
Tankyrase Inhibition Attenuates Cardiac Dilatation and Dysfunction in Ischemic Heart Failure
 , , ,
, , ,
Abstract
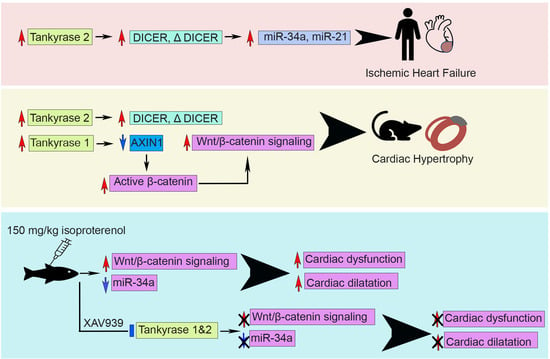
:
1. Introduction
2. Results
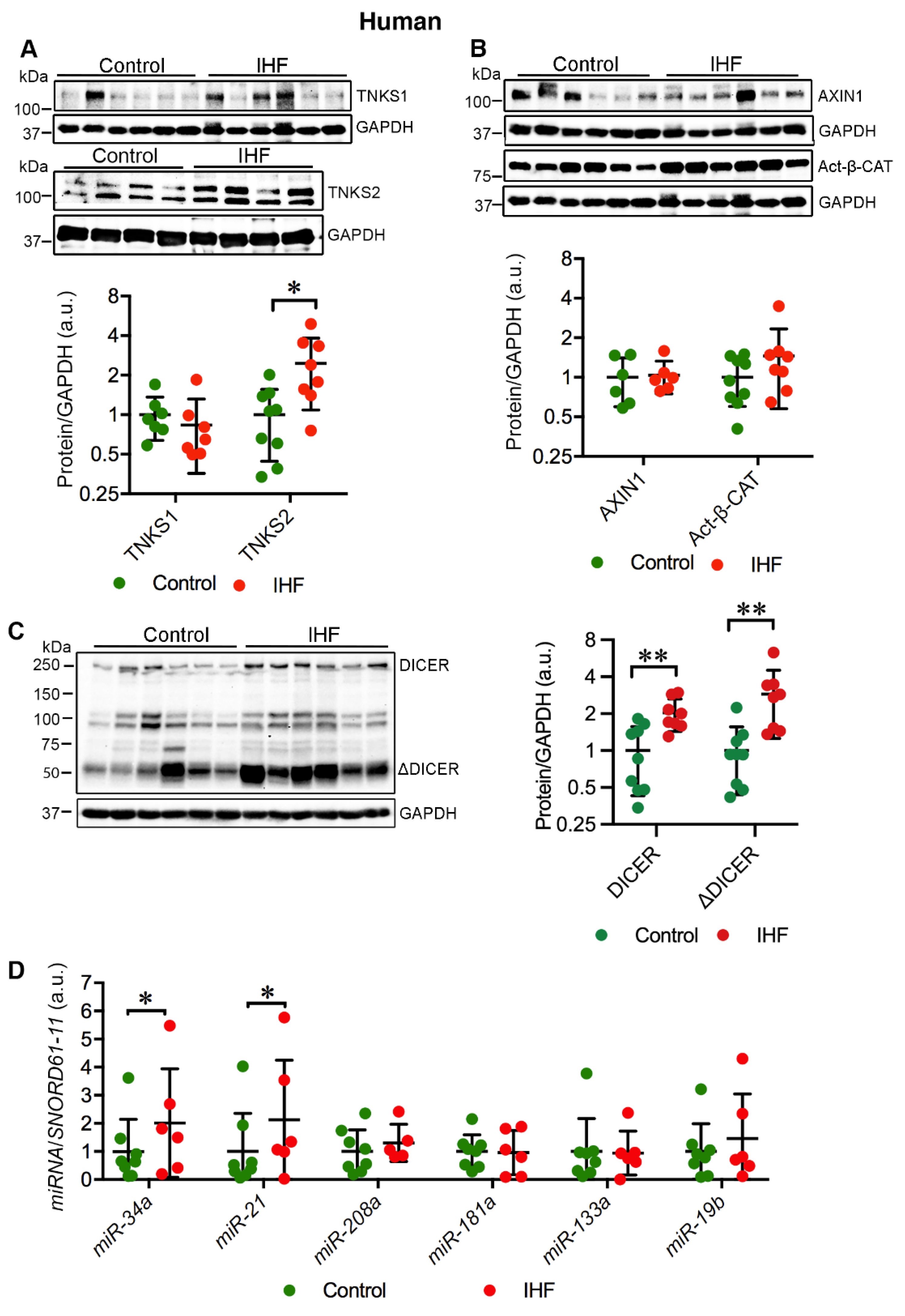
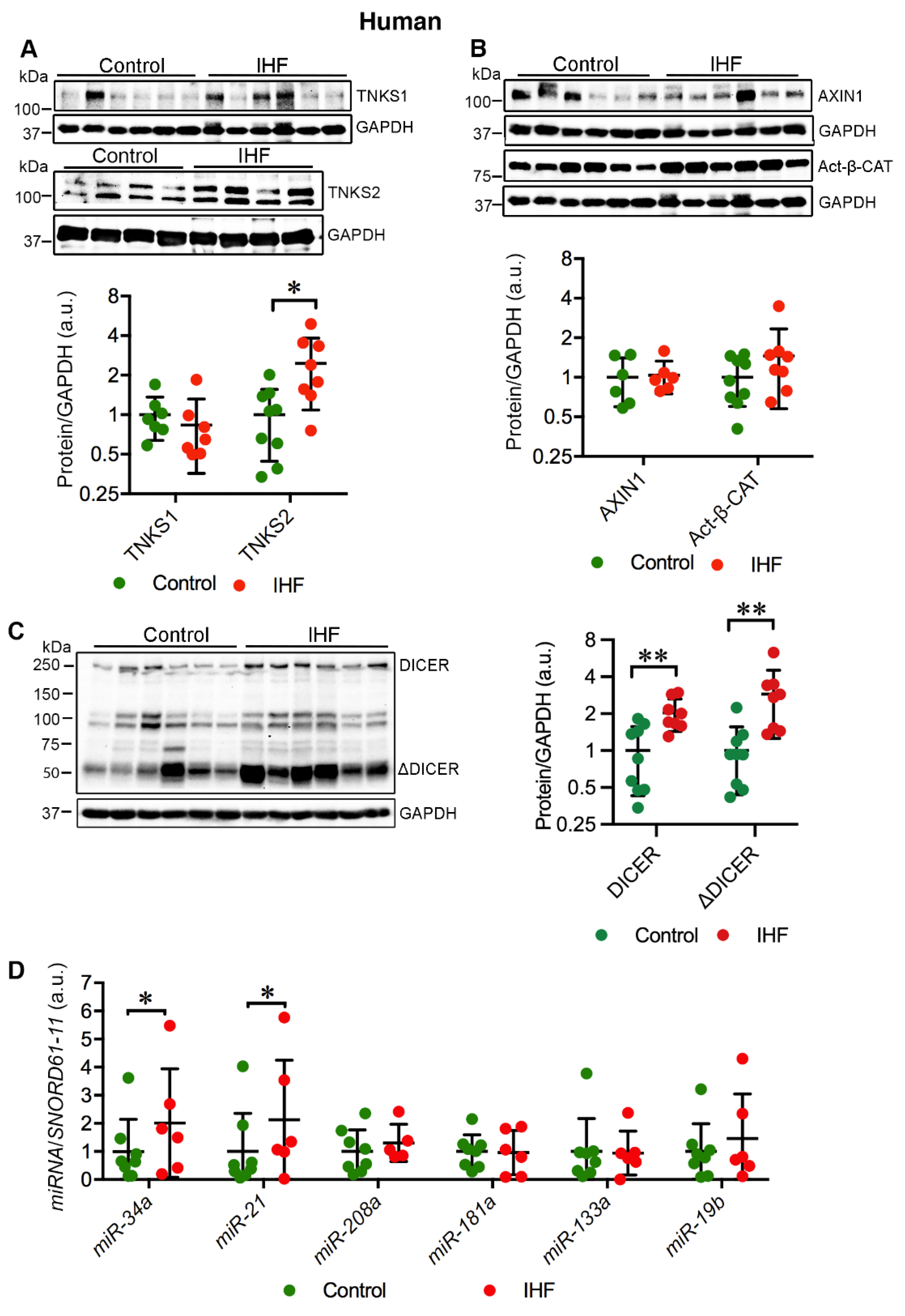
2.1. Cardiac TNKS2 and DICER Are Augmented in IHF Patients
2.2. Cardiac miR-34a-5p and miR-21-5p Are Upregulated in IHF Patients
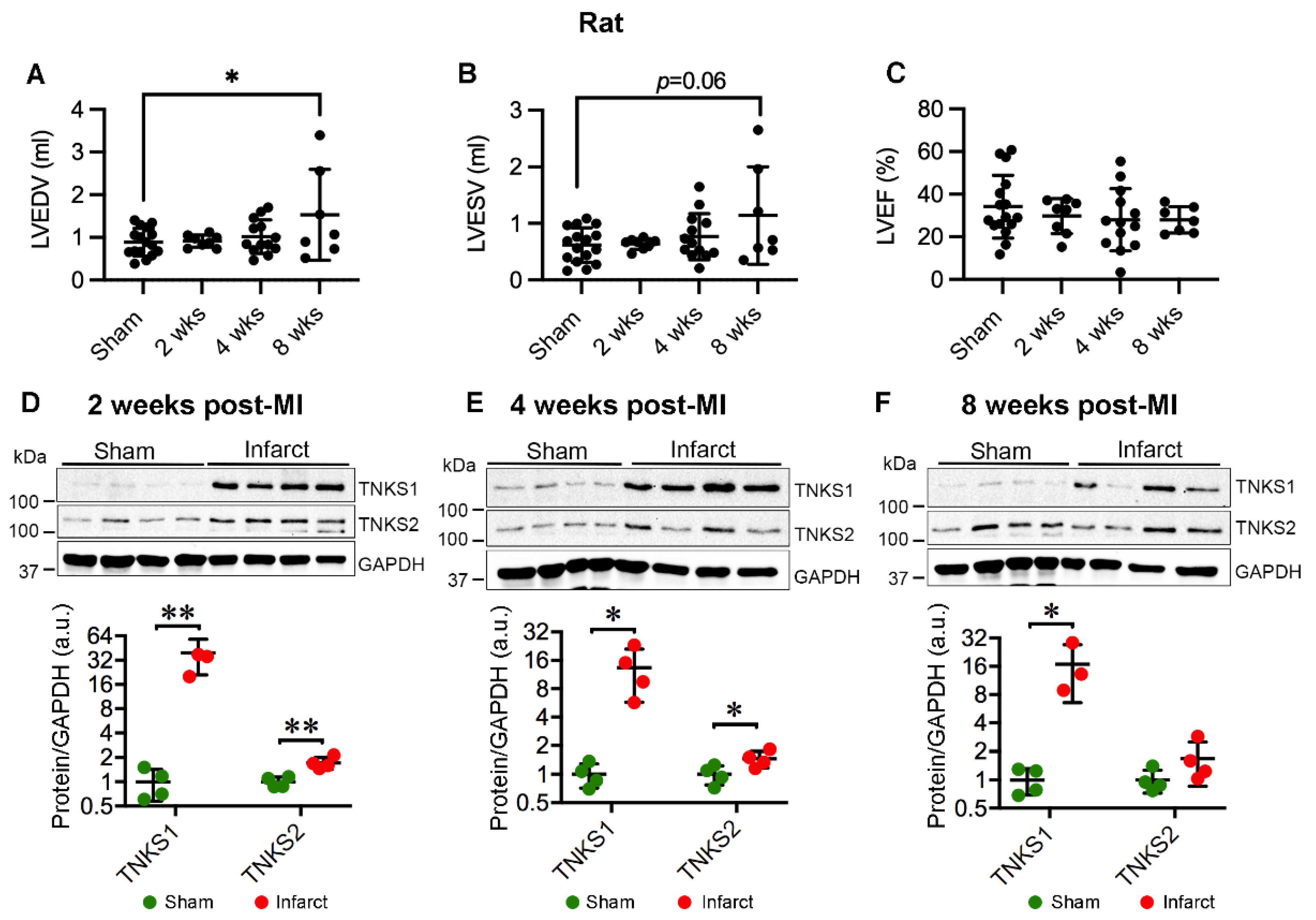
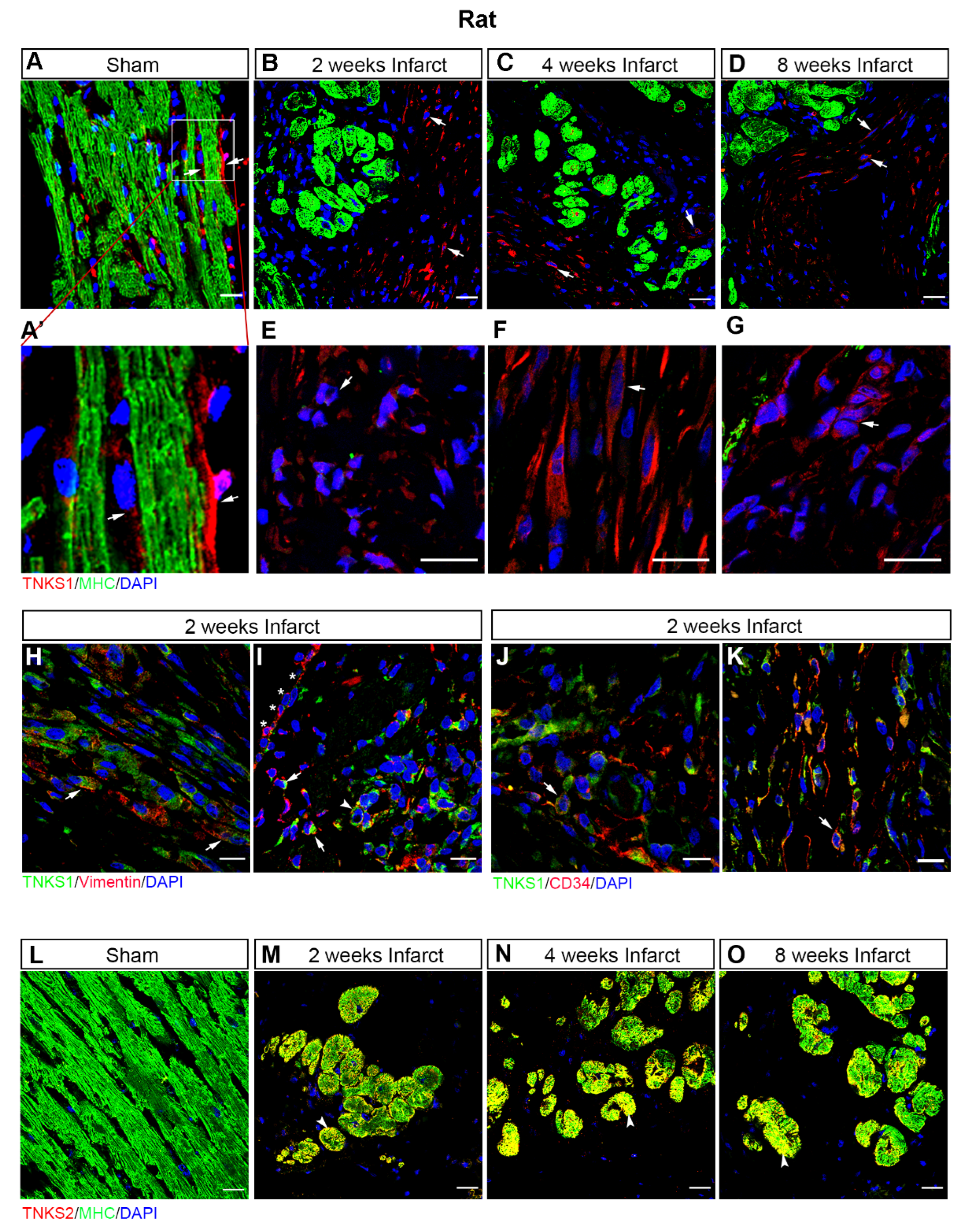
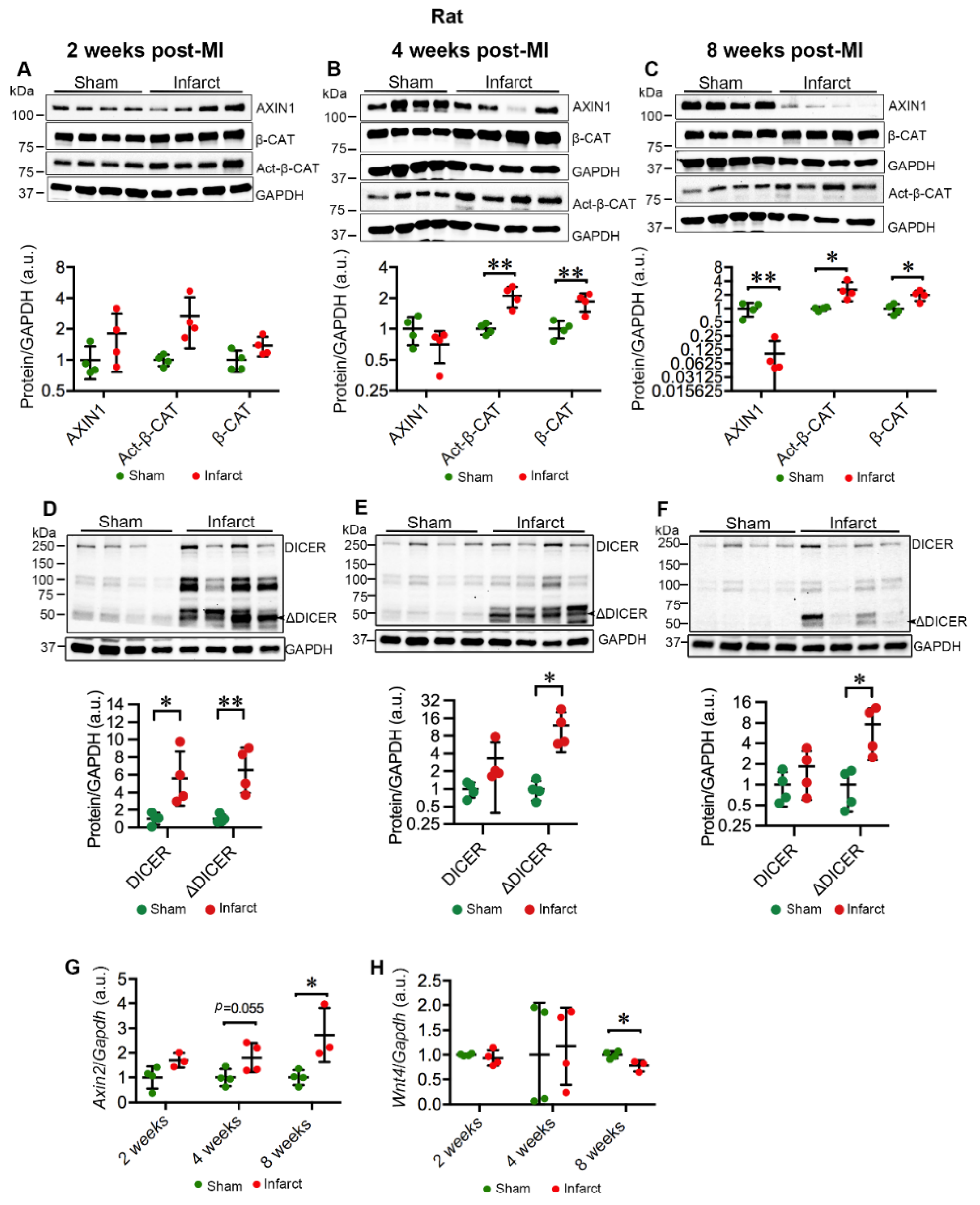
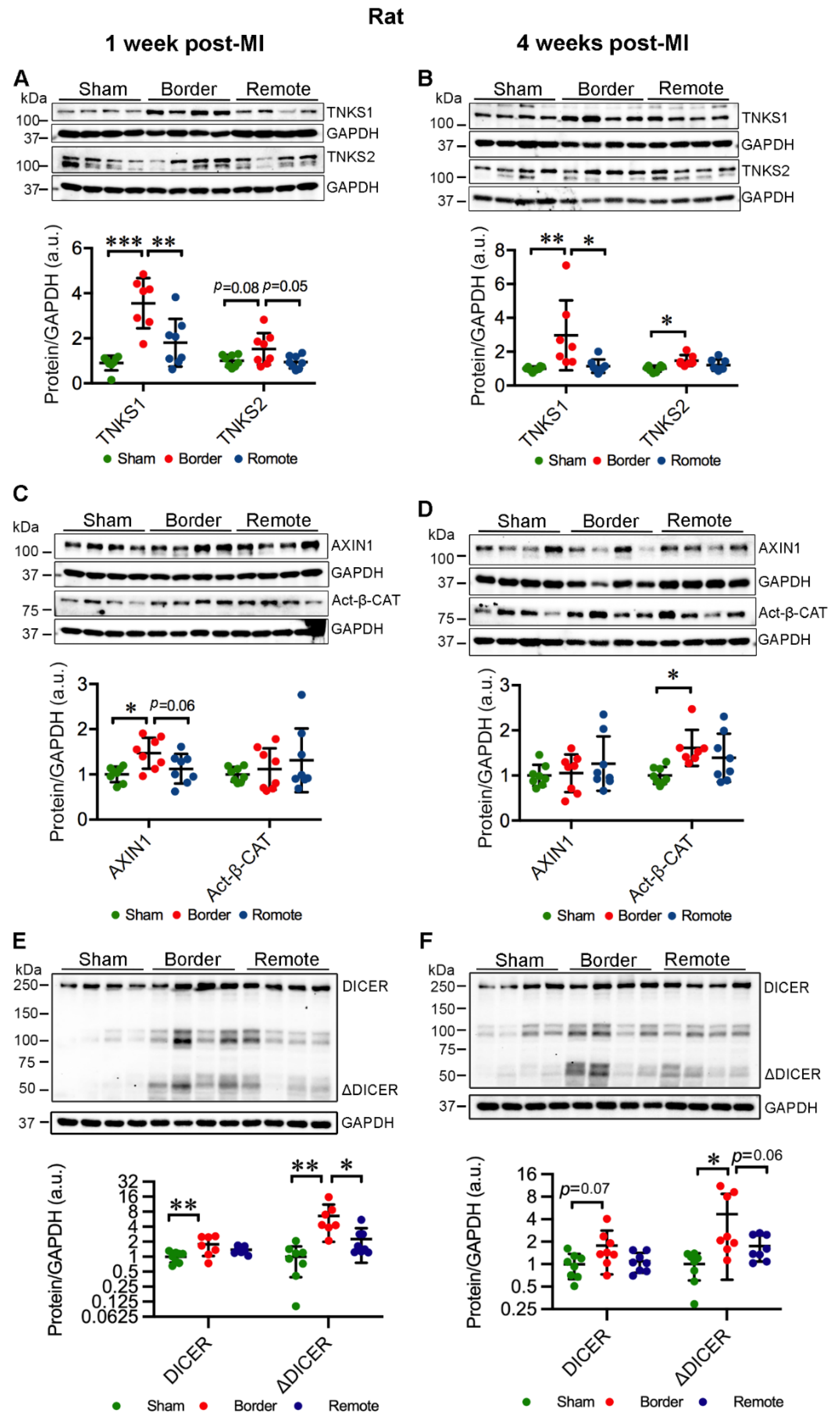
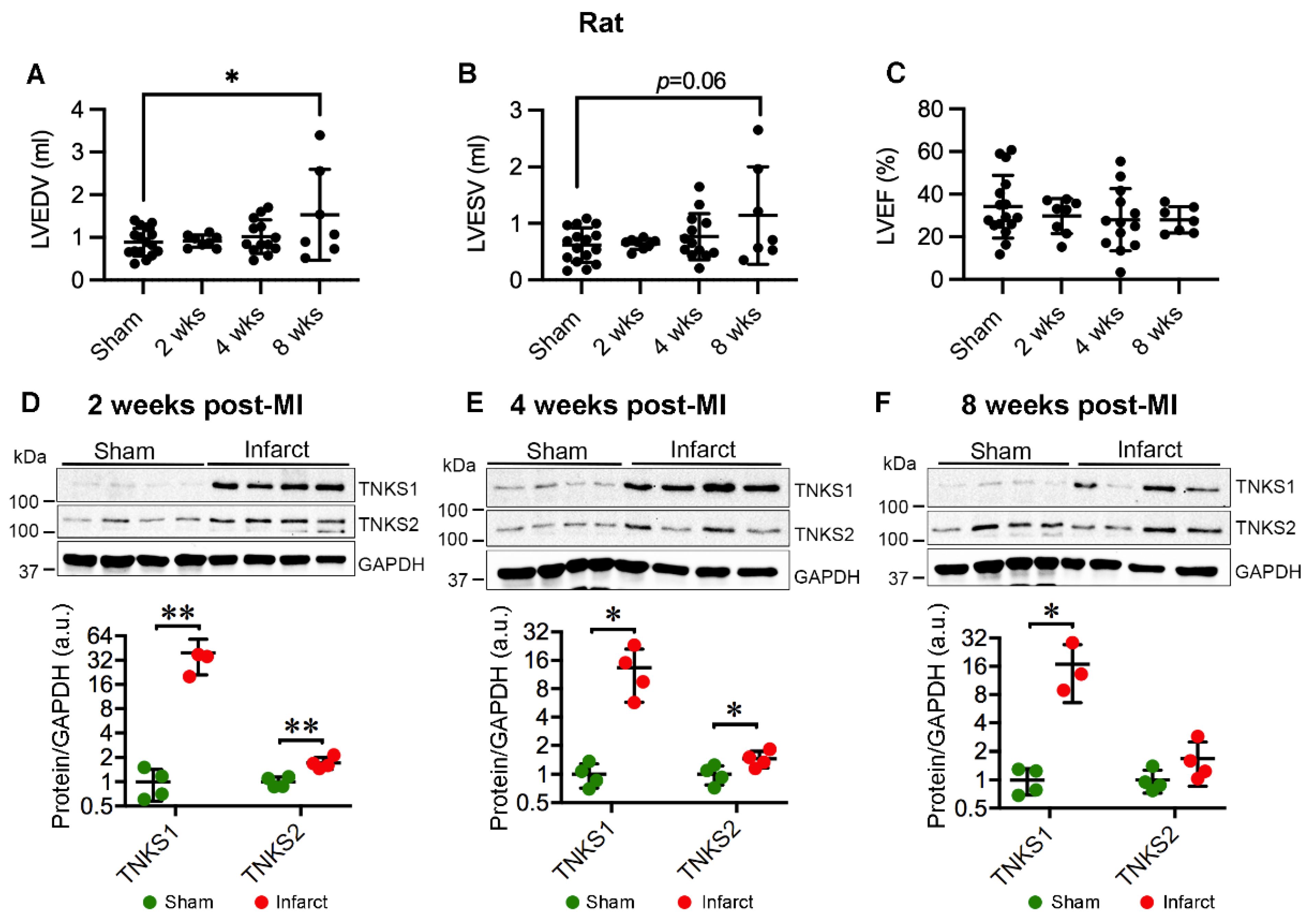
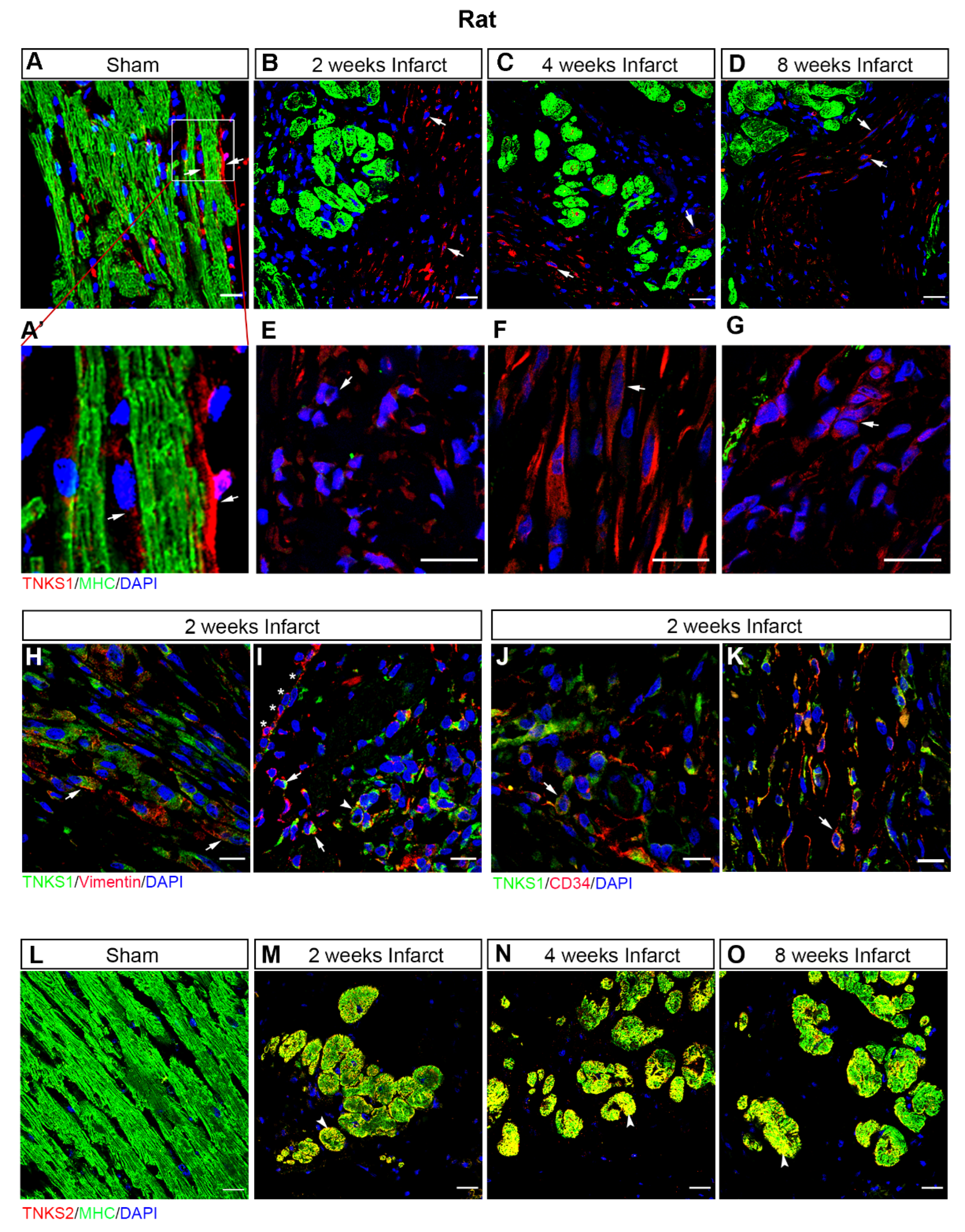
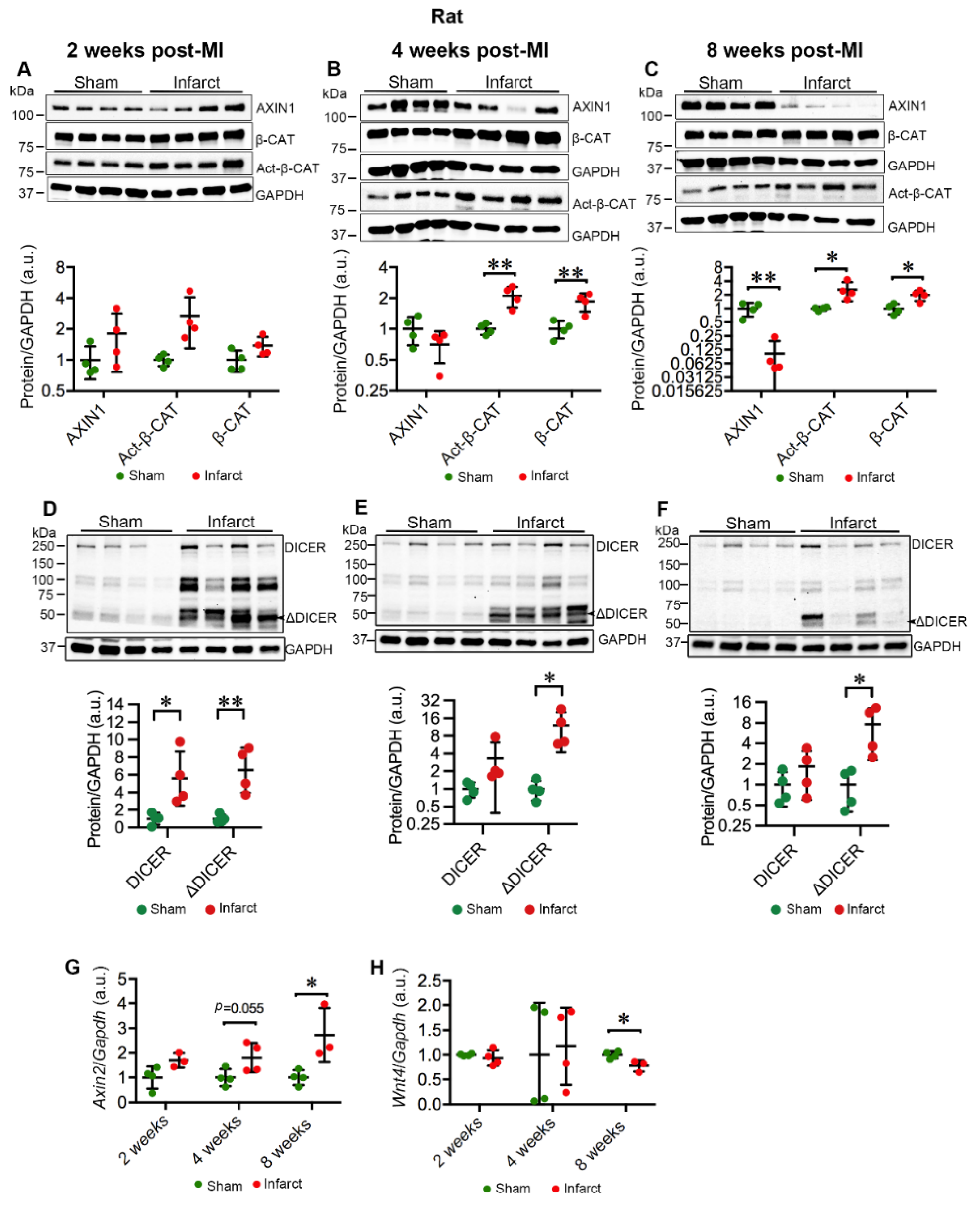
2.3. MI Stimulates the Expression and Activation of TNKSs in Rat Hearts
2.4. MI Activates Wnt/β-Catenin Signaling in Rat Hearts
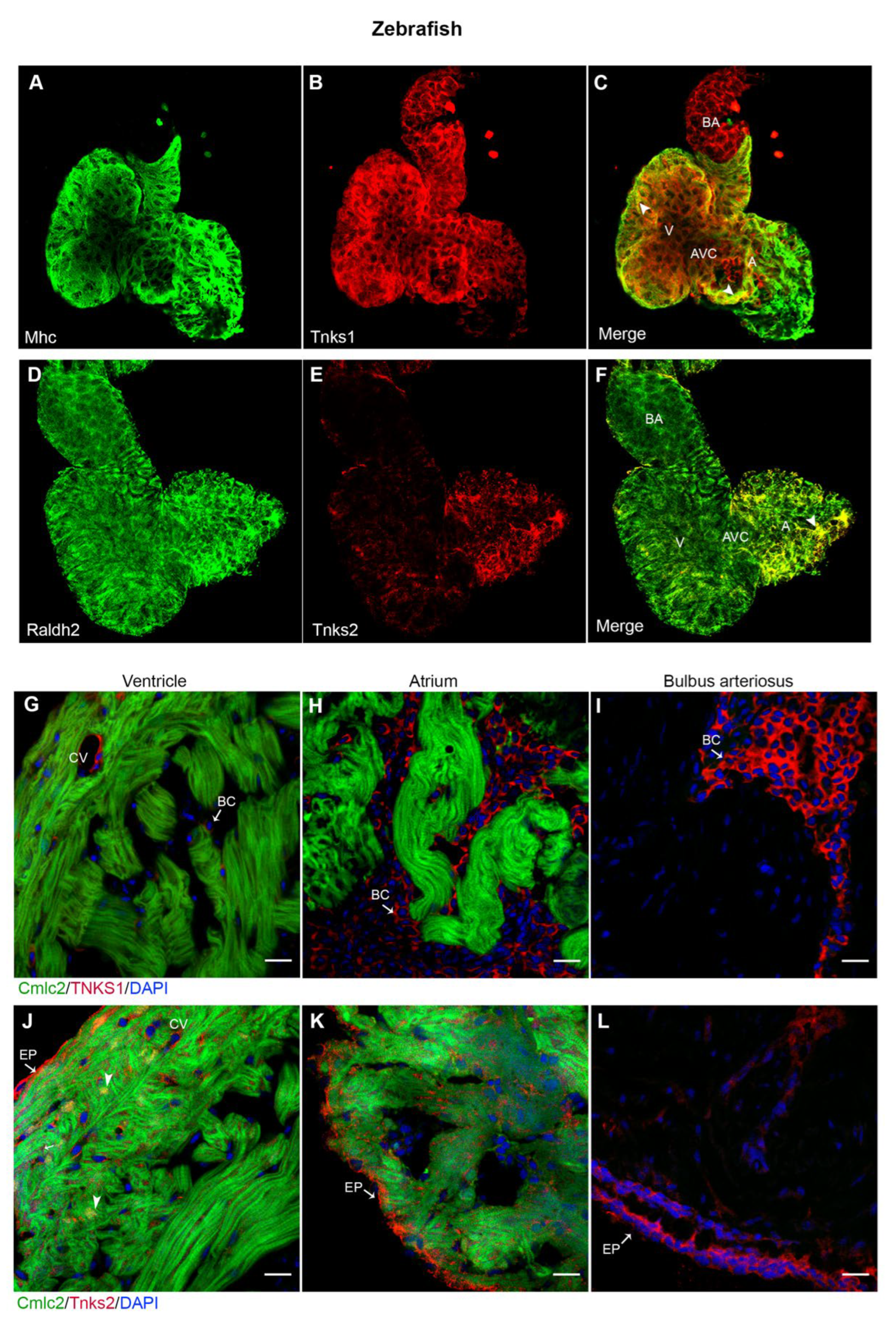
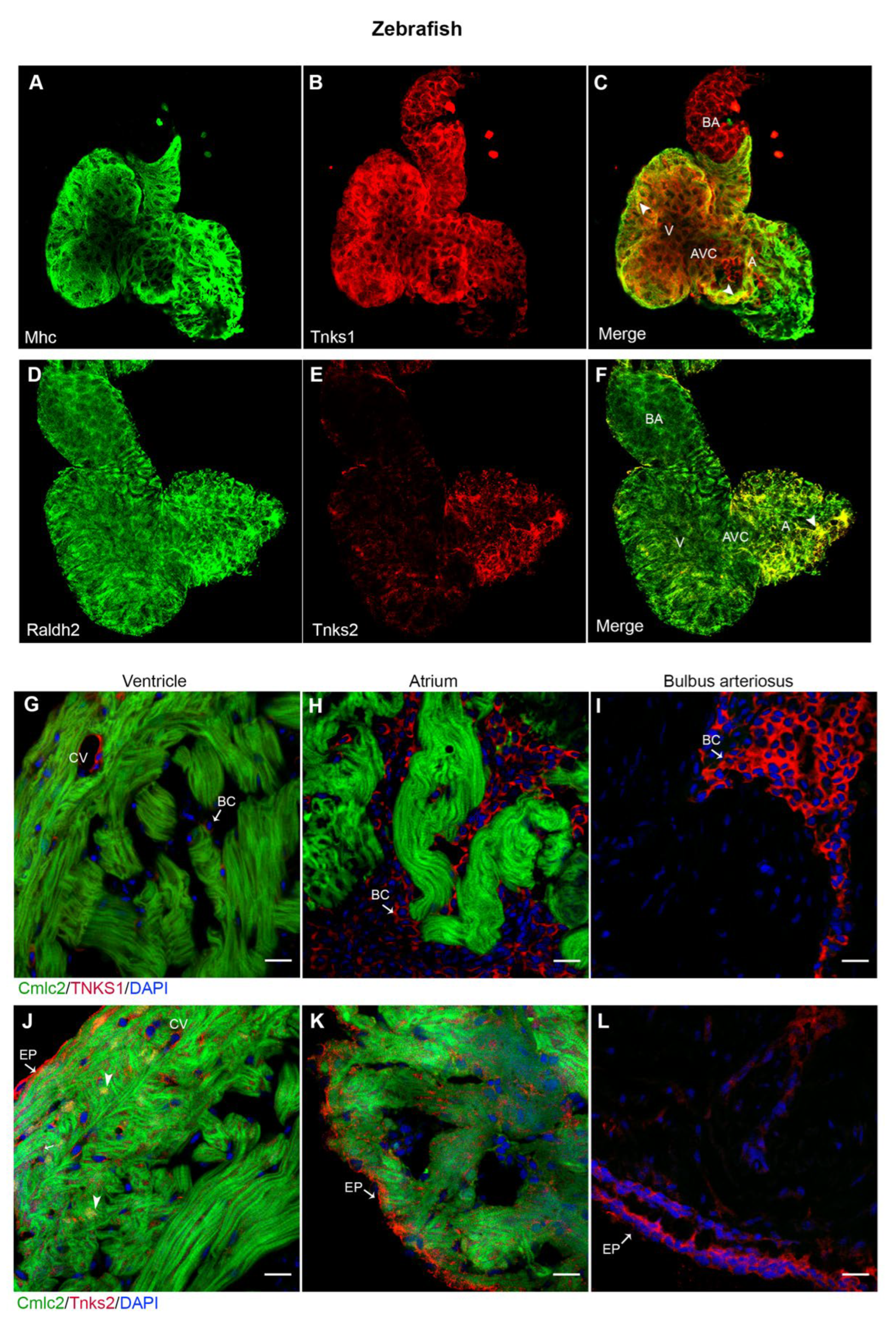
2.5. Tnks1 and Tnks2 Display Distinct Expression Pattern in Zebrafish Developing and Adult Heart
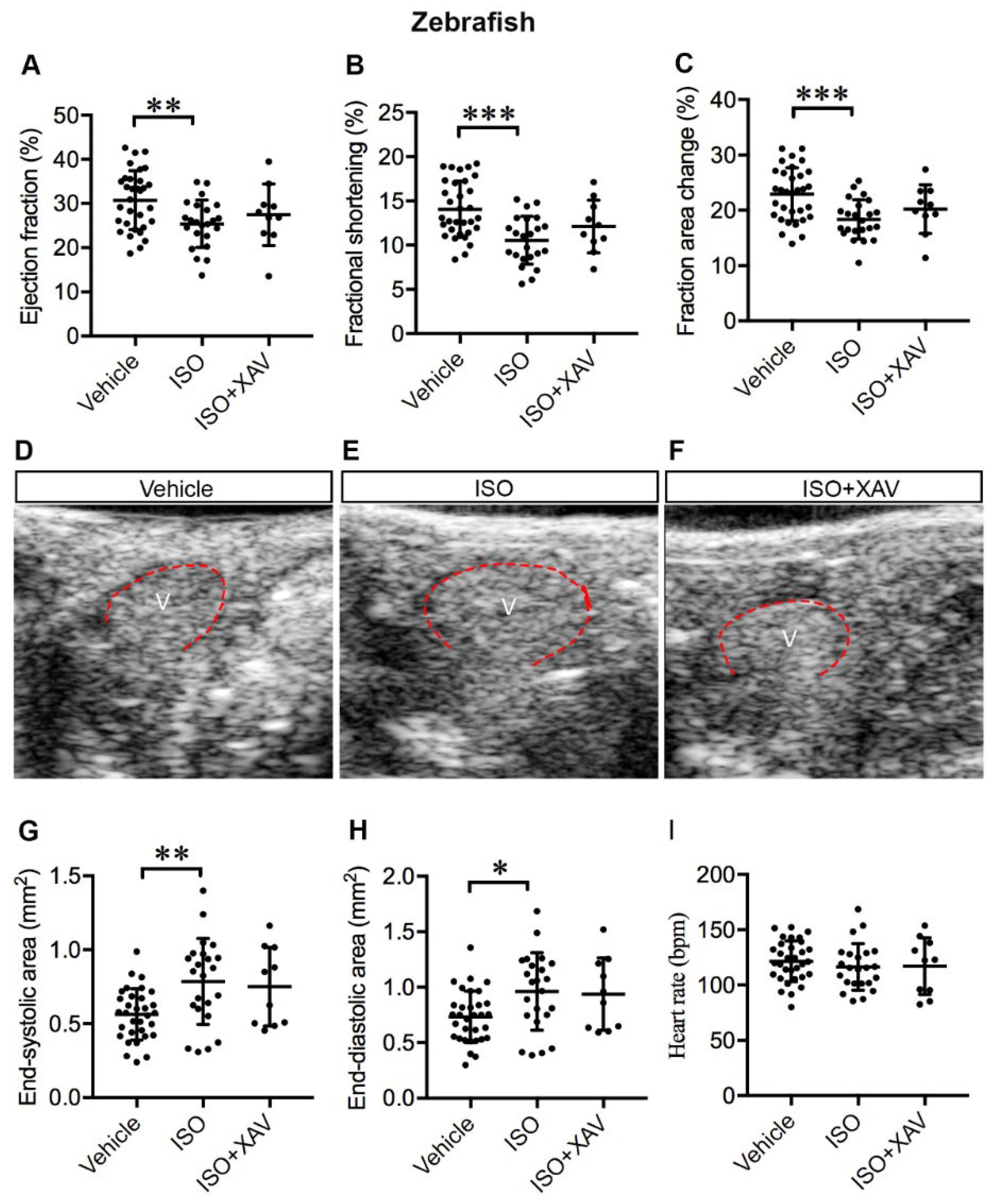
2.6. Tnks Inhibition Protects against ISO-Induced Cardiac Dysfunction in Adult Zebrafish
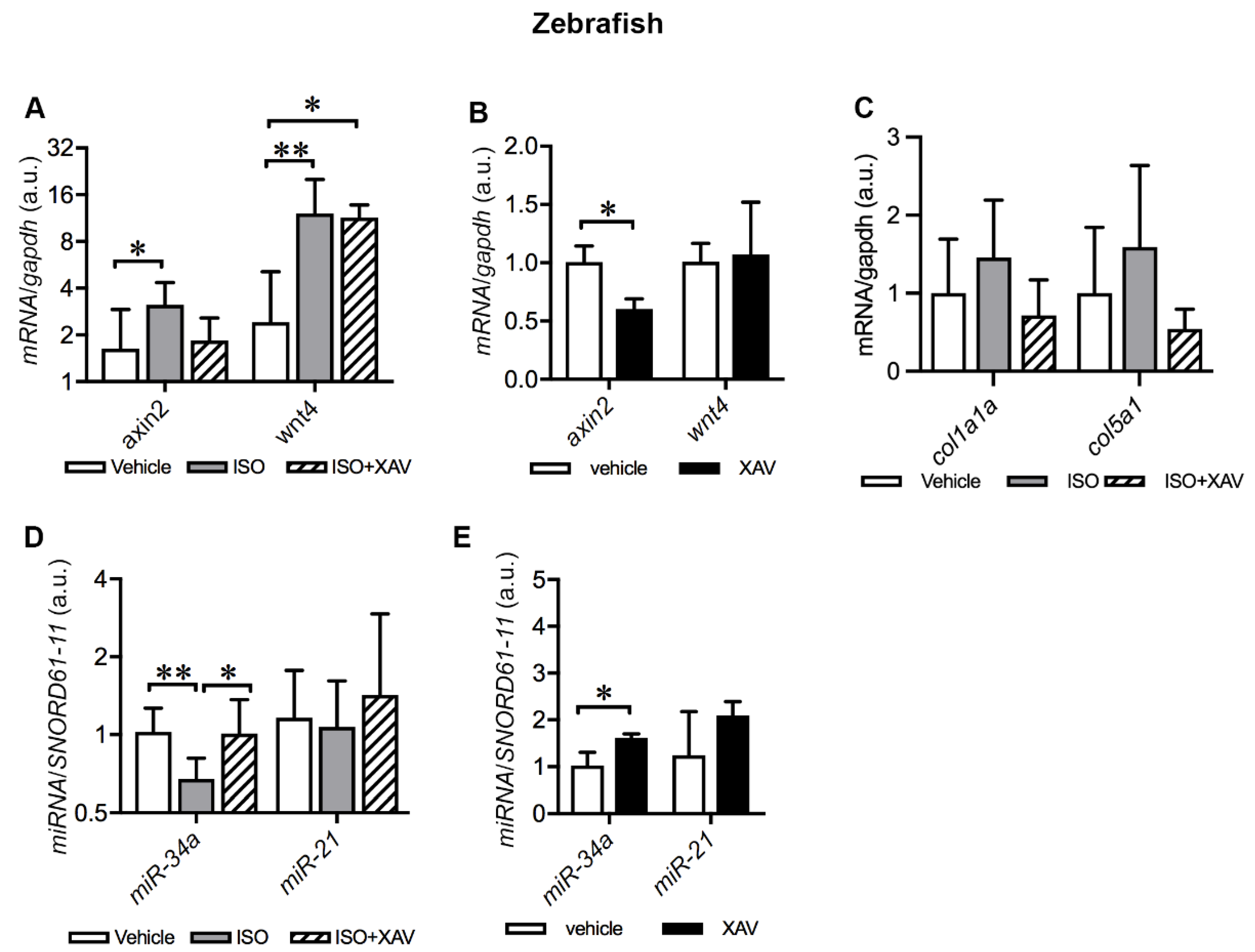
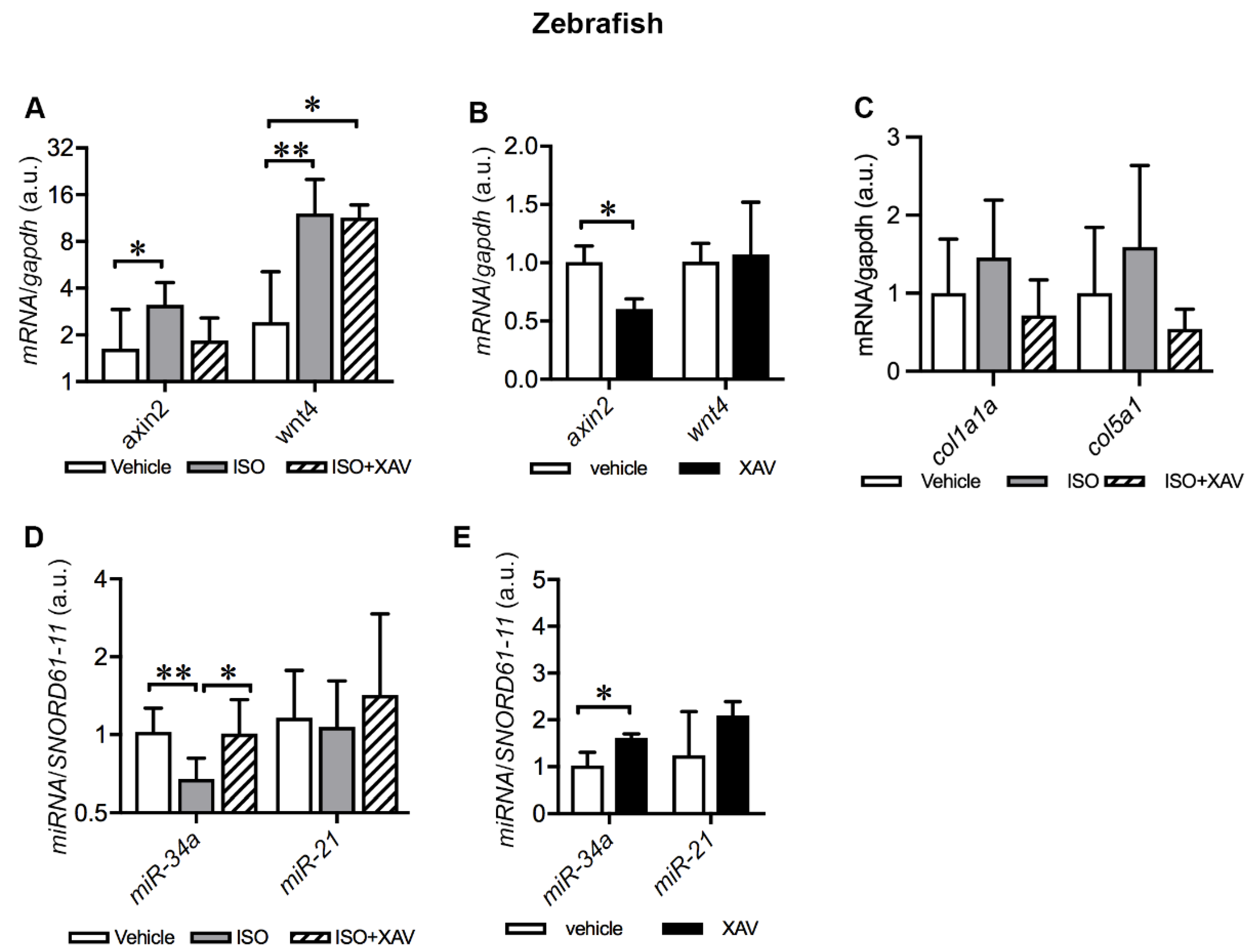
2.7. Tnks Inhibition Abrogates ISO-Induced Activation of Wnt Signaling in Adult Zebrafish Hearts
2.8. Tnks Inhibition Prevents ISO-Induced Dysregulation of miRNAs in Adult Zebrafish Hearts
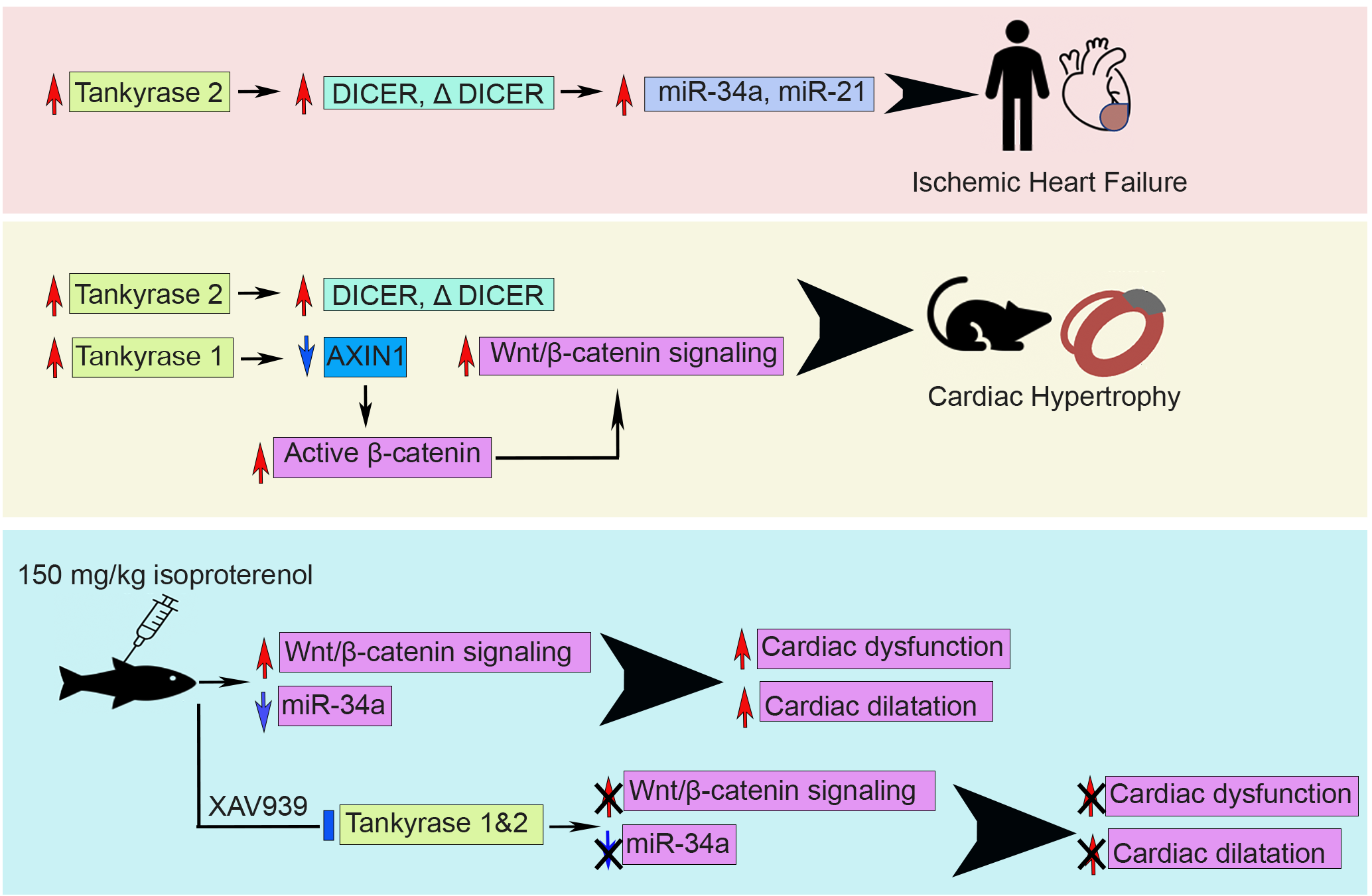
3. Discussion
4. Materials and Methods
4.1. Human IHF Patients
4.2. Rat MI Model
4.3. Zebrafish IHF Model and TNKS Inhibitor Treatment
4.4. Echocardiography
4.5. Western Blotting
4.6. RNA Extraction and Real-Time Quantitative RT-PCR
4.7. Immunohistochemistry
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Dwyer-Lindgren, L.; Bertozzi-Villa, A.; Stubbs, R.W.; Morozoff, C.; Naghavi, M.; Mokdad, A.H.; Murray, C.J.L. Trends and Patterns of Geographic Variation in Cardiovascular Mortality Among US Counties, 1980–2014. JAMA 2017, 317, 1976–1992. [Google Scholar] [CrossRef]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic Approaches for Cardiac Regeneration and Repair. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Dimmeler, S. MicroRNAs in Myocardial Infarction. Nat. Rev. Cardiol. 2015, 12, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. Mir-17-92 Cluster Is Required for and Sufficient to Induce Cardiomyocyte Proliferation in Postnatal and Adult Hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Dal Ferro, M.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional Screening Identifies MiRNAs Inducing Cardiac Regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Vergani-Junior, C.A.; Tonon-da-Silva, G.; Inan, M.D.; Mori, M.A. DICER: Structure, Function, and Regulation. Biophys. Rev. 2021, 13, 1081–1090. [Google Scholar] [CrossRef]
