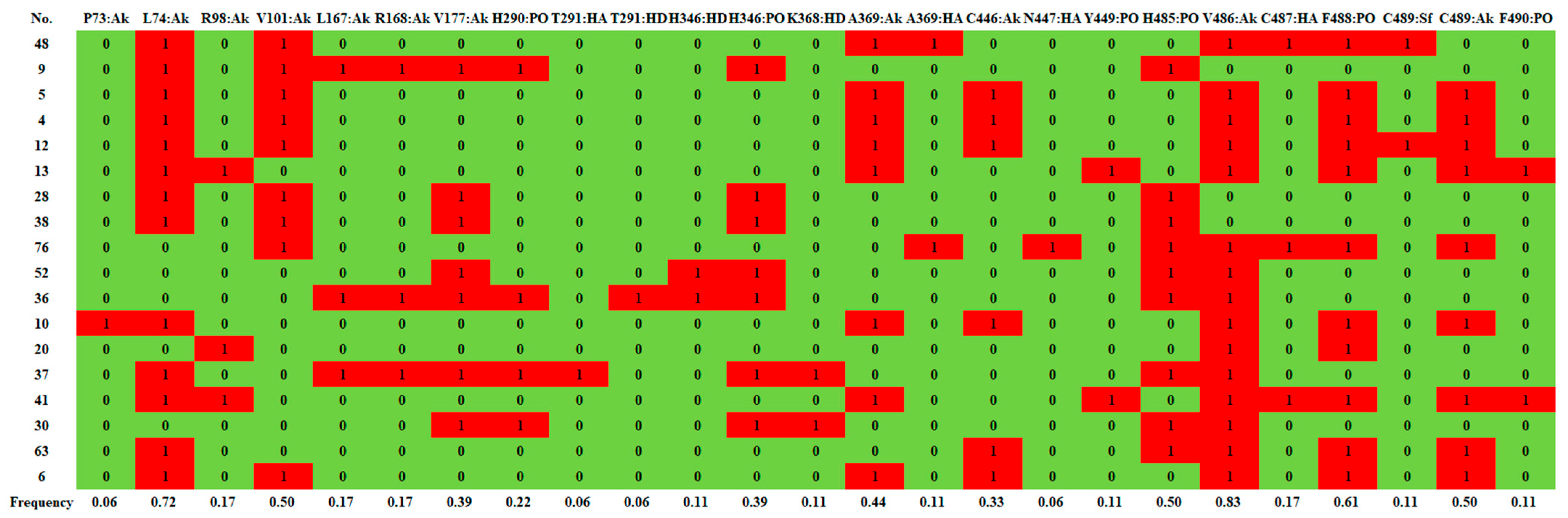
2.2. Reverse Docking, Structure Fingerprint, and Quantum Chemical Calculation Analysis
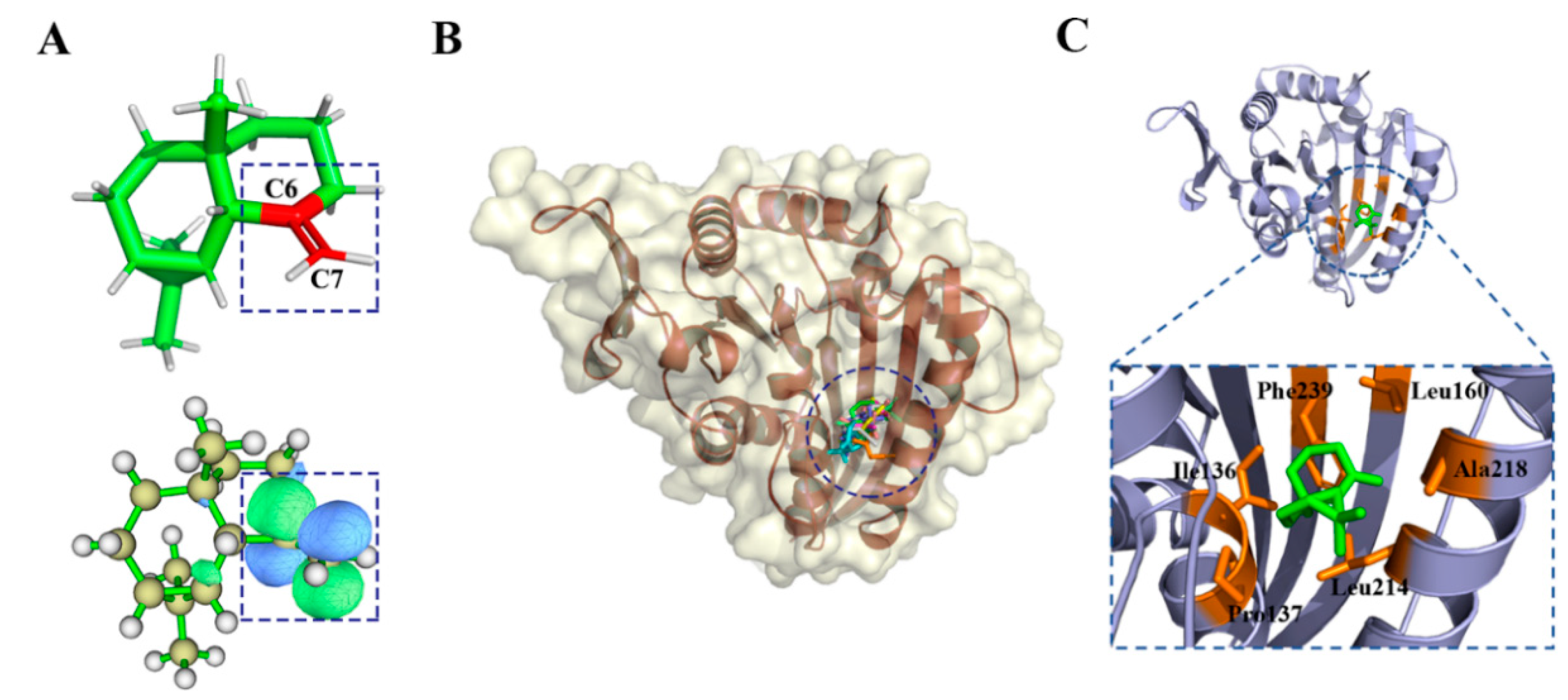
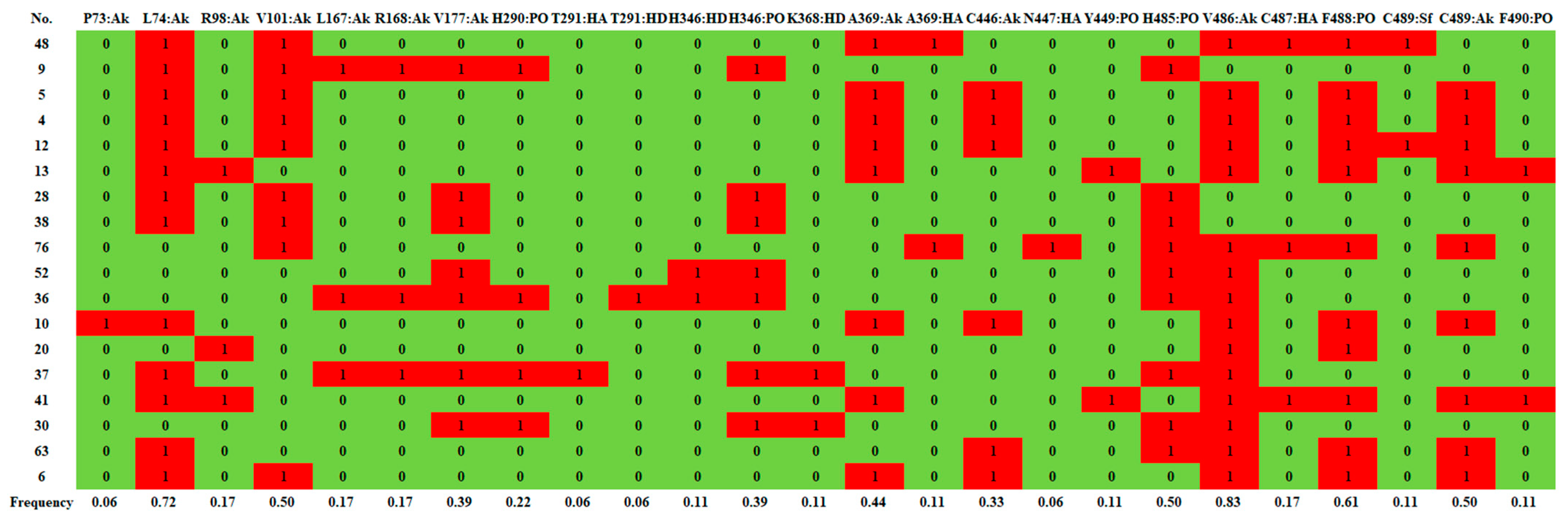
2.2.1. d-Limonene Group
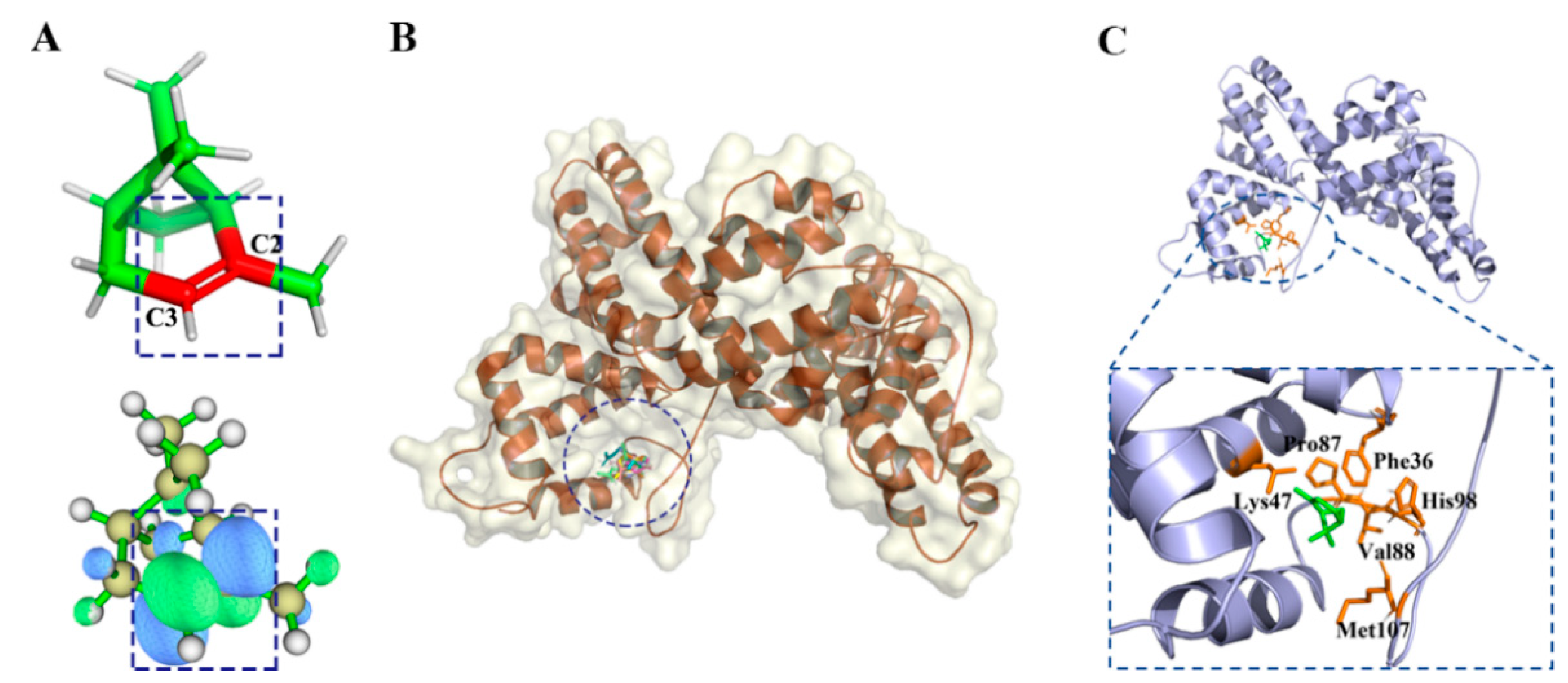
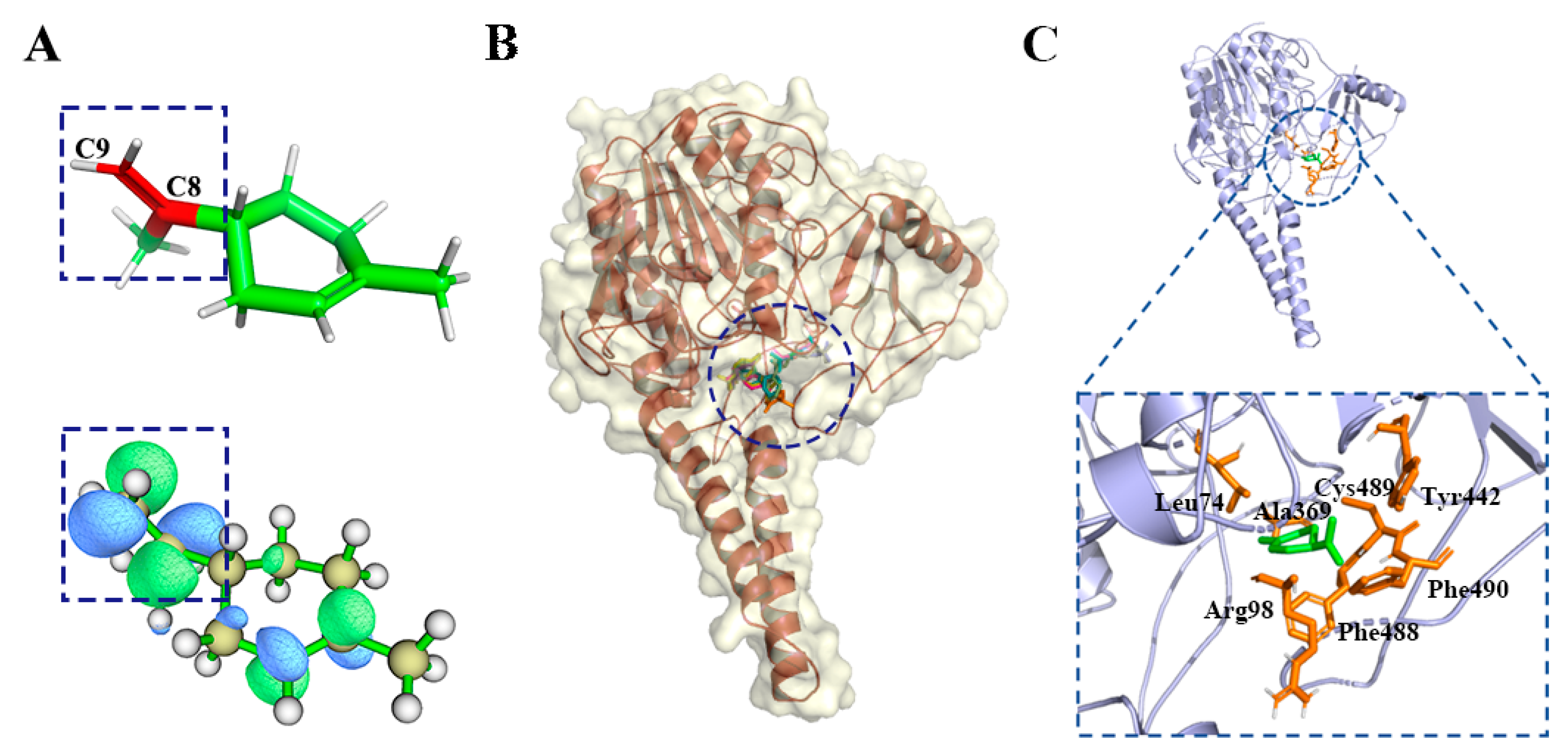
2.2.2. α-Pinene Group
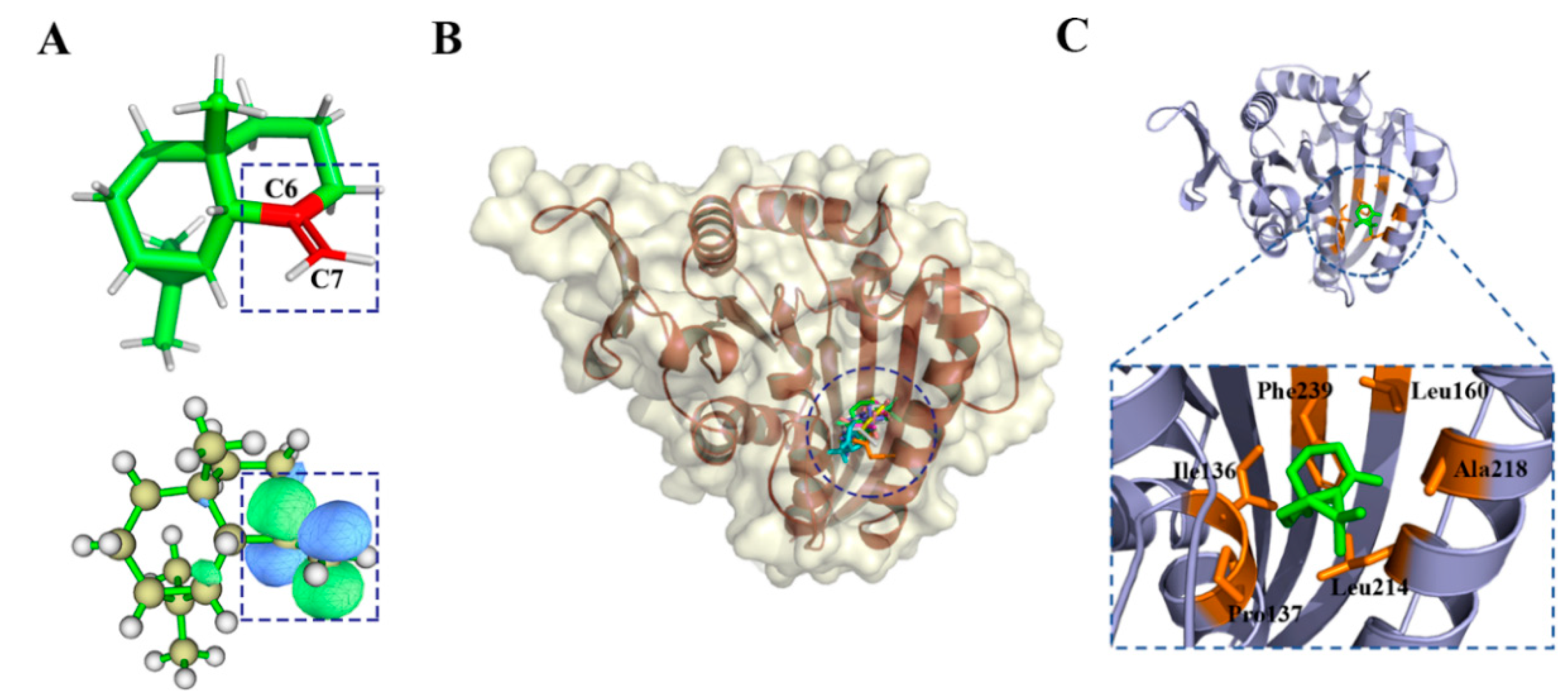
2.2.3. γ-Maaliene Group
3. Discussions
4. Materials and Methods
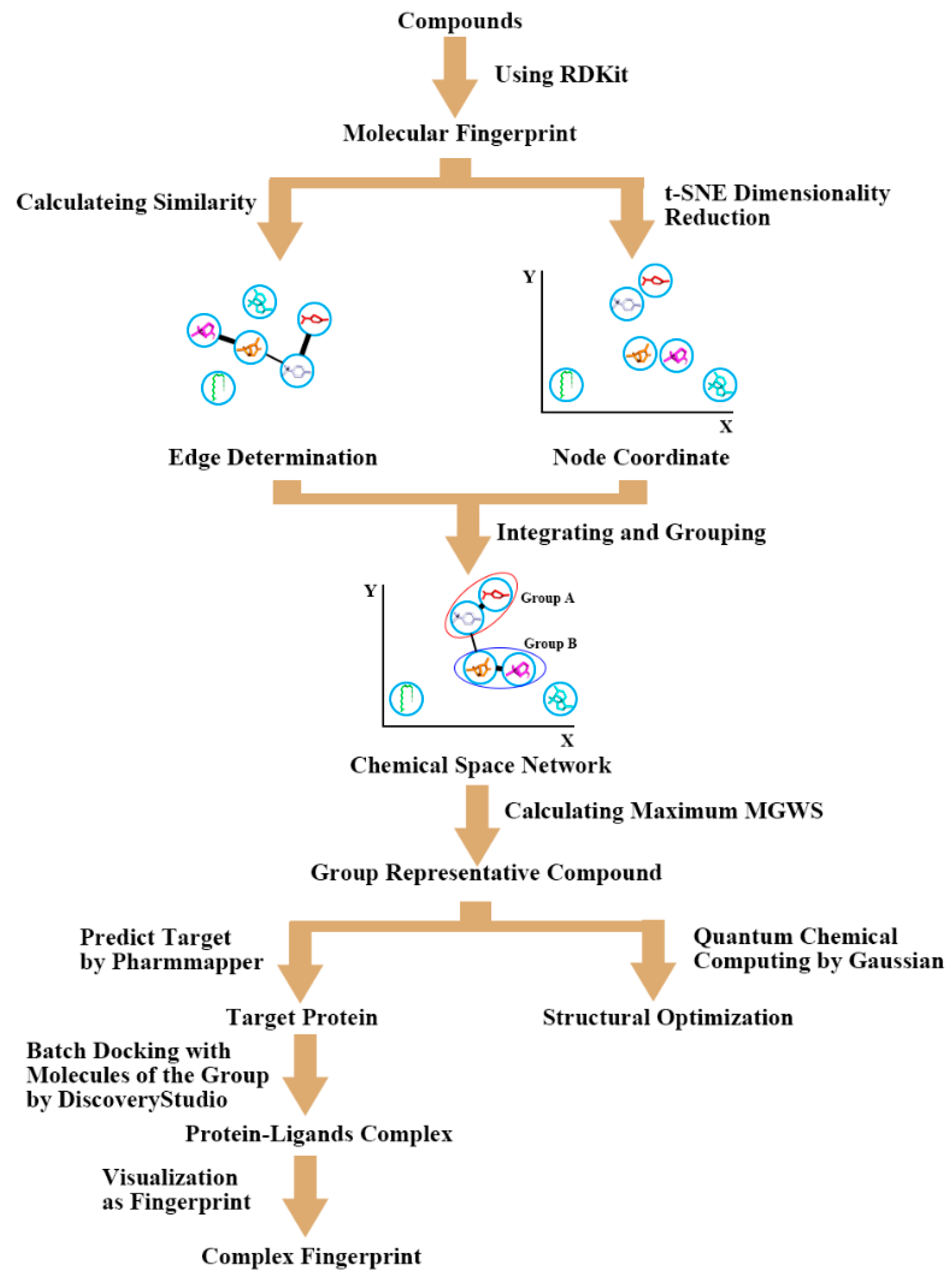
4.1. Cluster Analysis
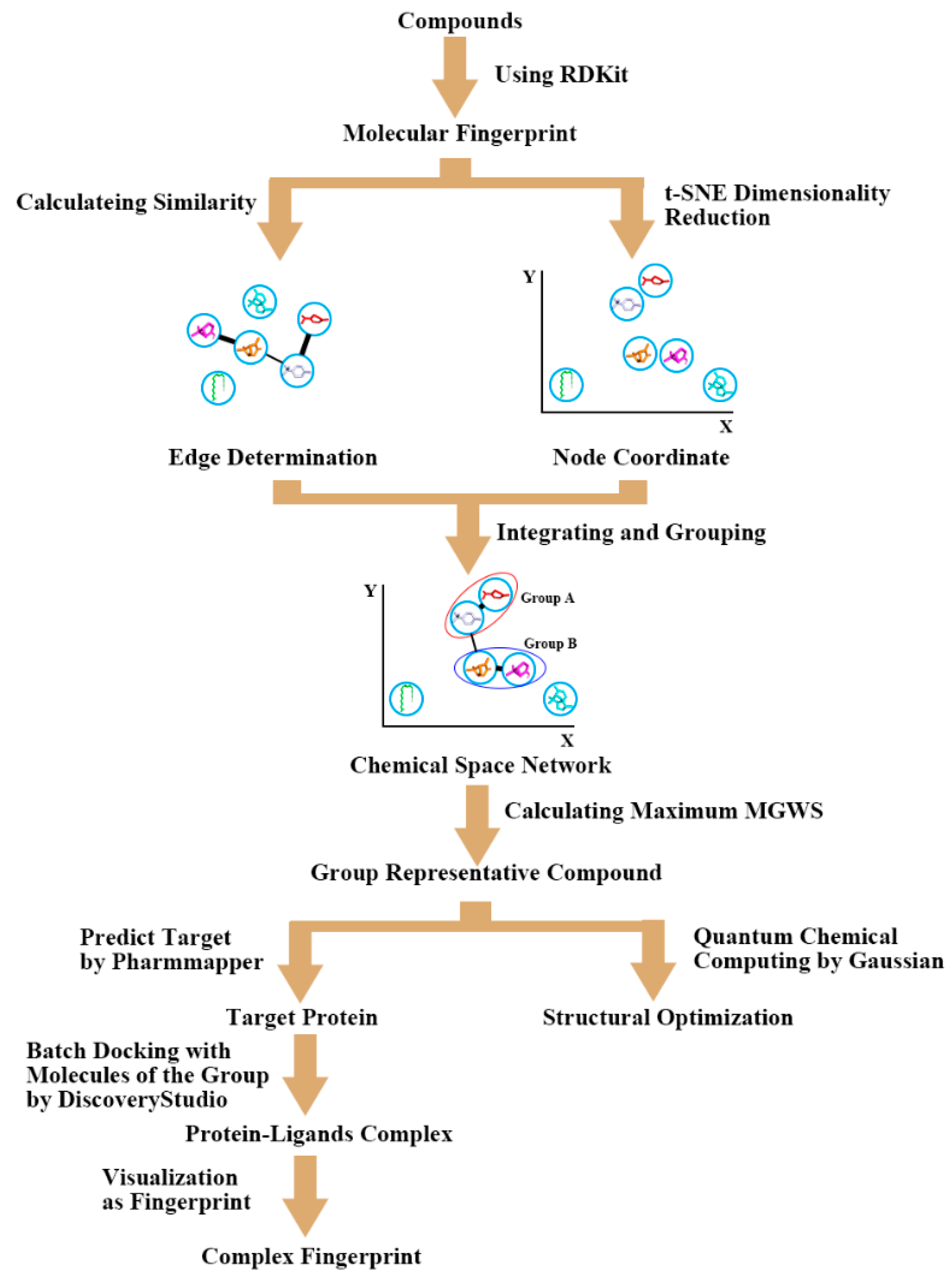
4.2. Group Docking and Structure Fingerprint Analysis
4.3. Quantum Chemical Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zamani, S.; Naderi, M.R.; Soleymani, A.; Nasiri, B.M. Sunflower (Helianthus annuus L.) biochemical properties and seed components affected by potassium fertilization under drought conditions. Ecotoxicol. Environ. Saf. 2020, 190, 110017. [Google Scholar] [CrossRef]
- Radonic, L.M.; Lewi, D.M.; López, N.E.; Hopp, H.E.; Escandón, A.S.; Bilbao, M.L. Sunflower (Helianthus annuus L.). Methods Mol. Biol. 2015, 1224, 47–55. [Google Scholar] [CrossRef]
- Lewi, D.M.; Hopp, H.E.; Escandón, A.S. Sunflower (Helianthus annuus L.). Methods Mol. Biol. 2006, 343, 291–297. [Google Scholar] [CrossRef]
- Smith, B.D. Eastern North America as an independent center of plant domestication. Proc. Natl. Acad. Sci. USA 2006, 103, 12223–12228. [Google Scholar] [CrossRef] [PubMed]
