
The Prostacyclin Analogue Iloprost Modulates CXCL10 in Systemic Sclerosis

 , ,
, ,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Iloprost Decreased Interferon (IFN)γ and Tumor Necrosis Factor (TNF)α-Induced CXCL10 Secretion in Human Endothelial Cells and Dermal Fibroblasts
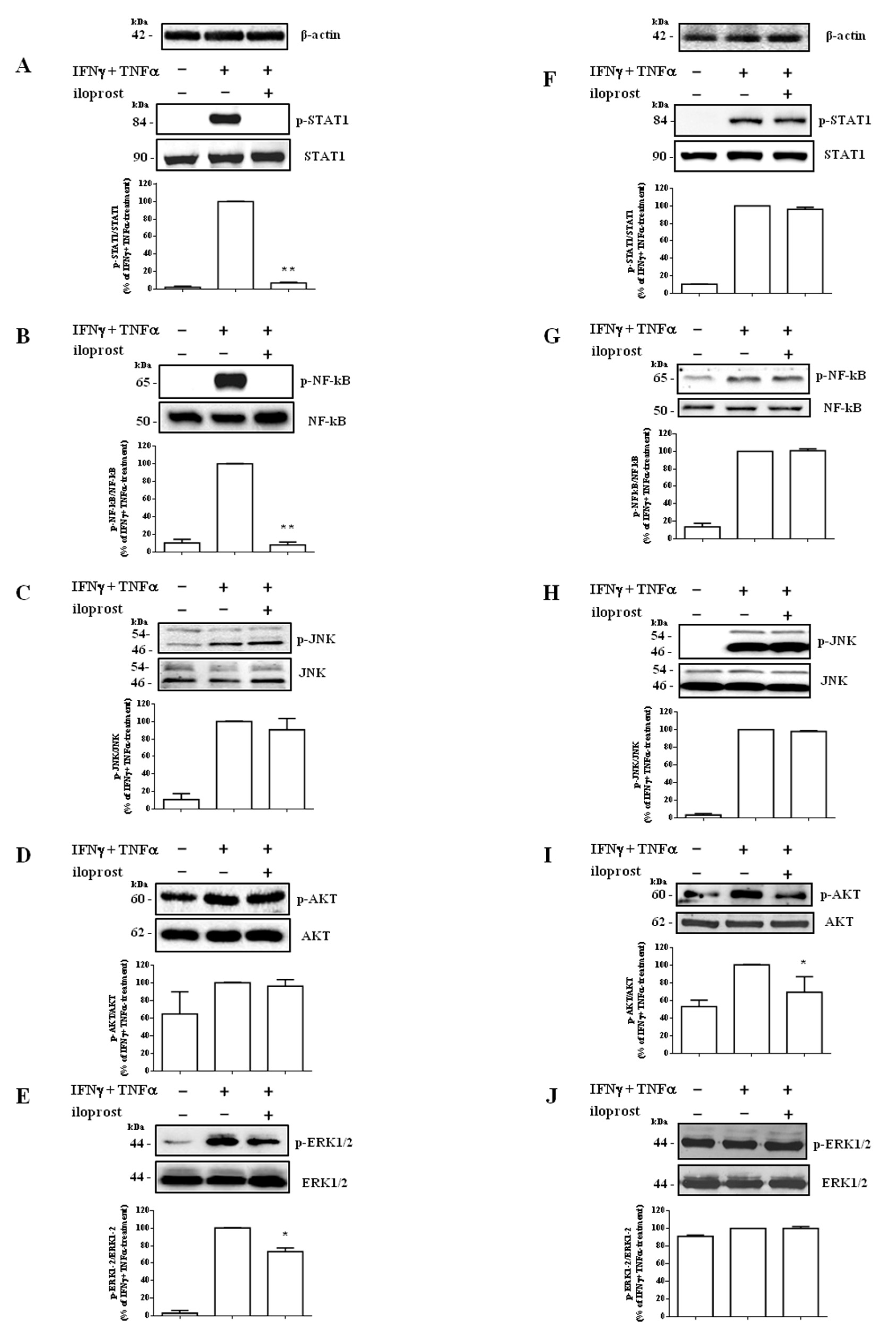
2.2. Iloprost Targeted IFNγ and TNFα-Dependent Signaling Only in Human Endothelial Cells
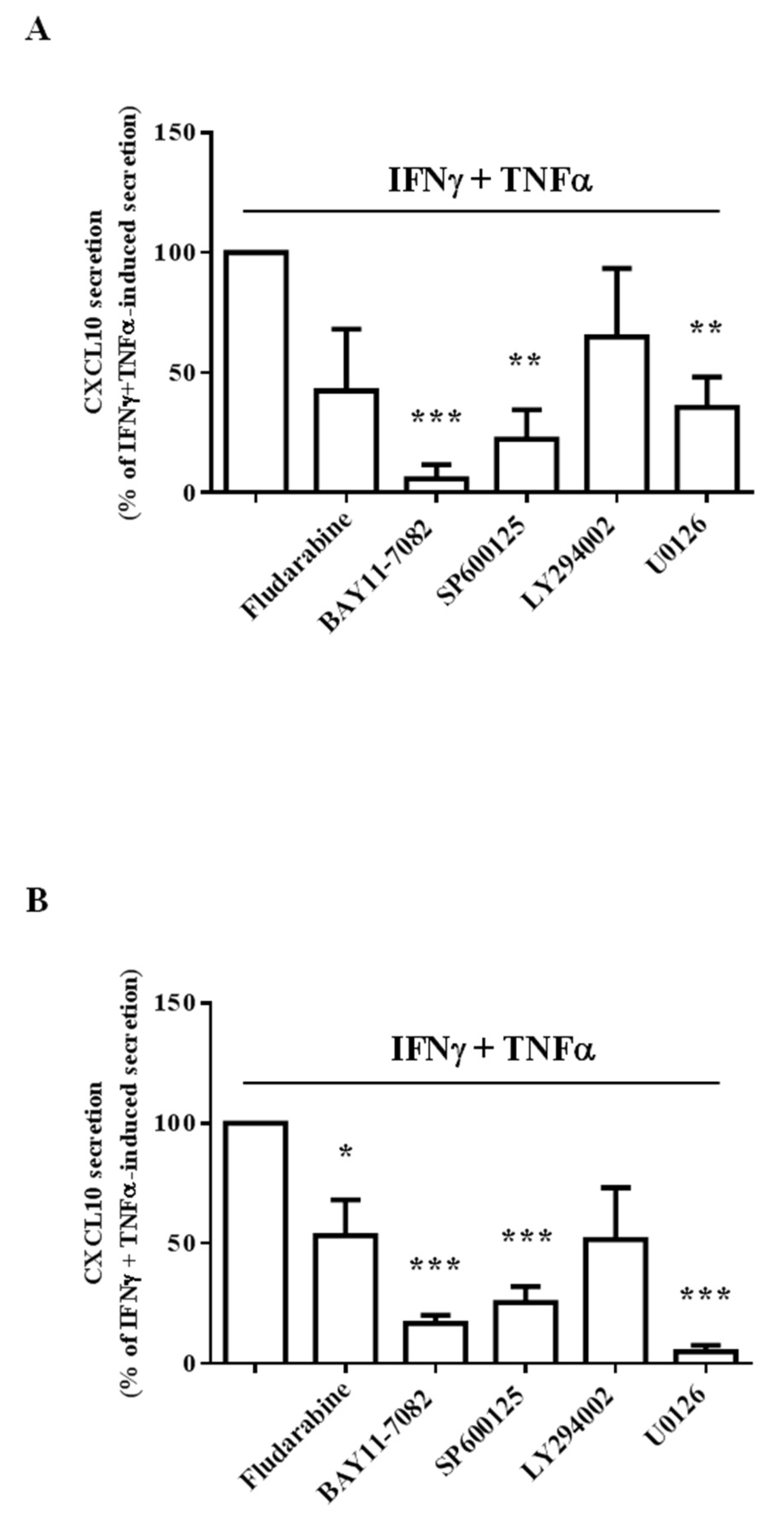
2.3. Different Intracellular Path Engagement in CXCL10 Protein Release by Human Endothelial Cells and Fibroblasts
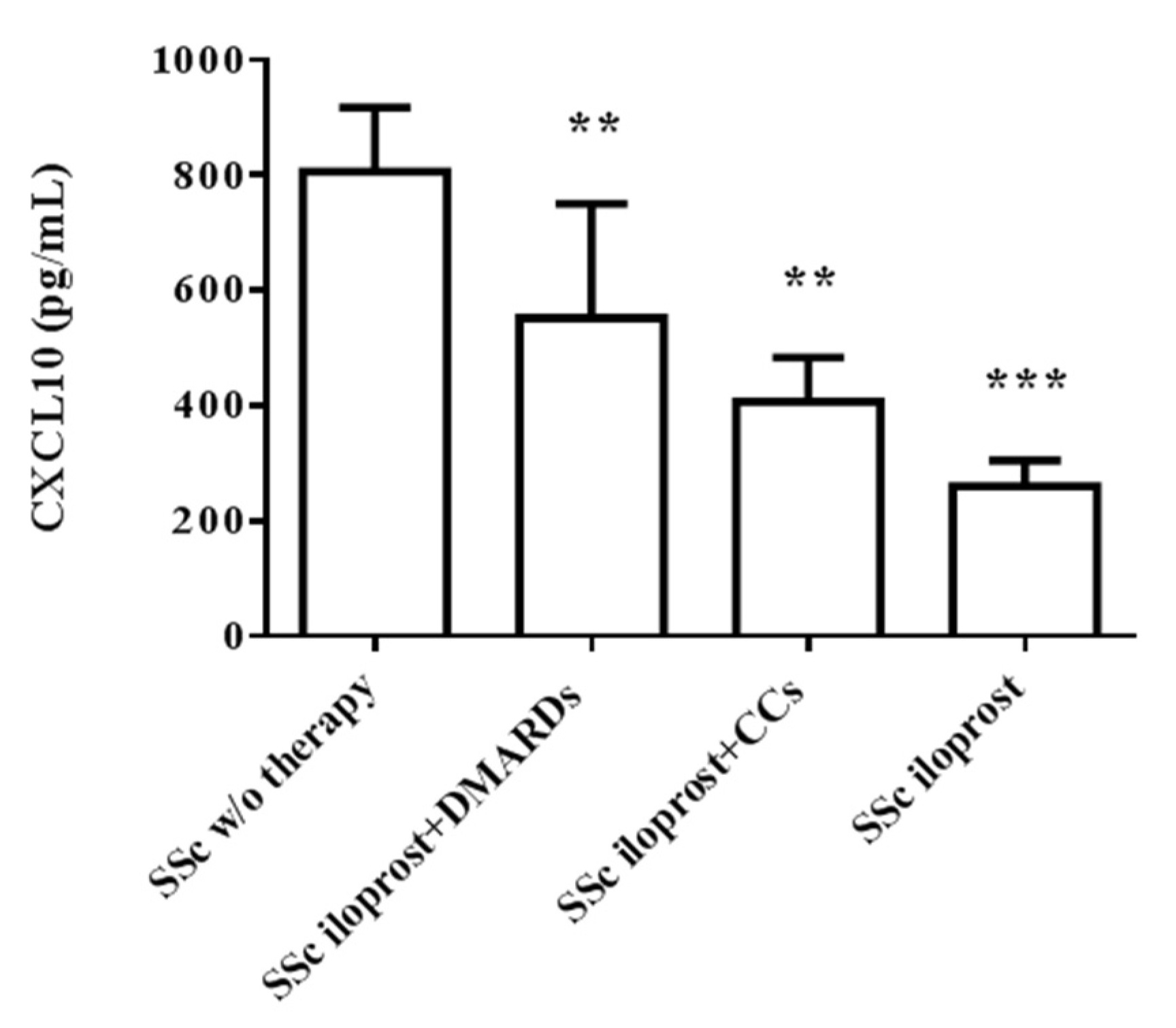
2.4. Iloprost Reduced Serum CXCL10 in SSc
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Chemicals
4.3. Cell Cultures
4.4. Enzyme-Linked ImmunoSorbent Assay (ELISA)
4.5. RNA Extraction, Reverse Transcription, and Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Investig. 2007, 117, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Chora, I.; Guiducci, S.; Manetti, M.; Romano, E.; Mazzotta, C.; Bellando-Randone, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M.; Soares, R. Vascular biomarkers and correlation with peripheral vasculopathy in systemic sclerosis. Autoimmun. Rev. 2015, 14, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K. Strategy for treatment of fibrosis in systemic sclerosis: Present and future. J. Dermatol. 2016, 43, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Pattanaik, D.; Brown, M.; Postlethwaite, A.E. Vascular involvement in systemic sclerosis (scleroderma). J. Inflamm. Res. 2011, 4, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Altorok, N.; Wang, Y.; Kahaleh, B. Endothelial dysfunction in systemic sclerosis. Curr. Opin. Rheumatol. 2014, 26, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Chora, I.; Romano, E.; Manetti, M.; Mazzotta, C.; Costa, R.; Machado, V.; Cortez, A.; Bruni, C.; Lepri, G.; Guiducci, S.; et al. Evidence for a Derangement of the Microvascular System in Patients with a Very Early Diagnosis of Systemic Sclerosis. J. Rheumatol. 2017, 44, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Mechanisms in the loss of capillaries in systemic sclerosis: Angiogenesis versus vasculogenesis. J. Cell. Mol. Med. 2010, 14, 1241–1254. [Google Scholar] [CrossRef]