- Basavarajappa, D.; Uebbing, S.; Kreiss, M.; Lukic, A.; Suess, B.; Steinhilber, D.; Samuelsson, B.; Radmark, O. Dicer Up-Regulation by Inhibition of Specific Proteolysis in Differentiating Monocytic Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 8573–8583. [Google Scholar] [CrossRef]
- Gross, T.J.; Powers, L.S.; Boudreau, R.L.; Brink, B.; Reisetter, A.; Goel, K.; Gerke, A.K.; Hassan, I.H.; Monick, M.M. A MicroRNA Processing Defect in Smokers’ Macrophages Is Linked to SUMOylation of the Endonuclease DICER. J. Biol. Chem. 2014, 289, 12823–12834. [Google Scholar] [CrossRef]
- Drake, M.; Furuta, T.; Man, K.S.; Gonzalez, G.; Liu, B.; Kalia, A.; Ladbury, J.; Fire, A.Z.; Skeath, J.B.; Arur, S. A Requirement for ERK Dependent Dicer Phosphorylation in Coordinating Oocyte-to-Embryo Transition in Caenorhabditis Elegans. Dev. Cell 2014, 31, 614–628. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.K.; Vyas, S.; Rood, J.E.; Bhutkar, A.; Sharp, P.A.; Chang, P. Poly(ADP-Ribose) Regulates Stress Responses and MicroRNA Activity in the Cytoplasm. Mol. Cell 2011, 42, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Ajima, R.; Bisson, J.A.; Helt, J.C.; Nakaya, M.A.; Habas, R.; Tessarollo, L.; He, X.; Morrisey, E.E.; Yamaguchi, T.P.; Cohen, E.D. DAAM1 and DAAM2 Are Co-Required for Myocardial Maturation and Sarcomere Assembly. Dev. Biol. 2015, 408, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Hulin, A.; Moore, V.; James, J.M.; Yutzey, K.E. Loss of Axin2 Results in Impaired Heart Valve Maturation and Subsequent Myxomatous Valve Disease. Cardiovasc. Res. 2016, 113, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/Betacatenin Injury Response Activates the Epicardium and Cardiac Fibroblasts to Promote Cardiac Repair. EMBO J. 2011, 31, 429–442. [Google Scholar] [CrossRef]
- Meyer, I.S.; Leuschner, F. The Role of Wnt Signaling in the Healing Myocardium: A Focus on Cell Specificity. Basic Res. Cardiol. 2018, 113, 44. [Google Scholar] [CrossRef]
- Oerlemans, M.I.; Goumans, M.J.; van Middelaar, B.; Clevers, H.; Doevendans, P.A.; Sluijter, J.P. Active Wnt Signaling in Response to Cardiac Injury. Basic Res. Cardiol. 2010, 105, 631–641. [Google Scholar] [CrossRef]
- Daskalopoulos, E.P.; Blankesteijn, W.M. Effect of Interventions in WNT Signaling on Healing of Cardiac Injury: A Systematic Review. Cells 2021, 10, 207. [Google Scholar] [CrossRef]
- Bertozzi, A.; Wu, C.-C.; Hans, S.; Brand, M.; Weidinger, G. Wnt/β-Catenin Signaling Acts Cell-Autonomously to Promote Cardiomyocyte Regeneration in the Zebrafish Heart. Dev. Biol. 2022, 481, 226–237. [Google Scholar] [CrossRef]