- Lawson, S.K.; Sharp, L.G.; Powers, C.N.; McFeeters, R.L.; Satyal, P.; Setzer, W.N. Essential Oil Compositions and Antifungal Activity of Sunflower (Helianthus) Species Growing in North Alabama. Appl. Sci. 2019, 9, 3179. [Google Scholar] [CrossRef]
- Shi, B.; Zhao, J. Recent progress on sunflower broomrape research in China. OCL 2020, 27, 30. [Google Scholar] [CrossRef]
- Serafini, M.; Peluso, I.; Raguzzini, A. Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 2010, 69, 273–278. [Google Scholar] [CrossRef]
- Wen, K.; Fang, X.; Yang, J.; Yao, Y.; Nandakumar, K.S.; Salem, M.L.; Cheng, K. Recent Research on Flavonoids and their Biomedical Applications. Curr. Med. Chem. 2021, 28, 1042–1066. [Google Scholar] [CrossRef] [PubMed]
- Cinelli, M.A.; Jones, A.D. Alkaloids of the Genus Datura: Review of a Rich Resource for Natural Product Discovery. Molecules 2021, 26, 2629. [Google Scholar] [CrossRef]
- Bhambhani, S.; Kondhare, K.R.; Giri, A.P. Diversity in Chemical Structures and Biological Properties of Plant Alkaloids. Molecules 2021, 26, 3374. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Tejada, S.; Setzer, W.N.; Gortzi, O.; Sureda, A.; Braidy, N.; Daglia, M.; Manayi, A.; Nabavi, S.M. Chlorogenic Acid and Mental Diseases: From Chemistry to Medicine. Curr. Neuropharmacol. 2017, 15, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.; Xiang, L. Pharmacological action and potential targets of chlorogenic acid. Adv. Pharmacol. 2020, 87, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Galúcio, C.S.; Souza, R.A.; Stahl, M.A.; Sbaite, P.; Benites, C.I.; Maciel, M.R.W. Physicochemical characterization of monoacylglycerols from sunflower oil. Procedia Food Sci. 2011, 1, 1459–1464. [Google Scholar] [CrossRef]