- Koenig, M.; Joyal, F.; Fritzler, M.J.; Roussin, A.; Abrahamowicz, M.; Boire, G.; Goulet, J.R.; Rich, E.; Grodzicky, T.; Raymond, Y.; et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: A twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum. 2008, 58, 3902–3912. [Google Scholar] [CrossRef]
- Antonelli, A.; Ferri, C.; Fallahi, P.; Ferrari, S.M.; Giuggioli, D.; Colaci, M.; Manfredi, A.; Frascerra, S.; Franzoni, F.; Galetta, F.; et al. CXCL10 (alpha) and CCL2 (beta) chemokines in systemic sclerosis-a longitudinal study. Rheumatology 2008, 47, 45–49. [Google Scholar] [CrossRef]
- Truchetet, M.E.; Brembilla, N.C.; Chizzolini, C. Current Concepts on the Pathogenesis of Systemic Sclerosis. Clin. Rev. Allergy Immunol. 2021; Online ahead of print. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front. Immunol. 2018, 15, 1970. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Corrado, A. The Th1 chemokine IP-10 in Systemic sclerosis. Clin. Ter. 2014, 165, e436–e441. [Google Scholar] [CrossRef] [PubMed]
- Crescioli, C.; Corinaldesi, C.; Riccieri, V.; Raparelli, V.; Vasile, M.; Del Galdo, F.; Valesini, G.; Lenzi, A.; Basili, S.; Antinozzi, C. Association of circulating CXCL10 and CXCL11 with systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Avouac, J.; Fransen, J.; Walker, U.A.; Riccieri, V.; Smith, V.; Muller, C.; Miniati, I.; Tarner, I.H.; Bellando Randone, S.; Cutolo, M.; et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: Results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann. Rheum. Dis. 2011, 70, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Valentini, G: Undifferentiated Connective Tissue Disease at risk for systemic sclerosis (SSc) (so far referred to as very early/early SSc or pre-SSc). Autoimmun. Rev. 2015, 14, 210–213. [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Vazirinejad, R.; Ahmadi, Z.; Kazemi Arababadi, M.; Hassanshahi, G.; Kennedy, D. The biological functions, structure and sources of CXCL10 and its outstanding part in the pathophysiology of multiple sclerosis. Neuroimmunomodulation 2014, 21, 322–330. [Google Scholar] [CrossRef]