- Gammons, M.; Bienz, M. Multiprotein Complexes Governing Wnt Signal Transduction. Curr. Opin. Cell Biol. 2017, 51, 42–49. [Google Scholar] [CrossRef]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase Inhibition Stabilizes Axin and Antagonizes Wnt Signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Yang, Y.; Ueberheide, B.; Smith, S. Whole Proteome Analysis of Human Tankyrase Knockout Cells Reveals Targets of Tankyrase-Mediated Degradation. Nat. Commun. 2017, 8, 2214. [Google Scholar] [CrossRef] [PubMed]
- Zee, R.Y.; Ridker, P.M.; Chasman, D.I. Genetic Variants in Eleven Telomere-Associated Genes and the Risk of Incident Cardio/Cerebrovascular Disease: The Women’s Genome Health Study. Clin. Chim. Acta 2011, 412, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Sung, Y.J.; Winkler, T.W.; de Las Fuentes, L.; Bentley, A.R.; Brown, M.R.; Kraja, A.T.; Schwander, K.; Ntalla, I.; Guo, X.; Franceschini, N.; et al. A Large-Scale Multi-Ancestry Genome-Wide Study Accounting for Smoking Behavior Identifies Multiple Significant Loci for Blood Pressure. Am. J. Hum. Genet. 2018, 102, 375–400. [Google Scholar] [CrossRef] [PubMed]
- Spoelstra, N.S.; Cittelly, D.M.; Christenson, J.L.; Gordon, M.A.; Elias, A.; Jedlicka, P.; Richer, J.K. Dicer Expression in Estrogen Receptor Positive versus Triple-Negative Breast Cancer: An Antibody Comparison. Hum. Pathol. 2016, 56, 40–51. [Google Scholar] [CrossRef]
- Segersvärd, H.; Lakkisto, P.; Hänninen, M.; Forsten, H.; Siren, J.; Immonen, K.; Kosonen, R.; Sarparanta, M.; Laine, M.; Tikkanen, I. Carbon Monoxide Releasing Molecule Improves Structural and Functional Cardiac Recovery after Myocardial Injury. Eur. J. Pharmacol. 2018, 818, 57–66. [Google Scholar] [CrossRef]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindstrom, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin Coordinates Fibroblast Proliferation and Keratinocyte Differentiation in Wound Healing via TGF-Beta-Slug Signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef]
- Villalobos, E.; Criollo, A.; Schiattarella, G.G.; Altamirano, F.; French, K.M.; May, H.I.; Jiang, N.; Nguyen, N.U.N.; Romero, D.; Roa, J.C.; et al. Fibroblast Primary Cilia Are Required for Cardiac Fibrosis. Circulation 2019, 139, 2342–2357. [Google Scholar] [CrossRef]
- Samman Tahhan, A.; Hammadah, M.; Kelli, H.M.; Kim, J.H.; Sandesara, P.B.; Alkhoder, A.; Kaseer, B.; Gafeer, M.M.; Topel, M.; Hayek, S.S.; et al. Circulating Progenitor Cells and Racial Differences. Circ. Res. 2018, 123, 467–476. [Google Scholar] [CrossRef]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental Myocardial Infarction Triggers Canonical Wnt Signaling and Endothelial-to-Mesenchymal Transition. Dis. Model. Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, M.; Wu, J.C.; Nusse, R. Fibrosis of the Neonatal Mouse Heart After Cryoinjury Is Accompanied by Wnt Signaling Activation and Epicardial-to-Mesenchymal Transition. J. Am. Heart Assoc. 2016, 5, e002457. [Google Scholar] [CrossRef]