- Aguirre, M.; Velasco, J.; Ruiz-Méndez, M.V. Characterization of sunflower oils obtained separately by pressing and subsequent solvent extraction from a new line of seeds rich in phytosterols and conventional seeds. OCL—Ol. Corps Gras Lipides 2014, 21, 5. [Google Scholar] [CrossRef]
- Liu, X.S.; Gao, B.; Dong, Z.D.; Qiao, Z.A.; Yan, M.; Han, W.W.; Li, W.N.; Han, L. Chemical Compounds, Antioxidant Activities, and Inhibitory Activities Against Xanthine Oxidase of the Essential Oils From the Three Varieties of Sunflower (Helianthus annuus L.) Receptacles. Front. Nutr. 2021, 8, 737157. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Gao, B.; Li, X.L.; Li, W.N.; Qiao, Z.A.; Han, L. Chemical Composition and Antimicrobial and Antioxidant Activities of Essential Oil of Sunflower (Helianthus annuus L.) Receptacle. Molecules 2020, 25, 5244. [Google Scholar] [CrossRef]
- Zhang, B.; Vogt, M.; Maggiora, G.M.; Bajorath, J. Comparison of bioactive chemical space networks generated using substructure- and fingerprint-based measures of molecular similarity. J. Comput. Aided Mol. Des. 2015, 29, 595–608. [Google Scholar] [CrossRef]
- Wu, M.; Vogt, M.; Maggiora, G.M.; Bajorath, J. Design of chemical space networks on the basis of Tversky similarity. J. Comput. Aided Mol. Des. 2016, 30, 1–12. [Google Scholar] [CrossRef]
- Vega de León, A.; Bajorath, J. Design of chemical space networks incorporating compound distance relationships. F1000Research 2016, 5, 2634. [Google Scholar] [CrossRef]