- Scolletta, S.; Buonamano, A.; Sottili, M.; Giomarelli, P.; Biagioli, B.; Vannelli, G.B.; Serio, M.; Romagnani, P.; Crescioli, C. CXCL10 release in cardiopulmonary bypass: An in vivo and in vitro study. Biomed. Aging Pathol. 2012, 2, 187–194. [Google Scholar] [CrossRef]
- Sagrinati, C.; Sottili, M.; Mazzinghi, B.; Borgogni, E.; Adorini, L.; Serio, M.; Romagnani, P.; Crescioli, C. Comparison between VDR analogs and current immunosuppressive drugs in relation to CXCL10 secretion by human renal tubular cells. Transpl. Int. 2010, 23, 914–923. [Google Scholar] [CrossRef]
- Crescioli, C.; Squecco, R.; Cosmi, L.; Sottili, M.; Gelmini, S.; Borgogni, E.; Sarchielli, E.; Scolletta, S.; Francini, F.; Annunziato, F.; et al. Immunosuppression in cardiac graft rejection: A human in vitro model to study the potential use of new immunomodulatory drugs. Exp. Cell Res. 2008, 314, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Sottili, M.; Cosmi, L.; Borgogni, E.; Sarchielli, E.; Maggi, L.; Francalanci, M.; Vannelli, G.B.; Ronconi, E.; Adorini, L.; Annunziato, F.; et al. Immunomodulatory effects of BXL-01-0029, a less hypercalcemic vitamin D analogue, in human cardiomyocytes and T cells. Exp. Cell Res. 2009, 315, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Corinaldesi, C.; Ross, R.L.; Abignano, G.; Antinozzi, C.; Marampon, F.; Di Luigi, L.; Buch, M.H.; Riccieri, V.; Lenzi, A.; Crescioli, C.; et al. Muscle Damage in Systemic Sclerosis and CXCL10: The Potential Therapeutic Role of PDE5 Inhibition. Int. J. Mol. Sci. 2021, 22, 2894. [Google Scholar] [CrossRef] [PubMed]
- Giannattasio, S.; Citarella, A.; Trocchianesi, S.; Filardi, T.; Morano, S.; Lenzi, A.; Ferretti, E.; Crescioli, C. Cell-Target-Specific Anti-Inflammatory Effect of Empagliflozin: In Vitro Evidence in Human Cardiomyocytes. Front. Mol. Biosci. 2022, 9, 879522. [Google Scholar] [CrossRef] [PubMed]