- Brown, D.R.; Samsa, L.A.; Qian, L.; Liu, J. Advances in the Study of Heart Development and Disease Using Zebrafish. J. Cardiovasc. Dev. Dis. 2016, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Elsner, D.; Schunkert, H.; Pfeifer, M.; Griese, D.; Bruckschlegel, G.; Muders, F.; Riegger, G.A.; Kromer, E.P. Development of Heart Failure Following Isoproterenol Administration in the Rat: Role of the Renin-Angiotensin System. Cardiovasc. Res. 1998, 37, 91–100. [Google Scholar] [CrossRef]
- Heather, L.C.; Catchpole, A.F.; Stuckey, D.J.; Cole, M.A.; Carr, C.A.; Clarke, K. Isoproterenol Induces in Vivo Functional and Metabolic Abnormalities: Similar to Those Found in the Infarcted Rat Heart. J. Physiol. Pharmacol. 2009, 60, 31–39. [Google Scholar] [PubMed]
- Sutton, M.G.S.J.; Sharpe, N. Left Ventricular Remodeling After Myocardial Infarction. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.M.; Gorman, J.H.; Moainie, S.L.; Guy, T.S.; Narula, N.; Narula, J.; St. John-Sutton, M.G.; Edmunds, L.H.; Gorman, R.C. Extension of Borderzone Myocardium in Postinfarction Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 1160–1167. [Google Scholar] [CrossRef]
- Pillai, J.B.; Isbatan, A.; Imai, S.; Gupta, M.P. Poly(ADP-Ribose) Polymerase-1-Dependent Cardiac Myocyte Cell Death during Heart Failure Is Mediated by NAD+ Depletion and Reduced Sir2α Deacetylase Activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef]
- Pillai, J.B.; Russell, H.M.; Raman, J.; Jeevanandam, V.; Gupta, M.P. Increased Expression of Poly(ADP-Ribose) Polymerase-1 Contributes to Caspase-Independent Myocyte Cell Death during Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H486–H496. [Google Scholar] [CrossRef]
- Molnár, A.; Tóth, A.; Bagi, Z.; Papp, Z.; Édes, I.; Vaszily, M.; Galajda, Z.; Papp, J.G.; Varró, A.; Szüts, V.; et al. Activation of the Poly(ADP-Ribose) Polymerase Pathway in Human Heart Failure. Mol. Med. 2006, 12, 143–152. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Stone, N.R.; Berry, E.C.; Radzinsky, E.; Huang, Y.; Pratt, K.; Ang, Y.S.; Yu, P.; Wang, H.; Tang, S.; et al. Chemical Enhancement of In Vitro and In Vivo Direct Cardiac Reprogramming. Circulation 2017, 135, 978–995. [Google Scholar] [CrossRef]