- Vass, M.; Kooistra, A.J.; Ritschel, T.; Leurs, R.; de Esch, I.J.; de Graaf, C. Molecular interaction fingerprint approaches for GPCR drug discovery. Curr. Opin. Pharmacol. 2016, 30, 59–68. [Google Scholar] [CrossRef]
- De las Heras, B.; Hoult, J.R. Non-cytotoxic inhibition of macrophage eicosanoid biosynthesis and effects on leukocyte functions and reactive oxygen species of two novel anti-inflammatory plant diterpenoids. Planta Med. 1994, 60, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Koba, K.; Yanagita, T. Health benefits of conjugated linoleic acid (CLA). Obes. Res. Clin. Pract. 2014, 8, e525–e532. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper server: A web server for potential drug target identification using pharmacophore mapping approach. Nucleic Acids Res. 2010, 38, W609–W614. [Google Scholar] [CrossRef]
- Hernandez-Guzman, F.G.; Higashiyama, T.; Pangborn, W.; Osawa, Y.; Ghosh, D. Structure of human estrone sulfatase suggests functional roles of membrane association. J. Biol. Chem. 2003, 278, 22989–22997. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Owen, C.P.; James, K.; Sampson, L.; Patel, C.K. Review of estrone sulfatase and its inhibitors--an important new target against hormone dependent breast cancer. Curr. Med. Chem. 2002, 9, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Verboven, C.; Rabijns, A.; De Maeyer, M.; Van Baelen, H.; Bouillon, R.; De Ranter, C. A structural basis for the unique binding features of the human vitamin D-binding protein. Nat. Struct. Biol. 2002, 9, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Fraley, M.E.; Garbaccio, R.M.; Arrington, K.L.; Hoffman, W.F.; Tasber, E.S.; Coleman, P.J.; Buser, C.A.; Walsh, E.S.; Hamilton, K.; Fernandes, C.; et al. Kinesin spindle protein (KSP) inhibitors. Part 2: The design, synthesis, and characterization of 2,4-diaryl-2,5-dihydropyrrole inhibitors of the mitotic kinesin KSP. Bioorganic Med. Chem. Lett. 2006, 16, 1775–1779. [Google Scholar] [CrossRef]
- Martin, L.J.; Bowen, M.T. Comparing Fingerprints for Ligand-Based Virtual Screening: A Fast and Scalable Approach for Unbiased Evaluation. J. Chem. Inf. Model. 2020, 60, 4536–4545. [Google Scholar] [CrossRef]
- Lovrić, M.; Molero, J.M.; Kern, R. PySpark and RDKit: Moving towards Big Data in Cheminformatics. Mol. Inf. 2019, 38, e1800082. [Google Scholar] [CrossRef]
- Coley, C.W.; Green, W.H.; Jensen, K.F. RDChiral: An RDKit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application. J. Chem. Inf. Model. 2019, 59, 2529–2537. [Google Scholar] [CrossRef] [PubMed]
- Kruger, F.; Stiefl, N.; Landrum, G.A. rdScaffoldNetwork: The Scaffold Network Implementation in RDKit. J. Chem. Inf. Model. 2020, 60, 3331–3335. [Google Scholar] [CrossRef]
- Bac, J.; Mirkes, E.M.; Gorban, A.N.; Tyukin, I.; Zinovyev, A. Scikit-Dimension: A Python Package for Intrinsic Dimension Estimation. Entropy 2021, 23, 1368. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- Linderman, G.C.; Steinerberger, S. Clustering with t-SNE, provably. SIAM J. Math. Data Sci. 2019, 1, 313–332. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; García García, D.; Lijffijt, J.; Santos-Rodríguez, R.; De Bie, T. Conditional t-SNE: More informative t-SNE embeddings. Mach. Learn. 2021, 110, 2905–2940. [Google Scholar] [CrossRef]