- Di Luigi, L.; Corinaldesi, C.; Colletti, M.; Scolletta, S.; Antinozzi, C.; Vannelli, G.B.; Giannetta, E.; Gianfrilli, D.; Isidori, A.M.; Migliaccio, S.; et al. Phosphodiesterase Type 5 Inhibitor Sildenafil Decreases the Proinflammatory Chemokine CXCL10 in Human Cardiomyocytes and in Subjects with Diabetic Cardiomyopathy. Inflammation 2016, 39, 1238–1252. [Google Scholar] [CrossRef]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef]
- Lai, Y.J.; Pullamsetti, S.S.; Dony, E.; Weissmann, N.; Butrous, G.; Banat, G.A.; Ghofrani, H.A.; Seeger, W.; Grimminger, F.; Schermuly, R.T. Role of the prostanoid EP4 receptor in iloprost-mediated vasodilatation in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 188–196. [Google Scholar] [CrossRef]
- Adderley, S.P.; Dufaux, E.A.; Sridharan, M.; Bowles, E.A.; Hanson, M.S.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Iloprost- and isoproterenol-induced increases in cAMP are regulated by different phosphodiesterases in erythrocytes of both rabbits and humans. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1617–H1624. [Google Scholar] [CrossRef]
- Newell, E.A.; Exo, J.L.; Verrier, J.D.; Jackson, T.C.; Gillespie, D.G.; Janesko-Feldman, K.; Kochanek, P.M.; Jackson, E.K. 2′,3′-cAMP, 3′-AMP, 2′-AMP and adenosine inhibit TNF-α and CXCL10 production from activated primary murine microglia via A2A receptors. Brain Res. 2015, 1594, 27–35. [Google Scholar] [CrossRef]
- Castro, S.V.; Jimenez, S.A. Biomarkers in systemic sclerosis. Biomark. Med. 2010, 4, 133–147. [Google Scholar] [CrossRef]
- Iwamoto, N.; Distler, O. Molecular targets for therapy in systemic sclerosis. Fibrogenesis Tissue Repair 2012, 5, S19. [Google Scholar] [CrossRef] [PubMed]
- Hummers, L.K. The current state of biomarkers in systemic sclerosis. Curr. Rheumatol. Rep. 2010, 12, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, Z.; Ye, D.; Feng, Y.; Liu, M.; Xu, Y.; Wang, M.; Zhang, J.; Liu, J.; Zhao, M.; et al. The Role of CXC Chemokines in Cardiovascular Diseases. Front. Pharmacol. 2022, 12, 765768. [Google Scholar] [CrossRef] [PubMed]