- Wang, M.; Hu, B.; Zhang, Y.L.; Shen, E.; Pan, X.Q. Effects of 3-Aminobenzamide on Ventricular Function in Infarct Heart Assessed by Quantitative Tissue Velocity Imaging. J. Cardiovasc. Med. 2016, 17, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Bartha, E.; Kiss, G.N.; Kalman, E.; Kulcsar, G.; Kalai, T.; Hideg, K.; Habon, T.; Sumegi, B.; Toth, K.; Halmosi, R. Effect of L-2286, a Poly(ADP-Ribose)Polymerase Inhibitor and Enalapril on Myocardial Remodeling and Heart Failure. J. Cardiovasc. Pharmacol. 2018, 52, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wahlberg, E.; Karlberg, T.; Kouznetsova, E.; Markova, N.; Macchiarulo, A.; Thorsell, A.G.; Pol, E.; Frostell, A.; Ekblad, T.; Oncu, D.; et al. Family-Wide Chemical Profiling and Structural Analysis of PARP and Tankyrase Inhibitors. Nat. Biotechnol. 2012, 30, 283–288. [Google Scholar] [CrossRef]
- Foulquier, S.; Daskalopoulos, E.P.; Lluri, G.; Hermans, K.C.M.; Deb, A.; Blankesteijn, W.M. WNT Signaling in Cardiac and Vascular Disease. Pharmacol. Rev. 2017, 70, 68–141. [Google Scholar] [CrossRef] [PubMed]
- Murakoshi, M.; Saiki, K.; Urayama, K.; Sato, T.N. An Anthelmintic Drug, Pyrvinium Pamoate, Thwarts Fibrosis and Ameliorates Myocardial Contractile Dysfunction in a Mouse Model of Myocardial Infarction. PLoS ONE 2013, 8, e79374. [Google Scholar] [CrossRef]
- Yang, Y.; Cheng, H.W.; Qiu, Y.; Dupee, D.; Noonan, M.; Lin, Y.D.; Fisch, S.; Unno, K.; Sereti, K.I.; Liao, R. MicroRNA-34a Plays a Key Role in Cardiac Repair and Regeneration Following Myocardial Infarction. Circ. Res. 2015, 117, 450–459. [Google Scholar] [CrossRef]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Treguer, K.; Carmona, G.; Bonauer, A.; et al. MicroRNA-34a Regulates Cardiac Ageing and Function. Nature 2013, 495, 107–110. [Google Scholar] [CrossRef]
- Lal, A.; Thomas, M.P.; Altschuler, G.; Navarro, F.; O’Day, E.; Li, X.L.; Concepcion, C.; Han, Y.C.; Thiery, J.; Rajani, D.K.; et al. Capture of MicroRNA-Bound MRNAs Identifies the Tumor Suppressor MiR-34a as a Regulator of Growth Factor Signaling. PLoS Genet. 2011, 7, e1002363. [Google Scholar] [CrossRef]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the Human Heart: A Clue to Fetal Gene Reprogramming in Heart Failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef]
- Adam, O.; Lohfelm, B.; Thum, T.; Gupta, S.K.; Puhl, S.L.; Schafers, H.J.; Bohm, M.; Laufs, U. Role of MiR-21 in the Pathogenesis of Atrial Fibrosis. Basic Res. Cardiol. 2012, 107, 278. [Google Scholar] [CrossRef]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 Contributes to Myocardial Disease by Stimulating MAP Kinase Signalling in Fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Hinkel, R.; Ramanujam, D.; Kaczmarek, V.; Howe, A.; Klett, K.; Beck, C.; Dueck, A.; Thum, T.; Laugwitz, K.L.; Maegdefessel, L.; et al. AntimiR-21 Prevents Myocardial Dysfunction in a Pig Model of Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2020, 75, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Lakkisto, P.; Siren, J.-M.; Kytö, V.; Forsten, H.; Laine, M.; Pulkki, K.; Tikkanen, I. Heme Oxygenase-1 Induction Protects the Heart and Modulates Cellular and Extracellular Remodelling after Myocardial Infarction in Rats. Exp. Biol. Med. 2011, 236, 1437–1448. [Google Scholar] [CrossRef]