- Le, T.; Winter, R.; Noé, F.; Clevert, D.A. Neuraldecipher—Reverse-engineering extended-connectivity fingerprints (ECFPs) to their molecular structures. Chem. Sci. 2020, 11, 10378–10389. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Morley, C.; Hutchison, G.R. Pybel: A Python wrapper for the OpenBabel cheminformatics toolkit. Chem. Cent. J. 2008, 2, 5. [Google Scholar] [CrossRef]
- Lehtola, S.; Dimitrova, M.; Fliegl, H.; Sundholm, D. Benchmarking Magnetizabilities with Recent Density Functionals. J. Chem. Theory Comput. 2021, 17, 1457–1468. [Google Scholar] [CrossRef]
- Sarkar, R.; Boggio-Pasqua, M.; Loos, P.F.; Jacquemin, D. Benchmarking TD-DFT and Wave Function Methods for Oscillator Strengths and Excited-State Dipole Moments. J. Chem. Theory Comput. 2021, 17, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, B.; Zimmerman, P.M.; Gavini, V. A Comparison of Exact and Model Exchange-Correlation Potentials for Molecules. J. Phys. Chem. Lett. 2021, 12, 12012–12019. [Google Scholar] [CrossRef]
- Han, W.; Zhu, J.; Wang, S.; Xu, D. Understanding the Phosphorylation Mechanism by Using Quantum Chemical Calculations and Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3565–3573. [Google Scholar] [CrossRef] [PubMed]
- Samuel, Y.; Garg, A.; Mulugeta, E. Synthesis, DFT Analysis, and Evaluation of Antibacterial and Antioxidant Activities of Sulfathiazole Derivatives Combined with In Silico Molecular Docking and ADMET Predictions. Biochem. Res. Int. 2021, 2021, 7534561. [Google Scholar] [CrossRef]