- Sahin, H.; Borkham-Kamphorst, E.; do O, N.T.; Berres, M.L.; Kaldenbach, M.; Schmitz, P.; Weiskirchen, R.; Liedtke, C.; Streetz, K.L.; Maedler, K.; et al. Proapoptotic effects of the chemokine, CXCL 10 are mediated by the noncognate receptor TLR4 in hepatocytes. Hepatology 2013, 57, 797–805. [Google Scholar] [CrossRef]
- Bellando Randone, S.; George, J.; Mazzotta, C.; Guiducci, S.; Furst, D.E.; Mor, A.; Matucci-Cerinic, M. Angiostatic and Angiogenic Chemokines in Systemic Sclerosis: An Overview. J. Scleroderma Relat. Disord. 2017, 2, 1–10. [Google Scholar] [CrossRef]
- Liu, X.; Mayes, M.D.; Tan, F.K.; Wu, M.; Reveille, J.D.; Harper, B.E.; Draeger, H.T.; Gonzalez, E.B.; Assassi, S. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum. 2013, 65, 226–235. [Google Scholar] [CrossRef]
- Scolletta, S.; Colletti, M.; Di Luigi, L.; Crescioli, C. Vitamin D receptor agonists target CXCL10: New therapeutic tools for resolution of inflammation. Mediat. Inflamm. 2013, 2013, 876319. [Google Scholar] [CrossRef]
- Romagnani, P.; Lasagni, L.; Annunziato, F.; Serio, M.; Romagnani, S. CXC chemokines: The regulatory link between inflammation and angiogenesis. Trends Immunol. 2004, 4, 201–209. [Google Scholar] [CrossRef]
- Antonelli, A.; Ferrari, S.M.; Giuggioli, D.; Ferrannini, E.; Ferri, C.; Fallahi, P. Chemokine (C-X-C motif) ligand (CXCL)10 in autoimmune diseases. Autoimmun. Rev. 2014, 13, 272–280. [Google Scholar] [CrossRef]
- Romagnani, P.; Crescioli, C. CXCL10: A candidate biomarker in transplantation. Clin. Chim. Acta 2012, 413, 1364–1373. [Google Scholar] [CrossRef]
- Crescioli, C. Chemokines and transplant outcome. Clin. Biochem. 2016, 49, 355–362. [Google Scholar] [CrossRef]
- Crescioli, C.; Cosmi, L.; Borgogni, E.; Santarlasci, V.; Gelmini, S.; Sottili, M.; Sarchielli, E.; Mazzinghi, B.; Francalanci, M.; Pezzatini, A.; et al. Methimazole inhibits CXC chemokine ligand 10 secretion in human thyrocytes. J. Endocrinol. 2007, 195, 145–155. [Google Scholar] [CrossRef]
- Aota, K.; Kani, K.; Yamanoi, T.; Nakashiro, K.; Ishimaru, N.; Azuma, M. Distinct Regulation of CXCL10 Production by Cytokines in Human Salivary Gland Ductal and Acinar Cells. Inflammation 2018, 41, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Eissner, G.; Multhoff, G.; Gerbitz, A.; Kirchner, S.; Bauer, S.; Haffner, S.; Sondermann, D.; Andreesen, R.; Holler, E. Fludarabine induces apoptosis, activation, and allogenicity in human endothelial and epithelial cells: Protective effect of defibrotide. Blood 2002, 100, 334–340. [Google Scholar] [CrossRef]