- Karlberg, T.; Markova, N.; Johansson, I.; Hammarstrom, M.; Schutz, P.; Weigelt, J.; Schuler, H. Structural Basis for the Interaction between Tankyrase-2 and a Potent Wnt-Signaling Inhibitor. J. Med. Chem. 2010, 53, 5352–5355. [Google Scholar] [CrossRef]
- Darbandi Azar, A.; Tavakoli, F.; Moladoust, H.; Zare, A.; Sadeghpour, A. Echocardiographic Evaluation of Cardiac Function in Ischemic Rats: Value of m-Mode Echocardiography. Res. Cardiovasc. Med. 2014, 3, e22941. [Google Scholar] [CrossRef]
- Wang, L.W.; Huttner, I.G.; Santiago, C.F.; Kesteven, S.H.; Yu, Z.-Y.; Feneley, M.P.; Fatkin, D. Standardized Echocardiographic Assessment of Cardiac Function in Normal Adult Zebrafish and Heart Disease Models. Dis. Model. Mech. 2017, 10, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Narumanchi, S.; Kalervo, K.; Perttunen, S.; Wang, H.; Immonen, K.; Kosonen, R.; Laine, M.; Ruskoaho, H.; Tikkanen, I.; Lakkisto, P.; et al. Inhibition of Let-7c Regulates Cardiac Regeneration after Cryoinjury in Adult Zebrafish. J. Cardiovasc. Dev. Dis. 2019, 6, 16. [Google Scholar] [CrossRef]
- Peinnequin, A.; Mouret, C.; Birot, O.; Alonso, A.; Mathieu, J.; Clarencon, D.; Agay, D.; Chancerelle, Y.; Multon, E. Rat Pro-Inflammatory Cytokine and Cytokine Related MRNA Quantification by Real-Time Polymerase Chain Reaction Using SYBR Green. BMC Immunol. 2004, 5, 3. [Google Scholar] [CrossRef]
- Mukherjee, D.; Wagh, G.; Mokalled, M.H.; Kontarakis, Z.; Dickson, A.L.; Rayrikar, A.; Günther, S.; Poss, K.D.; Stainier, D.Y.R.; Patra, C. Ccn2a Is an Injury-Induced Matricellular Factor That Promotes Cardiac Regeneration in Zebrafish. Development 2021, 148, dev193219. [Google Scholar] [CrossRef]
- Burkhalter, M.D.; Sridhar, A.; Sampaio, P.; Jacinto, R.; Burczyk, M.S.; Donow, C.; Angenendt, M.; Competence Network for Congenital Heart Defects Investigators; Hempel, M.; Walther, P.; et al. Imbalanced Mitochondrial Function Provokes Heterotaxy via Aberrant Ciliogenesis. J. Clin. Investig. 2019, 129, 2841–2855. [Google Scholar] [CrossRef] [Green Version]
- Paavola, J.; Alakoski, T.; Ulvila, J.; Kilpio, T.; Siren, J.; Perttunen, S.; Narumanchi, S.; Wang, H.; Lin, R.; Porvari, K.; et al. Vezf1 Regulates Cardiac Structure and Contractile Function. EBioMedicine 2020, 51, 102608. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Durand, E.M.; Yang, S.; Zhou, Y.; Zon, L.I. A CRISPR/Cas9 Vector System for Tissue-Specific Gene Disruption in Zebrafish. Dev. Cell 2015, 32, 756–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Features | Patients |