- Mora, J.R.; Kirby, A.J.; Nome, F. Theoretical study of the importance of the spectator groups on the hydrolysis of phosphate triesters. J. Org. Chem. 2012, 77, 7061–7070. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 2021, 23, 20323–20328. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | F36:PO | L47:Ak | S76:HD | S79:HD | P87:HA | P87:Ak | V88:Ak | H89:HD | H89:PO | M107:Ak | L110:Ak |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 51 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 |
| 19 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 |
| 31 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 |
| 14 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| 39 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 |
| 40 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 |
| 42 | 1 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| 27 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 1 |
| 32 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 |
| 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| No. | E116:HA | R119:Ak | I136:Ak | P137:Ak | L160:Ak | L172:Ak | Y211:PO | L214:HA | L214:Ak | E215:HD | A218:Ak |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 59 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| 68 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| 81 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 | 1 |
| 58 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| 65 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| 89 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 1 |
| 49 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 |
| 57 | 0 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
| 77 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Y.; Liu, K.; Han, L.; Han, W. Clustering Analysis, Structure Fingerprint Analysis, and Quantum Chemical Calculations of Compounds from Essential Oils of Sunflower (Helianthus annuus L.) Receptacles. Int. J. Mol. Sci. 2022, 23, 10169. https://doi.org/10.3390/ijms231710169
He Y, Liu K, Han L, Han W. Clustering Analysis, Structure Fingerprint Analysis, and Quantum Chemical Calculations of Compounds from Essential Oils of Sunflower (Helianthus annuus L.) Receptacles. International Journal of Molecular Sciences. 2022; 23(17):10169. https://doi.org/10.3390/ijms231710169
Chicago/Turabian StyleHe, Yi, Kaifeng Liu, Lu Han, and Weiwei Han. 2022. "Clustering Analysis, Structure Fingerprint Analysis, and Quantum Chemical Calculations of Compounds from Essential Oils of Sunflower (Helianthus annuus L.) Receptacles" International Journal of Molecular Sciences 23, no. 17: 10169. https://doi.org/10.3390/ijms231710169
APA StyleHe, Y., Liu, K., Han, L., & Han, W. (2022). Clustering Analysis, Structure Fingerprint Analysis, and Quantum Chemical Calculations of Compounds from Essential Oils of Sunflower (Helianthus annuus L.) Receptacles. International Journal of Molecular Sciences, 23(17), 10169. https://doi.org/10.3390/ijms231710169