- Oskolkova, O.; Sarich, N.; Tian, Y.; Gawlak, G.; Meng, F.; Bochkov, V.N.; Berdyshev, E.; Birukova, A.A.; Birukov, K.G. Incorporation of iloprost in phospholipase-resistant phospholipid scaffold enhances its barrier protective effects on pulmonary endothelium. Sci. Rep. 2018, 8, 879. [Google Scholar] [CrossRef] [PubMed]
- Clapp, L.H.; Gurung, R. The mechanistic basis of prostacyclin and its stable analogues in pulmonary arterial hypertension: Role of membrane versus nuclear receptors. Prostaglandins Other Lipid Mediat. 2015, 120, 56–71. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Stiles, J.K. The emerging role of CXCL10 in cancer. Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, J.; Wang, T.; Zhang, X.; Liu, D. CXCL10/CXCR3 axis promotes the invasion of gastric cancer via PI3K/AKT pathway-dependent MMPs production. Biomed. Pharmacother. 2016, 82, 479–488. [Google Scholar] [CrossRef]
- Callahan, V.; Hawks, S.; Crawford, M.A.; Lehman, C.W.; Morrison, H.A.; Ivester, H.M.; Akhrymuk, I.; Boghdeh, N.; Flor, R.; Finkielstein, C.V.; et al. The Pro-Inflammatory Chemokines CXCL9, CXCL10 and CXCL11 Are Upregulated Following SARS-CoV-2 Infection in an AKT-Dependent Manner. Viruses 2021, 13, 1062. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Chen, J.H.; Yeh, W.L. Pulmonary fibroblasts-secreted CXCL10 polarizes alveolar macrophages under pro-inflammatory stimuli. Toxicol. Appl. Pharmacol. 2019, 380, 114698. [Google Scholar] [CrossRef]
- Ortona, E.; Maselli, A.; Delunardo, F.; Colasanti, T.; Giovannetti, A.; Pierdominici, M. Relationship between redox status and cell fate in immunity and autoimmunity. Antioxid. Redox Signal. 2014, 21, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013, 65, 1953–1962. [Google Scholar] [CrossRef]