|---|---|
| No. of patients | 8 |
| Mean age (years) | 58 |
| Sex (female/male) | 1/7 |
| Past medical history | |
| Coronary artery disease | 8 |
| Ischemic heart failure | 8 |
| Previous medication | |
| ß-blocker, n (%) | 7 (87.5) |
| Statins, n (%) | 7 (87.5) |
| Angiotensin receptor inhibitors, n (%) | 5 (62.5) |
| Angiotensin-converting enzyme inhibitors, n (%) | 3 (37.5) |
| Phosphodiesterase type 5 inhibitors, n (%) | 6 (75) |
| Diuretics, n (%) | 7 (87.5) |
| Antithrombotic therapy, n (%) | 8 (100) |
| Proton pump inhibitors, n (%) | 8 (100) |
| Calcium, magnesium, potassium intake | 7 (87.5) |
| Antibody | Host | Manufacturer | Reference |
|---|---|---|---|
| TNKS | Rabbit polyclonal | Abcam® | ab86279, RRID:AB_1925488 |
| TNKS2 | Goat polyclonal | Abcam® | ab85690, RRID:AB_1861366 |
| AXIN1 | Rabbit monoclonal | Cell Signaling Technology | #2087, RRID:AB_2274550 |
| Active β-Catenin | Mouse monoclonal | EMD Millipore | #05-665, RRID:AB_309887 |
| β-Catenin | Mouse monoclonal | Sigma-Aldrich | C7207, RRID:AB_476865 |
| DICER | Rabbit monoclonal | Abcam® | ab259327 |
| DICER | Rabbit polyclonal | Abcam® | ab227518 |
| RALDH2 | Rabbit polyclonal | Bioss Antibodies | BS-3676R, RRID:AB_10856810 |
| MHC | Mouse monoclonal | EMD Millipore | 05-716, RRID:AB_309930 |
| Vimentin | Mouse monoclonal | Lab Vision | MS-129-P1, RRID:AB_63350 |
| CD34 | Goat polyclonal | R&D system | AF4117, RRID:AB_2074613 |
| GAPDH | Rabbit polyclonal | Abcam® | Ab9485, RRID:AB_307275 |
| HRP-rabbit IgG | Goat polyclonal | Jackson ImmunoResearch | 111-035-003, RRID:AB_2313567 |
| HRP-mouse IgG | Goat polyclonal | Jackson ImmunoResearch | 115-035-044, RRID:AB_2338503 |
| HRP-goat IgG | Rabbit polyclonal | Jackson ImmunoResearch | 305-035-003, RRID:AB_2339400 |
| Alexa Fluor® 488-mouse IgG | Goat polyclonal | Invitrogen | A-11001, RRID:AB_2534069 |
| Alexa Fluor® 488-rabbit IgG | Goat polyclonal | Invitrogen | A-11008, RRID:AB_143165 |
| AlexaFluor® 594-rabbit IgG | Goat polyclonal | Invitrogen | A-11012, RRID:AB_2534079 |
| AlexaFluor® 546-goat IgG | Donkey polyclonal | Invitrogen | A-11056, RRID:AB_2534103 |
| Target | Species | Primer Sequences | Reference |
|---|---|---|---|
| Axin2 | Rattus norvegicus | Forward 5′-CAGCAAAACTCTCCGGGCCA-3′ Reverse 5′-GCGTCGCTGGATAACTCGCT-3′ | This study |
| Wnt4 | Rattus norvegicus | Forward 5′-GGACAGTACACGGGGTCAGC-3′ Reverse 5′-CCTGCCAGCCTCGTTGTTGT-3′ | This study |
| Gapdh | Rattus norvegicus | Forward 5′-TCTTGTGCAGTGCCAGCCTC-3′ Reverse 5′-CAAGAGAAGGCAGCCCTGGT-3′ | This study |
| CypA | Rattus norvegicus | Forward 5′-TATCTGCACTGCCAAGACTGAGTG-3′ Reverse 5′-CTTCTTGCTGGTCTTGCCATTCC-3′ | [58] |