- van den Hoogen, F.; Khanna, D.; Fransen, J.; Johnson, S.R.; Baron, M.; Tyndall, A.; Matucci-Cerinic, M.; Naden, R.P.; Medsger, T.A., Jr.; Carreira, P.E.; et al. 2013 classification criteria for systemic sclerosis: An American college of rheumatology/European league against rheumatism collaborative initiative. Ann. Rheum. Dis. 2013, 72, 1747–1755. [Google Scholar] [CrossRef]
- Giannattasio, S.; Corinaldesi, C.; Colletti, M.; Di Luigi, L.; Antinozzi, C.; Filardi, T.; Scolletta, S.; Basili, S.; Lenzi, A.; Morano, S.; et al. The phosphodiesterase 5 inhibitor sildenafil decreases the proinflammatory chemokine IL-8 in diabetic cardiomyopathy: In vivo and in vitro evidence. J. Endocrinol. Investig. 2019, 42, 715–725. [Google Scholar] [CrossRef]
- Vischer, U.M. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J. Thromb. Haemost. 2006, 4, 1186–1193. [Google Scholar] [CrossRef]
- Ema, M.; Yokomizo, T.; Wakamatsu, A.; Terunuma, T.; Yamamoto, M.; Takahashi, S. Primitive erythropoiesis from mesodermal precursors expressing VE-cadherin, PECAM-1, Tie2, endoglin, and CD34 in the mouse embryo. Blood 2006, 108, 4018–4024. [Google Scholar] [CrossRef]
- Mannelli, M.; Ferruzzi, P.; Luciani, P.; Crescioli, C.; Buci, L.; Corona, G.; Serio, M.; Peri, A. Cushing’s syndrome in a patient with bilateral macronodular adrenal hyperplasia responding to cisapride: An in vivo and in vitro study. J. Clin. Endocrinol. Metab. 2003, 88, 4616–4622. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Di Luigi, L.; Sottili, M.; Antinozzi, C.; Vannelli, G.B.; Romanelli, F.; Riccieri, V.; Valesini, G.; Lenzi, A.; Crescioli, C. The vitamin D receptor agonist BXL-01-0029 as a potential new pharmacological tool for the treatment of inflammatory myopathies. PLoS ONE 2013, 8, e77745. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.; Boorsma, D.M.; van Beek, P.J.; Nieboer, C.; Stoof, T.J.; Willemze, R.; Tensen, C.P. Differential expression of CXCR3 targeting chemokines CXCL10, CXCL9, and CXCL11 in different types of skin inflammation. J. Pathol. 2001, 194, 398–405. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value |
|---|---|
| Sex, female/male | 2/23 |
| Age, mean ± SD, years | 60 ± 13.3 |
| Disease duration, mean ± SD, years | 19 ± 15.3 |
| Form (limited SSc/diffuse SSc) | 14/11 |
| Raynaud Phenomenon (n/%) | 25/100 |
| Digital Ulcers (n/%) | 14/56 |
| Interstitial Lung Disease (n/%) | 6/24 |
| Pulmonary Arterial Hypertension (n/%) | 1/4 |
| Anti-nuclear Antibodies (ANA) (n/%) | 25/100 |
| Anti-topoisomerase I Antibodies (anti-Scl70) (n/%) | 12/48 |
| Anti-centromere Antibodies (ACA) (n/%) | 8/32 |
| Drug therapy (in combination with iloprost) | |
| DMARDs (n/%) | 9/36 |
| Hydroxychloroquine (n/%) | 1/4 |
| Azathioprine (n/%) | 3/12 |
| Cyclosporine (n/%) | 3/12 |
| Methotrexate (n/%) | 2/8 |
| Corticosteroids (n/%) | 9/36 |
| Vasoactive drugs (n/%) | 25/100 |
| Iloprost (single therapy) (n/%) | 7/28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colasanti, T.; Stefanantoni, K.; Fantini, C.; Corinaldesi, C.; Vasile, M.; Marampon, F.; Di Luigi, L.; Antinozzi, C.; Sgrò, P.; Lenzi, A.; et al. The Prostacyclin Analogue Iloprost Modulates CXCL10 in Systemic Sclerosis. Int. J. Mol. Sci. 2022, 23, 10150. https://doi.org/10.3390/ijms231710150
Colasanti T, Stefanantoni K, Fantini C, Corinaldesi C, Vasile M, Marampon F, Di Luigi L, Antinozzi C, Sgrò P, Lenzi A, et al. The Prostacyclin Analogue Iloprost Modulates CXCL10 in Systemic Sclerosis. International Journal of Molecular Sciences. 2022; 23(17):10150. https://doi.org/10.3390/ijms231710150
Chicago/Turabian StyleColasanti, Tania, Katia Stefanantoni, Cristina Fantini, Clarissa Corinaldesi, Massimiliano Vasile, Francesco Marampon, Luigi Di Luigi, Cristina Antinozzi, Paolo Sgrò, Andrea Lenzi, and et al. 2022. "The Prostacyclin Analogue Iloprost Modulates CXCL10 in Systemic Sclerosis" International Journal of Molecular Sciences 23, no. 17: 10150. https://doi.org/10.3390/ijms231710150
APA StyleColasanti, T., Stefanantoni, K., Fantini, C., Corinaldesi, C., Vasile, M., Marampon, F., Di Luigi, L., Antinozzi, C., Sgrò, P., Lenzi, A., Riccieri, V., & Crescioli, C. (2022). The Prostacyclin Analogue Iloprost Modulates CXCL10 in Systemic Sclerosis. International Journal of Molecular Sciences, 23(17), 10150. https://doi.org/10.3390/ijms231710150