| axin2 | Danio rerio | Forward 5′-CAGAAGTGGCCTTGGGGCAT-3′ Reverse 5′-TGGCAGCTGGAGGAGACTGT-3′ | This study |
| wnt4 | Danio rerio | Forward 5′-GCCATCGACGAGTGCCAGTA-3′ Reverse 5′-ATGCAGCTTCCCTCGTACCTTG-3′ | This study |
| col1a1a | Danio rerio | Forward 5′-TATTGG TGG TCA GCGTGGTA-3′ Reverse 5′-TCCTGG AGT ACC CTCACGAC-3′ | [59] |
| col5a1 | Danio rerio | Forward 5′-GATCCCAACCAGGGCTGCTC-3′ Reverse 5′-GGAGGTGAGTCTGGCCCCTT-3′ | This study |
| Gapdh | Danio rerio | Forward 5′-CAGGCATAATGGTTAAAGTTGGTA-3′ Reverse 5′-CATGTAATCAAGGTCAATGAATGG-3′ | [60] |
| ef1α1a | Danio rerio | Forward 5′-GAGACGCGGCCATTGTGGAA-3′ Reverse 5′-ACCGTCTGACGCATGTCACG-3′ | This study |
| miR-34a-5p | Homo sapiens | 5′-UGGCAGUGUCUUAGCUGGUUGU-3′ | Qiagen:MS00003318 |
| miR-21-5p | Homo sapiens | 5′-UAGCUUAUCAGACUGAUGUUGA-3′ | Qiagen:MS00009079 |
| miR-181a-5p | Homo sapiens | 5′-AACAUUCAACGCUGUCGGUGAGU-3′ | Qiagen:MS00008827 |
| miR-208a-3p | Homo sapiens | 5′-AUAAGACGAGCAAAAAGCUUGU-3′ | Qiagen:MS00003794 |
| miR-133a-3p | Homo sapiens | 5′-UUUGGUCCCCUUCAACCAGCUG-3′ | Qiagen:MS00031423 |
| miR-19b-3p | Homo sapiens | 5′-UGUGCAAAUCCAUGCAAAACUGA-3′ | Qiagen:MS00031584 |
| RNU6-2_11 | Homo sapiens | - | Qiagen:MS00033740 |
| SNORD61_11 | Homo sapiens | - | Qiagen:MS00033705 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Segersvärd, H.; Siren, J.; Perttunen, S.; Immonen, K.; Kosonen, R.; Chen, Y.-C.; Tolva, J.; Laivuori, M.; Mäyränpää, M.I.; et al. Tankyrase Inhibition Attenuates Cardiac Dilatation and Dysfunction in Ischemic Heart Failure. Int. J. Mol. Sci. 2022, 23, 10059. https://doi.org/10.3390/ijms231710059
Wang H, Segersvärd H, Siren J, Perttunen S, Immonen K, Kosonen R, Chen Y-C, Tolva J, Laivuori M, Mäyränpää MI, et al. Tankyrase Inhibition Attenuates Cardiac Dilatation and Dysfunction in Ischemic Heart Failure. International Journal of Molecular Sciences. 2022; 23(17):10059. https://doi.org/10.3390/ijms231710059
Chicago/Turabian StyleWang, Hong, Heli Segersvärd, Juuso Siren, Sanni Perttunen, Katariina Immonen, Riikka Kosonen, Yu-Chia Chen, Johanna Tolva, Mirjami Laivuori, Mikko I. Mäyränpää, and et al. 2022. "Tankyrase Inhibition Attenuates Cardiac Dilatation and Dysfunction in Ischemic Heart Failure" International Journal of Molecular Sciences 23, no. 17: 10059. https://doi.org/10.3390/ijms231710059
APA StyleWang, H., Segersvärd, H., Siren, J., Perttunen, S., Immonen, K., Kosonen, R., Chen, Y.-C., Tolva, J., Laivuori, M., Mäyränpää, M. I., Kovanen, P. T., Sinisalo, J., Laine, M., Tikkanen, I., & Lakkisto, P. (2022). Tankyrase Inhibition Attenuates Cardiac Dilatation and Dysfunction in Ischemic Heart Failure. International Journal of Molecular Sciences, 23(17), 10059. https://doi.org/10.3390/ijms231710059